

Abstract
Hepatosplenic T-cell lymphoma is a rare form of non-Hodgkin lymphoma, which carries a poor prognosis. We report our single-institution experience in the management of hepatosplenic T-cell lymphoma (HSTCL)- in 14 patients (pts) among whom 7 who remain alive (50%) and in remission at a median follow-up of 66 months. More frequent long-term survival was seen in those treated with a non-CHOP (cyclophosphamide/doxorubicin/vincristine/prednisone) induction and consolidative stem cell transplant (SCT).
Introduction
Hepatosplenic T-cell lymphoma is a rare form of extranodal non-Hodgkin lymphoma, first recognized as a distinct entity in the Revised European-American Lymphoma classification. Typical presentation includes lymphomatous infiltration of spleen and liver, and peripheral lymphadenopathy is rarely seen. The prognosis is almost uniformly poor, and there are no prospective studies of treatment of HSTCL.
Patients and Methods
For this report, we conducted a retrospective review of all pts who underwent treatment for HSTCL at our institution. Individual chart review was performed to report clinical presentation, management, and outcome.
Results
We identified 14 pts with HSTCL managed at our center, 7 of which remain alive with median follow-up of 65.6 months. Six of 7 received alternative induction chemotherapy regimens such as ICE (ifosfamide, carboplatin, etoposide) or IVAC (ifosfamide, etoposide, high-dose cytarabine) as opposed to CHOP and all surviving pts had proceeded to undergo either autologous or allogeneic SCT.
Conclusion
Our results suggest that use of non-CHOP induction regimen and early use of high dose therapy and SCT consolidation may translate to improved survival for pts with HSTCL.
Keywords: Peripheral, TCR gamma-delta
Introduction
Described first by Farcet et al in 1990,1 hepatosplenic T-cell lymphoma (HSTCL) is a distinct lymphoma entity with unique clinicopathologic features and poor clinical outcome, which has been recognized in the revised European-American Lymphoma classification in 19942 and in the subsequent World Health Organization classifications.3–7
Hepatosplenic T-cell lymphoma can occur at any age but is most often seen in teenagers or young adults, with a strong male predominance. 1,8–12 It is an extremely rare lymphoma making up <5% of peripheral T-cell lymphomas.5,12 Immunocompromised patients are overrepresented, with reports of HSTCL developing during long-term immunosuppression after solid-organ transplant13–15 and in the setting of other immune dysregulation including malignancy and infection.16,17 The importance of iatrogenic immunosuppression as a contributor to lymphomagenesis has become particularly relevant in light of increased incidence of HSTCL in patients with chronic inflammatory diseases after treatment with immunosuppressants, specifically agents blocking tumor necrosis factor (TNF)-α and/or thiopurine agents.18–24
Occurrence is predominant in young male adults, who typically present with hepatosplenomegaly and peripheral blood cytopenias, especially thrombocytopenia. B-symptoms are common, whereas peripheral lymphadenopathy is usually absent. Patients are most frequently diagnosed after splenectomy and/or liver biopsy, although bone marrow biopsy with an appropriate immunophenotype in this clinical setting might be sufficient to make the diagnosis.4,25 On pathologic review, neoplastic cells are commonly found in the red pulp of the spleen and show a preference to infiltrate the splenic, hepatic, and bone marrow sinusoids.11,26 The immunophenotype typically is a CD4−/CD8− T-cell with CD2+ and CD3+ expression. Other markers such as CD5, CD25, TIA-1, and granzyme B are usually absent. NK cell markers, such as CD56 and CD16 might be expressed.4,5,12,25,26 The malignant cells most often express a γ/δ T-cell phenotype as can be demonstrated by flow cytometry, and as such β F-1 staining is not found.5,11 Reports have described similar clinical presentations with tumor cells expressing an αβ-phenotype,27,28 and are considered an immunophenotypic variant of the same disease entity in the World Health Organization classification. The T-cell receptor (TCR) γ gene is always clonally rearranged4,5,12,25,26; the T-cell β gene might be rearranged as well.4 Cytogenetic evaluation frequently demonstrates isochromosome 7q although this is not specific for this disease.29–32
