

Abstract
Cathepsin K is a cysteine peptidase primarily located in osteoclasts, cells involved in normal growth and remodeling of bone but that are also responsible for bone loss in osteolytic diseases such as osteoporosis. In vivo imaging of cathepsin K may provide a method to assess changes in osteoclast numbers in such disease states. To that end, two high-affinity and selective cathepsin K inhibitors were radiolabeled with carbon-11. In vivo microPET imaging studies demonstrated uptake and prolonged retention of radioactivity in actively growing or remodeling bone regions (e.g., distal ulnar, carpal, distal and proximal humeral, distal femur, proximal tibia, tail vertebrae). Uptake into bone could be blocked by pre- or co-injection of unlabeled ligand, supporting a specific and saturable binding mechanism for radiotracer localization. These proof-of-concept studies indicate that radiolabeled cathepsin K inhibitors may have potential as in vivo imaging radiotracers for assessing changes of osteoclast numbers in osteolytic diseases.
Keywords: Bone, Osteoclast, Cathepsin
1. Introduction
Cathepsin K is one of a family of cysteine peptidases with a primary physiological function of cleavage of type I and type II collagen [1]. The enzyme is highly expressed in osteoclasts, multinucleated cells that are principally involved in resorption of bone in normal and pathological conditions. Deficiencies of cathepsin K cause a rare genetic disease, pycnodysotosis, whereas excessive function of osteoclasts (and expression of cathepsin K) produces bone loss and such diseases as osteoporosis [2]. The proteolytic activity of cathepsin K has also been demonstrated in numerous other diseases, including examples in oncology [3,4] and cardiovascular disease [5].
A very large number of inhibitors of cathepsin K have now been synthesized and evaluated both in vitro and in vivo, with several compounds in current clinical trials [6]. In addition to the therapeutic potential of cathepsin K inhibitors, the high expression of the enzyme in osteoclasts may also provide a unique method for non-invasive imaging of biochemical changes in diseases involving up- or down-regulation of osteoclast numbers and/or activity. The potential of targeting cathepsin K was demonstrated both in vitro and in vivo using a near-infrared reporter probe attached to a cathepsin K activatable substrate [7], which provided an imaging method based on enzymatic activity.
Current clinical tools for non-invasive imaging of skeletal diseases include anatomical imaging methods, such as radiography, dual-energy x-ray absorptiometry, MRI and CT, but these methods lack sensitivity for detecting early biochemical or cellular changes that occur before significant bone losses. The standard nuclear medicine imaging method using γ-camera imaging of 99mTc-methylene diphosphonate (MDP) requires incorporation of the radiopharmaceutical into the bone via the action of osteoblasts, and is poor at detecting osteolytic lesions. Positron emission tomography (PET) imaging of [18F]fluoride ion incorporation into bone has developed as a potentially better imaging method than traditional MDP bone scans [8], but the mechanism of incorporation of the radiotracer is non-specific and provides no biochemical information relating to cellular activities (osteoclasts or osteoblasts). Recently, research efforts into the development of osteoclast-specific imaging agents have targeted the avb3 integrin as a marker of cell numbers, with successful demonstration in rodents of localization of a copper-64 labeled integrin into areas of increased cellular responses including osteoclasts and inflammatory cells [9].
We report here the carbon-11 labeling of two candidate high-affinity cathepsin K inhibitors (Fig. 1) and in vivo evaluation of the whole-body biodistribution and cathepsin K specific uptake into normal bone. Our preliminary results, and a similar study from others have been previously presented in abstract form [10,11].
Fig. 1.
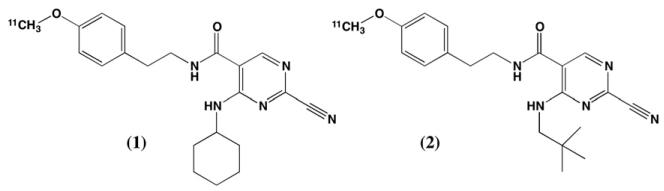
Structures of carbon-11 labeled cathepsin K inhibitors.
2. Materials and methods
2.1. General
Organic and inorganic reagents and solvents (ACS grade) were purchased from commercially available sources and used without purification. Authentic unlabeled samples of compounds 1 and 2 were prepared by minor modifications of the published procedures [12]. Thin layer chromatography (TLC) was performed on glass plates pre-coated with 0.25 mm thickness of silica gel (60 Å) with fluorescent indicator (EMD). Automated flash chromatography was performed using a Biotage SP1 system with Isolute silica cartridges (Biotage). All 1H and 13C NMR spectra were obtained using a Varian 400 MHz Mercury plus instrument at ambient temperatures in chloroform-d (CDCl3) or methanol-d (CD3OD). High resolution mass spectra were obtained on a Micromass Autospec in the ESI-TOF mode. Analytical (Luna C8(2) 100x2 mm) and semi-preparative (Luna C18 150x10 mm) HPLC columns were obtained from Phenomenex (Phenomenex). Radiochemical syntheses were done using a General Electric Medical Systems TRACERLab FX C Pro synthesis system. For radiochemical syntheses, Sep-Pak C18 1 cc cartridges were purchased from Waters and conditioned with ethanol (10 mL) and water (10 mL) prior to use.
2.2. Chemistry
2.2.1. N-(4-(benzyloxy)phenethyl)-2,4-dichloropyrimidine-5-carboxamide (5b)
To a solution of 2-(4-(benzyloxy)phenyl)ethanamine hydrochloride (3.55 mmol) and diisopropylethylamine (7.46 mmol) in 10 ml of dichloromethane was added 2,4-dichloropyrimidine-5-carbonyl chloride (3.55 mmol), and the resulting suspension was stirred at ambient temperature for 24 h. The solvent was removed under reduced pressure, and the resulting oil was purified using flash chromatography on silica gel (dichloromethane/methanol 0-5%) to afford 5b (0.870 g, 61%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 7.35-7.19 (m, 5H), 6.41 (broad s, 1H), 3.78 (dt, 2H, J = 7.5, 6.3, Hz), 2.95 (t, 2H, J = 6.3Hz). 13C NMR (101 MHz, CDCl3) δ 161.51 (1C), 161.23 (1C), 158.27 (1C), 157.73 (1C), 136.83 (1C), 130.18 (1C), 129.72 (2C), 128.57 (2C), 127.98 (1C), 127.40 (2C), 126.97 (1C), 115.23 (2C), 70.04 (1C), 41.58 (1C), 34.20 (1C).
2.2.2. N-(4-(benzyloxy)phenethyl)-2-chloro-4-(cyclohexylamino) pyrimidine-5-carboxamide (7c)
To a stirring solution of (5b) (1.745 mmol) in 5 ml of anhydrous tetrahydrofuran was added cyclohexylamine (2.095 mmol), and the resulting suspension was stirred at ambient temperature for 24 h. The solution was filtered to remove the undissolved salts, and the filtrate was concentrated in vacuo to produce a crude yellow oil. Purification of the oil was performed using flash chromatography on silica gel (ethyl acetate/hexanes 0-30%) to afford 7c (1.260 mmol, 75%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.78 (d, J = 7.9 Hz, 1H), 7.99 (s, 1H), 7.44-7.27 (m, 4H), 6.99 (dd, J = 78.1, 8.6 Hz, 4H), 6.40 (s, 1H), 5.01 (s, 2H), 4.13-4.00 (m, 1H), 3.58 (q, J = 6.8 Hz, 2H), 2.82 (t, J = 6.9 Hz, 2H), 2.00-1.90 (m, 2H), 1.72 (dd, J = 13.3, 4.1 Hz, 2H), 1.61 (dd, J = 12.3, 3.5 Hz, 1H), 1.48-1.18 (m, 5H). 13C NMR (101 MHz, CDCl3) δ 166.06 (1C), 162.43 (1C), 160.84 (1C), 157.64 (1C), 154.63 (1C), 136.89 (1C), 130.64 (1C), 129.70 (2C), 128.55 (2C), 127.96 (1C), 127.46 (2C), 115.13 (2C), 106.81 (1C), 70.04 (1C 4H), 49.04 (1C), 40.99 (1C), 34.57 (s, 4H), 32.36 (2C), 25.53 (1C), 24.44 (2C).