In the literature, the prognosis of HSTCL is almost uniformly poor, and no prospective trials investigating treatment approaches are reported. Most of the published data consists of case reports and series, with 2 larger single-institution series focused on treatment outcome, demonstrating exceedingly poor long-term therapeutic results with a CHOP (cyclophosphamide/doxorubicin/vincristine/prednisone)-based regimen.25,33Anecdotal activity of several other chemotherapy regimens has been reported in form of case reports.34–39 Several authors have published experiences with high-dose therapy (HDT) autologous or allogeneic stem cell transplantation (SCT),11,12,25,33,40–46 and a 2007 collection of published case reports of HSTCL treated with allogeneic stem cell transplantation suggests a better outcome for that approach.47
Patients and Methods
To investigate our center's experience in the management of HSTCL, we conducted a search using our T-cell lymphoma and bone marrow transplant databases. Included in this report were all patients treated at Memorial Sloan-Kettering Cancer Center with a diagnosis of HSTCL, for whom follow-up information was available. This report summarizes our single-center experience with 14 consecutive patients treated between the years of 1994 and 2012. We reviewed each patient's records for characteristics of initial clinical presentation, the immunohistochemistry of lymphomatous cells, treatment regimen, and responses. Sufficient data to calculate an International Prognostic Index (IPI)48 and prognostic index for peripheral T-cell lymphoma (PIT)49 were available for 12 of 14 subjects with 2 patients missing lactate dehydrogenase (LDH) values at time of diagnosis. Kaplan–Meier curves were calculated to determine overall survival (OS) and progression-free survival (PFS). Log-rank χ2 test was used to compare the effect of clinical variables on survival.
Results
Patient Characteristics
All patients were male with a median age of 36 years (range, 12–59 years, see also Table 1). All subjects had stage IV disease with hepatomegaly and/or splenomegaly. Ten of 14 cases (71%) had documented bone marrow involvement, 10 of 12 patients (91%) had elevated LDH, and all but 1 had B symptoms. Thrombocytopenia was present at diagnosis in 9 of 14 patients (64%), anemia in 12 of 14 patients (86%), and leukopenia in 6 of 14 patients (43%). Transaminases and/or alkaline phosphatase were elevated in 10 of 14 patients (71%). Eight of 14 cases (57%) had previous autoimmune disease: 2 with ulcerative colitis, 4 with Crohn's disease, and 2 with juvenile rheumatoid arthritis. Three patients had received both anti-TNFα therapy (2, infliximab; 1, adalimumab) and 6-mercaptopurine (6-MP); 3 had been treated only with 6-MP. Risk stratification per IPI and PIT are summarized in Table 2.
Table 1. Clinical Presentation.
| Patient Number | Age/Sex | HM | SM | LAN | B-Sx | Hgb | Pltl | WBC | LDH | LFT | BM | EBV | Comorbidities | IS-Therapy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 46/M | Yes | Yes | No | Yes | Yes | Yes | No | Yes | Yes | No | Yes | None | No |
| 2 | 34/M | Yes | Yes | No | Yes | Yes | No | Yes | Yes | Yes | Yes | NA | UC | 6-MP |
| 3 | 51/M | Yes | Yes | No | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | HTN, DM, Emphysema | No |
| 4 | 23/M | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | No | NA | jRA | Unclear |
| 5 | 37/M | Yes | Yes | No | Yes | No | Yes | No | Yes | Yes | No | NA | UC Seminoma | 6-MP |
| 6 | 59/M | Yes | No | Yes | Yes | Yes | No | No | NA | NA | No | NA | PVD | No |
| 7 | 12/M | No | Yes | Yes | Yes | Yes | No | Yes | No | No | Yes | No | Asthma | No |
| 8 | 19/M | No | Yes | No | No | Yes | No | Yes | Yes | Yes | Yes | NA | None | No |
| 9 | 48/M | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | NA | jRA | No |
| 10 | 53/M | No | Yes | Yes | Yes | Yes | No | Yes | NA | NA | Yes | NA | Depression | No |
| 11 | 50/M | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | NA | CD | Infliximab, 6-MP |
| 12 | 22/M | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | NA | CD | 6-MP |
| 13 | 27/M | Yes | Yes | No | Yes | Yes | Yes | No | Yes | Yes | Yes | NA | CD | Adalimumab, 6-MP |
| 14 | 18/M | Yes | Yes | No | Yes | Yes | Yes | Yes | No | No | Yes | NA | CD | Infliximab, 6-MP |
Listed for all 14 patients are clinical, laboratory, and pathologic variables at the time of diagnosis, comorbidities, and previous immunosuppressive therapy.