2.2.3. N-(4-(benzyloxy)phenethyl)-2-chloro-4-(neopentylamino) pyrimidine-5-carboxamide (7d)
The product was obtained from 5b and neopentylamine (0.598 mmol) in a similar manner as described for the preparation and purification of 7c, affording the pure compound 7d (0.393 mmol, 79%). 1H NMR (400 MHz, CDCl3) δ 9.08 (t, J = 5.6 Hz, 1H), 8.02 (s, 1H), 7.43-7.27 (m, 4H), 7.09 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 6.69 (t, J = 5.6 Hz, 1H), 5.00 (s, 2H), 3.59 (q, J = 6.8 Hz, 2H), 3.32 (d, J = 5.8 Hz, 2H), 2.82 (t, J = 7.0 Hz, 2H), 0.99 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 166.14 (1C), 162.28 (1C), 162.24 (1C), 157.59 (1C), 154.67 (1C), 136.90 (1C), 130.76 (s, 1H), 129.73 (2C), 128.56 (2C), 127.96 (1C), 127.48 (2C), 115.07 (2C), 106.86 (1C), 70.04 (1C), 52.07 (1C), 41.05 (1C), 34.56 (1C), 31.84 (1C), 27.41 (3C).
2.2.4. N-(4-(benzyloxy)phenethyl)-2-cyano-4-(cyclohexylamino) pyrimidine-5-carboxamide (8a)
A mixture of 7c (0.433 mmol), sodium cyanide (0.866 mmol), and 1,4-diazabicyclo[2.2.2]octane (DABCO) (0.866 mmol) in anhydrous dimethylsulfoxide (2 mL) was heated in a CEM microwave at 80 °C for 10 minutes. After cooling to ambient temperature, the solution was diluted with 8 mL of water and the precipitate filtered. The precipitate was dissolved in dicloromethane and purified by flash chromatography (ethyl acetate/hexanes 0-50%) to afford pure 8a (0.294 mmol, 68%) as a white powder. 1H NMR (400 MHz, CDCl3) δ 8.75 (d, J = 7.4 Hz, 1H), 8.12 (s, 1H), 7.49-7.28 (m, 5H), 7.12 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.18 (s, 1H), 5.05 (s, 2H), 4.14-3.97 (m, 1H), 3.63 (q, J = 6.5 Hz, 2H), 2.85 (t, J = 6.7 Hz, 2H), 2.03-1.91 (m, 2H), 1.75 (dd, J = 9.3, 4.0 Hz, 2H), 1.69-1.59 (m, 1H), 1.52-1.19 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 165.44 (1C), 159.49 (1C), 157.75 (1C), 152.78 (1C), 145.87 (1C), 136.83 (1C), 130.29 (1C), 129.66 (2C), 128.56 (2C), 127.98 (1C), 127.44 (2C), 115.77 (1C), 115.25 (2C), 109.63 (1C), 70.04 (1C), 60.37 (1C), 49.35 (1C), 41.02 (1C), 34.41 (1C), 32.27 (1C), 29.67 (2C), 25.47 (1C), 24.47 (1C).
2.2.5. N-(4-(benzyloxy)phenethyl)-2-cyano-4-(neopentylamino) pyrimidine-5-carboxamide (8b)
The product was obtained from 7d (0.376 mmol) in a similar manner as described for the preparation and purification of 8a, affording the pure compound 8b (0.293 mmol, 78%). 1H NMR (400 MHz, CDCl3) δ 9.03 (t, J = 5.5 Hz, 1H), 8.13 (s, 1H), 7.44-7.27 (m, 5H), 7.11 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 6.45 (t, J = 5.6 Hz, 1H), 5.01 (s, 2H), 3.63 (q, J = 6.6 Hz, 2H), 3.33 (d, J = 6.0 Hz, 2H), 2.85 (t, J = 6.9 Hz, 2H), 0.98 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 165.55 (1C), 160.93 (1C), 157.66 (1C), 152.88 (1C), 145.66 (1C), 136.87 (1C), 130.51 (1C), 129.73 (2C), 128.57 (2C), 127.98 (1C), 127.45 (2C), 115.79 (1C), 115.17 (2C), 109.72 (1C), 70.04 (1C), 52.01 (1C), 41.14 (1C), 34.44 (1C), 31.97 (1C), 27.35 (3C).
2.2.6. 2-cyano-4-(cyclohexylamino)-N-(4-hydroxyphenethyl) pyrimidine-5-carboxamide (9a)
To a solution of 8a (0.439 mmol) in dry dichloromethane (3 mL) under N2 atmosphere was added boron tribromide (1 M in dichloromethane, 0.439 mmol) dropwise at room temperature. The reaction mixture was monitored by TLC; upon completion (approximately 2 hours), ice-water was added, and the mixture was extracted three times with ethyl acetate. The combined organic layers were washed with water (20 mL), brine (20 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure to afford a crude yellow oil. The oil was purified by flash chromatography (methanol/dichloromethane 0-5%) to afford compound 9a (0.068 mmol, 16%). 1H NMR (400 MHz, CD3OD) δ 8.36 (s, 1H), 7.03 (d, J = 8.4 Hz, 2H), 6.70 (d, J = 8.5 Hz, 2H), 4.05-3.96 (m, J = 13.2, 9.6, 3.7 Hz, 1H), 3.54-3.44 (m, J = 13.4, 5.7 Hz, 2H), 2.76 (t, J = 7.3 Hz, 2H), 1.98-1.90 (m, J = 12.3, 3.3 Hz, 2H), 1.81-1.69 (m, J = 9.1, 4.2 Hz, 2H), 1.68-1.56 (m, J = 8.6, 4.1 Hz, 1H), 1.51-1.22 (m, 6H). 13C NMR (101 MHz, CD3OD) δ 165.74 (1C), 159.36 (1C), 155.57 (1C), 153.33 (1C), 145.05 (1C), 129.23 (2C), 114.83 (2C), 110.29 (1C), 49.06 (1C), 41.17 (1C), 34.15 (1C), 31.79 (2C), 25.20 (1C), 24.14 (2C). HRMS (ESI) calc. for C20H23N5O2 (M + H)+ 366.1925, found 366.1926.
2.2.7. 2-cyano-N-(4-hydroxyphenethyl)-4-(neopentylamino) pyrimidine-5-carboxamide (9b)
The product was obtained from 8b (0.371 mmol) in a similar manner as described for the preparation and purification of 9a, affording the pure compound 9b (0.043 mmol, 38%). 1H NMR (400 MHz, CDCl3) δ 8.99 (t, J = 5.4 Hz, 1H), 8.09 (s, 1H), 7.04 (d, J = 8.4 Hz, 2H), 6.75 (d, J = 8.4 Hz, 2H), 6.31 (t, J = 5.4 Hz, 1H), 5.64 (s, 1H), 3.68-3.56 (m, 2H), 3.33 (d, J = 6.0 Hz, 2H), 2.82 (t, J = 6.7 Hz, 2H), 0.97 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 165.46 (1C), 160.90 (1C), 154.68 (1C), 152.64 (1C), 130.07 (1C), 129.89 (2C), 115.72 (2C), 115.57 (1C), 109.66 (1C), 52.02 (1C), 41.10 (1C), 34.33 (1C), 31.97 (1C), 27.32 (3C). HRMS (ESI) calc. for C19H23N5O2 (M + H)+354.1925, found 354.1921.