Abbreviations: 6-MP = 6-mercaptopurine; BM = bone marrow involvement; B-Sx = presence of B-symptoms; CD = Crohn's disease; DM = diabetes mellitus; EBV = evidence of Ebstein Barr virus in pathologic specimen; Hgb = decreased hemoglobin; HM = hepatomegaly; HTN = hypertension; IS-Therapy = prior immunosuppressive therapy other than steroids; jRA = juvenile rheumatoid arthritis; LAN = lymphadenopathy; LDH = elevated lactate dehydrogenase; LFT = abnormal liver function tests; M = male; NA = not assessed; Pltl = decreased platelet count; PVD = peripheral vascular disease; SM = splenomegaly; UC = ulcerative colitis; WBC = abnormal leukocyte count.
Table 2. Treatment Summary and Response to Therapyb.
| Patient Number |
Time to Treatment Start (Months) |
Removal of Spleen |
Induction Regimen |
Response Induction |
Further Regimen (Pre-Txp) |
Auto or Allo |
Status at Time of Txp |
Conditioning Regimen | CD34+
Dose (× 106/kg) |
Response to Txp |
Relapse, After Txp (Months) |
Further Treatment Post Txp |
Response to Further Rx |
IPI | PIT | OS
From Induction (Months) |
Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 10 | Yes | ICE × 3 | CR | None | Allo | CR | Melphalan, fludarabine, campath | 9.4 | CR | 6 | DLI | CR | 3 (HI) | 1 | 50.5 | Alive |
| 2 | 2 | No | IVAC × 4 | CR | None | Auto | CR | Etoposide, cyclophosphamide, TBI | 10 | CR | — | — | NA | 3 (HI) | 2 | 57.7 | Alive |
| 3 | 6 | Yes | IVAC × 4 | POD | EPOCH × 2 | Allo | CR | TBI, thiotepa, cyclophosphamide | 27.4 | CR | — | — | NA | 3 (HI) | 3 | 9.9 | DOT |
| 4 | 6 | Yes | ICE × 3 | PR | None | Allo | PR | Bus/mel/flu, splenic XRT, equine ATG | 14.3 | SD | 3 | Allo | CR | 2 (LI) | 1 | 66.0 | Alive |
| 5 | 4 | Yes | CHOP × 2 | POD | ICE × 5 | Auto | CR | BEAM | 2.58 | CR | 5 | CIE | Uncleara | 3 (HI) | 2 | 21.7 | DOD |
| 6 | 1 | Yes | CHOP × 1 | PR | ICE × 4 | Auto | CR | Cyclophosphamide, carmustine, etoposide | 2.8 | CR | 25 | CHOP × 3, Allo | PR | NAb | NAb | 59.2 | DOT |
| 7 | 3 | Yes | Pentostatin | POD | NYII–POD; topotecan/vinorelbine/thiotepa | Allo | PR | Thiotepa, cyclophosphamide, TBI, ATG | 9.91 | CR | 3 | TLR7 agonist, fludarabin cyclophosphamide, DLI | POD | 1 (L) | 0 | 13.1 | DOD |
| 8 | 4 | Yes | 2CDA × 2 | POD | CHOP × 3-POD; ICE × 2 | Allo | PR | TBI, cyclophosphamide | Unclear | CR | — | — | NA | 1 (L) | 2 | 149.6 | Alive |
| 9 | 2 | No | CHOP × 4 | PR | ICE-POD; EPOCH × 2 | — | NA | NA | NA | NA | NA | NA | NA | 4 (H) | 3 | 5.9 | DOD |
| 10 | 4 | Yes | CHOP × 6 | CR | ICE | Auto | CR | BEAM | 4.5 | CR | — | — | NA | NAb | NAb | 23 | Alive |
| 11 | 1 | No | ICE × 4 | PR | EPOCH × 1-POD; pentostatin × 4 | — | NA | NA | NA | NA | NA | NA | NA | 3 (HI) | 2 | 4 | DOD |
| 12 | 1 | No | ICE × 4 | POD | None | — | NA | NA | NA | NA | NA | NA | NA | 3 (HI) | 2 | 5.7 | DOD |
| 13 | 2 | No | IVAC × 4 | CR | Campath × 1 | Allo | CR | Thiotepa, cyclophosphamide, TBI | 8.02 | CR | NA | NA | NA | 3 (HI) | 2 | 4.7 | Alive |
| 14 | 4 | No | Pediatric ALL induction then IVAC × 2 | CR | None | Allo | CR | Etoposide, TBI | 8.01 | NRE | NA | NA | NA | 2 (LI) | 1 | 3 | Alive |