2.3. Radiochemical syntheses
2.3.1. Syntheses of 2-cyano-4-(cyclohexylamino)-N-(4-[11C] methoxyphenethyl)-pyrimidine-5-carboxamide ([11C]1) and 2-cyano-N-(4-[11C]methoxyphenethyl)-4-(neopentylamino)pyrimidine-5-carboxamide ([11C]2)
All steps (Fig. 1) were performed using the integrated functions of the TRACERLab FX C Pro synthesis system. [11C]CO2 from the cyclotron (GE PETtrace 6) was converted by gas-phase methods to [11C]methyl iodide. The reaction of gaseous [11C]CH3I with 2-cyano-4-(cyclohexylamino)-N-(4-hydroxyphenethyl)pyrimidine-5-carboxamide (9a) or 2-cyano-N-(4-hydroxyphenethyl)-4-(neopentylamino) pyrimidine-5-carboxamide (9b) took place in the FX C Pro reaction vessel. The vessel was charged with 1.0 mg of precursor (9a or 9b), 10 μL of 7.5 M NaOH, and 100 μL of dimethylformamide. [11C]CH3I was delivered from the methyl iodide synthesis unit and bubbled into the reaction vessel at a fixed flow rate of 15 mL/min for 3 min; then the flow was stopped, and the solution was stirred for an additional 2 min at ambient temperatures. The reaction mixture was diluted with 1 mL of 50/50 acetonitrile/water and injected into a preparatory HPLC column (Phenomenex Luna C18(2), 10 × 150 mm, eluted with 60% acetonitrile, 40% H2Om 20 mM NH4OAc at 6 mL/min). The fractions corresponding to [11C]1 or [11C]2 (typically eluting out between 9.5 and 11.5 min) were collected and transferred into a dilution flask containing sterile water (50 mL). The resulting solution was transferred through a Waters C18 1 cc Sep-Pak to collect the desired product. The C18 Sep-Pak was then washed with sterile water (10 mL) to remove unwanted hydrophilic impurities and residual acetonitrile to waste. [11C]1 or [11C]2 were eluted off into a collection vial with ethanol for injection, USP (0.5 mL) and 0.9% sodium chloride for injection, USP (4.5 mL). The final formulation (5 mL) was then passed into a sterile vial to provide [11C]1 (2.47 ± 0.84 GBq, n = 15) or [11C]2 (2.26 ± 0.81 GBq, n = 5). Radiochemical purities (N99%) and specific activities ([11C]1; 560 ± 180 GBq/μmol: [11C]2; 488 ± 129 GBq/μmol) were determined using analytical HPLC (45% acetonitrile: 25% H2O, 20 mM NH4OAc, oven 30 ° C, 254 nm, flow rate: 1.0 mL/min, RT = 5-6 min, RCP ≥ 99.7%. Based on approx. 3 Ci of [11C]CO2, overall radiochemical yields were 2-3% (end-of-synthesis) and were not optimized.
2.4. Biology
2.4.1. Rat biodistribution studies
The whole body biodistribution of radioactivity after intravenous injection of [11C]1 was determined in rats. Animals (N = 6, female CD rats) were lightly anesthetized (isoflurane) and injected via the tail vein with 1.85-4.44 MBq of radioligand formulated in 5% ethanol/saline. At selected times (5 and 40 min), groups (N = 3) of animals were anesthetized, euthanized (decapitation) and samples of tissues removed. Tissue samples were weighed, counted for carbon-11, and the data calculated as % injected dose/g.
2.4.2. MicroPET imaging studies
Studies were done using imaging with Concorde Microsystems R4 and P4 PET scanners. Rats were anesthetized with isoflurane and maintained on 1% isoflurane throughout the imaging period. The animals were placed on their dorsal side in the scanner, and body temperature was maintained using a heating pad. A catheter was inserted into the tail vein for administration of radioactive solutions. Following a measured transmission scan, solutions of radioligands ([11C]1 or ([11C]2, 37-185 MBq in 0.5-1 mL volumes) were injected via the catheter,followed by a 1 ml flush of saline. For the blocking study, a 2 mg/kg dose of unlabeled compound 2 was administered 10 minutes prior to the injection of the radiolabeled [11C]2. Emission data was collected for 60 minutes (12 × 5 min fames). The emission data was corrected for decay, dead time and random coincidences, then reconstructed using iterative ordered subset expectation maximization-maximum a posteriori (OSEM-MAP) yielding a reconstructed image resolution of approximately 1.4 mm. Volumetric regions of interest (ROIs) were defined using hand-drawn circular ROIs placed on the summed images, and the time-activity curves determined for each ROI (ASI Pro VM software: Siemens Medical Systems, Malvern, PA).
3. Results and discussion
The family of cysteine proteases known as the cathepsins (11 members: cathepsins B, C, F, H, K, L, O, S, V, W and X) have widespread functions in the mammalian body and are involved in both normal and pathological biochemical processes. Only recently have the cathepsins become of interest as potential targets for development of disease-specific in vivo imaging agents [9,13-16]. The clear involvement of cathepsin K in osteolytic bone diseases, and the potential importance of new cathepsin K inhibitors as treatment for osteoporosis, prompted the current preliminary study into the potential of radiolabeling cathepsin k inhibitors.
The target compounds (1 and 2) were selected from a large number of potential cathepsin K inhibitors which have now been described in the literature [12,17-20]. The selection was based on synthetic accessibility to the required precursors for radiolabeling, high apparent affinities for human cathepsin K (in vitro IC50 values of 0.022 nM and 0.003 nM for 1 and 2, respectively), and 10- to 100-fold selectivities over cathepsins L and S [12].
The syntheses of the authentic ligands 1 and 2, and the required precursors for carbon-11 labeling 9a and 9b, were done following slight modifications of the published methods (scheme 1). The syntheses began with commercially available 2,4-dichloropyrimidine-5-carbonyl chloride (3) which underwent amide bond formation with 2-(4-methoxyphenyl)ethylamine (4a) or 2-(4-(benzyloxy)phenyl)ethanamine hydrochloride (4b), in the presence of diiopropylethyl amine, to afford compounds 5a and 5b. Subsequent reaction of 5a or 5b with cyclohexylamine or neopentyl amine providing compounds 7a-d in good yields. The 2-substituted pyrimidines (7a-d) were then converted into the corresponding cyanopyrimidines by displacement of the 4-chloro substituent with cyanide. The reaction was performed using microwave heating techniques in the presence of DABCO (1,4-diazabicyclooctane) and NaCN and yielded cyanides 1, 2, 8a, and 8b. As a final step in preparation for carbon-11 labeling, the nitriles 8a-b underwent debenzylation in the presence of 1 M boron tribromide in dichloromethane to yield the necessary desmethyl precursors 9a and 9b. Careful chromatographic purifications provided the desmethyl compounds in low yield.
Scheme 1.

(I) CH2Cl2, rt, overnight. (II) THF, rt, overnight. (III) NaCN, DABCO, DMSO, microwave 80 °C 30 min. (IV) 1.0 M BBr3/CH2Cl2, rt., 20 min.
The radiochemical syntheses of [11C]1 and [11C]2 were done using the automated Tracerlab FXcPro system and standard reaction conditions used for alkylation reactions with [11C]methyl iodide. Emphasis was placed on preparation of radiochemically pure (N99%) and high specific activity products, and synthesis yields were low and not optimized.
The in vivo biological properties of these potential radioligands was first evaluated by determining the whole-body biodistribution of [11C]1 in rats (Table 1). No unusual early uptake (5 min) or significant retention (40 min) was observed for any specific organ in the rat body. Highest concentrations were observed in abdominal organs (e.g., liver) and very low levels in muscle and bone (a large bone sample of the femur in its entirety).
Table 1.
Tissue biodistribution of radioligand [11C]1 in female rats following intravenous injection.
| 5 min |
40 min |
|||
|---|---|---|---|---|
| avg | s.d. | avg | s.d. | |
| Brain | 0.43 | 0.05 | 0.11 | 0.0015 |
| Heart | 1.23 | 0.18 | 0.19 | 0.02 |
| Lung | 0.68 | 0.09 | 0.16 | 0.019 |
| Liver | 3.02 | 0.21 | 0.68 | 0.033 |
| Pancreas | 1.68 | 0.19 | 0.56 | 0.069 |
| Spleen | 1.03 | 0.079 | 0.22 | 0.015 |
| Adrenal | 3.57 | 0.86 | 0.51 | 0.053 |
| Kidney | 2.03 | 0.17 | 0.41 | 0.042 |
| Muscle | 0.11 | 0.008 | 0.11 | 0.024 |
| Bone | 0.26 | 0.018 | 0.14 | 0.028 |
| Blood | 0.24 | 0.007 | 0.11 | 0.015 |
The potential for imaging the osteoclasts in bone was then evaluated in normal rats using microPET imaging; representative images of localization of [11C]1 in the rat are shown in Fig. 2 (equivalent results were obtained for [11C]2, data not shown). As an aid in identification of appropriate skeletal features, a bone scan using [18F]fluoride ion was completed in several animals following the [11C]radiotracer study. Consistent with the biodistribution data, high early uptake was observed in the liver and low in muscle, with steadily increasing concentrations in the bladder (Fig. 3A). The uptake and clearance from the heart and brain are consistent with a moderately lipophilic radiotracer. For both radioligands, localization of radioactivity in actively growing bone regions (e.g., distal ulnar, carpal, distal and proximal humeral, distal femur, proximal tibia, tail vertebrae) was clearly evident, reaching levels 2 to 3 times higher than in non-target regions such as muscle (Fig. 3B). The lack of clearance of radioactivity from the bone regions is consistent with the irreversible nature of binding of these cathepsin K inhibitors.