Listed for all 14 patients are initial chemotherapy regimens with response; further management including subsequent chemotherapy and transplant approach; risk stratification per IPI and PIT; overall survival, and vital status at the time of analysis. Time to treatment start indicates time interval between the onset of symptoms and start of induction.
Abbreviations: 2CDA = 2-chlorodeoxyadenosine/cladribine; ALL = acute lymphoblastic leukemia; ATG = antithymocyte globulin; BEAM = carmustine, etoposide, ara-C, melphalan; Bus/mel/flu = busulfan, melphalan, fludarabine; CHOP = cyclophosphamide/doxorubicin/vincristine/prednisone; CIE = cyclophosphamide, idarubicin, etoposide; CR = complete response; DLI = donor leukocyte infusion; DOD = died of disease; DOT = treatment-related death; EPOCH = etoposide, vincristine, doxorubin, cyclophosphamide, prednisone; H = high; HD = high dose; HI = high intermediate; ICE = ifosfamide, carboplatin, etoposide; IPI = International Prognostic Index; IVAC = ifosfamide, etoposide, high dose cytarabine; L = low; LI = low intermediate; MTX = methotrexate; NRE = no response evaluable; NYII = high dose cytarabinec, daunorubicin, PEG asparaginase, cyclophosphamide, HD MTX, vincristine; OS = overall survival; PEG = pegylated; PIT = index for peripheral T-cell lymphoma; POD = progression of disease; PR = partial response; Rx = treatment; TBI = total body irradiation; TLR7 = toll-like receptor 7; Txp = transplant.
Response unclear, treatment received at outside institution.
Incomplete clinical information to fully determine score.
Clinical Outcomes
Responses to induction regimens were CHOP (complete remission [CR], 1; partial response [PR], 2; progression of disease [POD], l), ICE (ifosfamide, carboplatin, etoposide)/IVAC (ifosfamide, etoposide, high-dose cytarabine) (CR, 4; PR, 2; POD, 2), and pentostatin/2-CDA (POD, 2). Three patients induced with CHOP received ICE as second-line therapy, 2 of 3 achieved a CR. One patient received ICE as consolidation after obtaining a CR to CHOP before proceeding to SCT. Eleven of 14 patients achieved at least a PR and proceeded to HDT-SCT. Four patients received an autologous SCT, and 8 patients an allogeneic SCT (1 as a second graft after relapse from autologous SCT). At the time of this report, 7 of 14 patients are alive, 3–149 months from the time of diagnosis; the 7 surviving patients all underwent HDT-SCT. Six of these remain in remission, 1 relapsed <1 year after allogeneic SCT, had been re-treated successfully with donor leukocyte infusions, but has since again relapsed. A total of 8 patients received ICE or IVAC as part of initial therapy and 5 of 8 are alive. Only 2 of 6 patients remain alive among those treated with other initial regimens and both surviving patients received ICE as part of consolidation before HDT-SCT. After autologous-SCT, 2 of 4 patients relapsed at 5 and 35 months. Moreover, after allogeneic-SCT 2 of 7 patients relapsed at 3 and 6 months. Two of 8 patients undergoing allo-SCT died of treatment-related toxicities. With a median follow-up time of 66 months, median PFS and OS for the entire cohort are 13.3 months (range, 2.4–148) and 59 months (range, 4–150 months), respectively (Figure 1).