Fig. 2.

Summed microPET images (0-60 min) for (A) cathepsin K radioligand [11C]1 and (B) [18F]fluoride ion in normal rat.
Fig. 3.

Representative tissue time vs. radioactivity curves for cathepsin K radioligand uptake and distribution in normal rat. Panel 3A: tissue kinetics in non-target regions of brain, heart, liver and muscle, and clearance into bladder; panel 3B: uptake and retention of radioactivity into selected target bone areas relative to non-specific distribution into muscle; panel 3C: reductions of bone uptake and retention after pre-administration of blocking dose (2 mg/kg) of unlabeled cathepsin K inhibitor; panel 3D: comparison of uptake and retention of cathepsin K radioligand in bone following intravenous injection of high and low specific activity preparations.
That the bone uptake of [11C]1 and [11C]2 represented saturable, high-affinity binding was demonstrated in two different ways. The administration of a blocking dose of unlabeled ligand 2, ten minutes prior to the injection of [11C]2, resulted in a reduction of bone uptake of radioactivity to essentially levels equivalent to background in non-target tissues (Fig. 3C). Uptake and retention of radioactivity in the non-target areas were unaltered by dosing with unlabeled drug. In a slightly different experiment, studies were done using high (414 GBq/μmol) and low (10.8 GBq/μmol) specific activity preparations of radioligand [11C]1. The administration of the low specific activity dose, representing co-injection of 14 micrograms/kg of unlabeled 1, produced a similar nearly full block of the uptake and retention of the radioligand into the target bone regions (Fig. 3D). Thus, for both [11C]1 and [11C]2, the localization of radioactivity into specific bone regions appears to represent specific binding that can be readily competed for by pre- or co-injection of unlabeled compound.
4. Conclusions
These studies add to our preliminary findings (Rodnick et al. 2013) and together with the similar results obtained by others in rabbits and non-human primate (Bennacef et al. 2013) provide evidence that radiolabeled cathepsin K inhibitors may have potential as in vivo imaging agents. The uptake in bone tissues, which can be blocked with cold ligand, is most likely due to osteoclasts as the normal tissue distribution of cathepsin K is predominantly in those cells. Further evaluation of these radioligands will require utilization of an in vivo model of increased osteoclast activity, such as the ovariectomized rat or primate models of osteoporosis.
Acknowledgments
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Institutes of Biomedical Imaging and Bioengineering, under Award Numbers 1R21AR061594-01 and T32EB005172-02. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank the staff of the University of Michigan Cyclotron and Radiochemistry Facility for assistance in the syntheses of the radioligands.
References
- [1].Novinec M, Lenarčič B. Cathepsin K: a unique collagenolytic cysteine peptidase. Biol Chem. 2013;394:1163–79. doi: 10.1515/hsz-2013-0134. [DOI] [PubMed] [Google Scholar]
- [2].Troeberg L, Nagase H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim Biophys Acta. 1824;2012:133–45. doi: 10.1016/j.bbapap.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ishida M, Kojima F, Okabe H. Cathepsin k expression in basal cell carcinoma. J Eur Acad Dermatol Venereol. 2012;27:128–30. doi: 10.1111/j.1468-3083.2011.04436.x. [DOI] [PubMed] [Google Scholar]
- [4].Husmann K, Muff R, Bolander ME, Sarkar G, et al. Cathepsins and osteosarcoma: expression analysis identifies cathepsin K as an indicator of metastasis. Mol Carcinog. 2008;47:66–73. doi: 10.1002/mc.20362. [DOI] [PubMed] [Google Scholar]
- [5].Jaffer FA, Kim D-E, Quinti L, Tung C-H, et al. Optical visualization of cathepsin K activity in atherosclerosis with a novel, protease-activatable fluorescence sensor. Circulation. 2007;115:2292–8. doi: 10.1161/CIRCULATIONAHA.106.660340. [DOI] [PubMed] [Google Scholar]
- [6].Stoch SA, Wagner JA. Cathepsin K inhibitors: a novel target for osteoporosis therapy. Nature. 2008;83:172–6. doi: 10.1038/sj.clpt.6100450. [DOI] [PubMed] [Google Scholar]
- [7].Kozloff KM, Quinti L, Patntirapong S, Haushka PV, et al. Non-invasive optical detection of cathepsin K-mediated fluorescence reveals osteoclast activity in vitro and in vivo. Bone. 2009;44:190–8. doi: 10.1016/j.bone.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li Y, Schiepers C, Lake R, Dadparvar S, et al. Clinical utility of 18F-fluoride PET/CT in benign and malignant bone diseases. Bone. 2012;50:128–39. doi: 10.1016/j.bone.2011.09.053. [DOI] [PubMed] [Google Scholar]
- [9].Wadas TJ, Deng H, Sprague JE, et al. Targeting the avb3 integrin for small-animal PET/CT of osteolytic bone metastases. J Nucl Med. 2009;50:1873–80. doi: 10.2967/jnumed.109.067140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rodnick ME, Shao X, Kozloff KM, et al. Syntheses of radiolabeled cathepsin k inhibitors as potential osteoclast imaging agents. J Labelled Compd Radiopharm. 2013;56:S422. [Google Scholar]
- [11].Bennacef I, Rubins D, Riffel K, et al. Towards the imaging of cathepsin K by PET. J Labelled Compd Radiopharm. 2013;56:S49. [Google Scholar]
- [12].Altmann E, Aicholz R, Betschart C, et al. 2-Cyano-pyrimidines: a new chemotype for inhibitors of the cysteine protease cathepsin k. J Med Chem. 2007;50:591–4. doi: 10.1021/jm0613525. [DOI] [PubMed] [Google Scholar]
- [13].Edem PE, Valiant JF. Technetium-labeled cathepsin B inhibitors as molecular imaging agents. Nucl Med Biol. 2010;37:681. abstract. [Google Scholar]
- [14].Ren G, Blum G, Verdoes M, Liu H, et al. Non-invasive imaging of cysteine cathepsin activity in solid tumors using a 64-Culabeled activity-based probe. PLoS ONE. 2011:e28029. doi: 10.1371/journal.pone.0028029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Loser R, Bergmann R, Frizler M, et al. Synthesis and radiopharmacological characterization of a fluorine-18-labelelled azapeptide nitrile as a potential PET tracer for in vivo imaging of cysteine cathepsins. ChemMedComm. 2013;8:1330–44. doi: 10.1002/cmdc.201300135. [DOI] [PubMed] [Google Scholar]
- [16].Ogbomo SM, Shi W, Wagh NK, et al. 177Lu-lLabeled HPMA copolymers utilizing cathepsin B and S cleavable linkers: synthesis, characterization and preliminary in vivo investigation in a pancreatic cancer model. Nucl Med Biol. 2013;40:606–17. doi: 10.1016/j.nucmedbio.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Altmann E, Betschart C, Haykawa K, et al. Heteroaryl nitrile derivatives. WO 2004/020441 A1
- [18].Robichaud J, Bayly CI, Black WC, Desmarais S, et al. Beta-substituted cyclohex-anecorbaxamide cathepsin K inhibitors: modification of the 1,2-disubstituted aromatic core. Bioorg Med Chem Lett. 2007;17:3146–51. doi: 10.1016/j.bmcl.2007.03.028. [DOI] [PubMed] [Google Scholar]
- [19].Teno N, Miyake T, Ehara T, et al. Novel Scaffolds for cathepsin K inhibitors. Bioorg Med Chem Lett. 2007;17:6096–100. doi: 10.1016/j.bmcl.2007.09.047. [DOI] [PubMed] [Google Scholar]
- [20].Teno N, Irie O, Miyake T, Gohda K, et al. New chemotypes for cathepsin K inhibitors. Bioorg Med Chem Lett. 2008;18:5833–6. doi: 10.1016/j.bmcl.2008.03.036. [DOI] [PubMed] [Google Scholar]