Figure 1.
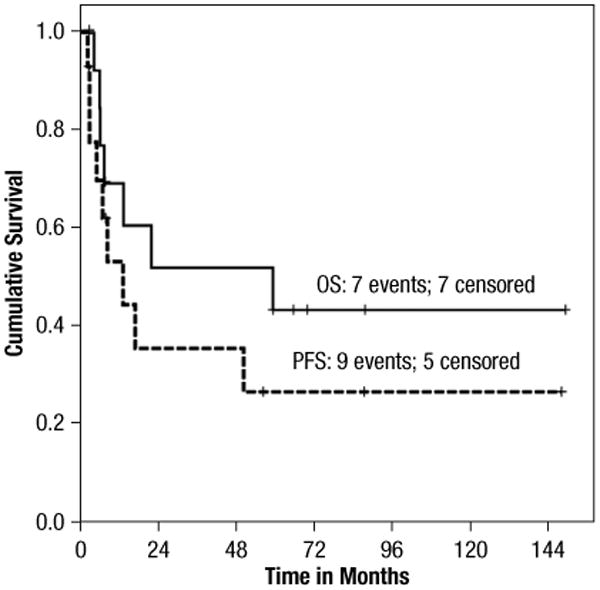
Progression-Free Survival and Overall Survival. Kaplan-Meier Curve of Progression-Free Survival and Overall Survival in Our Cohort
Complete information to calculate respective prognostic indexes (IPI or PIT) were available for 12 of 14 patients. A correlation with outcome could not be found for either of the 2. Three of 4 patients with an IPI of low to low-intermediate risk (0–2 factors) remain alive compared with 3 of 8 patients with IPI high-intermediate to high-risk disease (≥3 factors) (P = .267) (Figure 2). For the PIT, all 12 patients had at least 1 risk factor: 6 of 10 patients with a PIT of 1–2 are alive versus 0 of 2 patients for PIT of ≥3 (P = .117).
Figure 2.
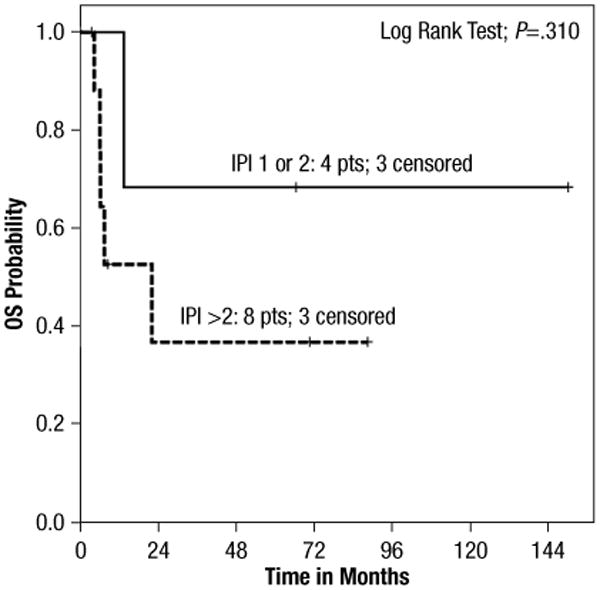
Survival per IPI. Kaplan-Meyer Curve Stratified in Groups with International Prognostic Index (IPI) 0–2 Versus 3–5 Demonstrating No Significant Difference in Overall Survival
Discussion
Management of HSTCL is challenging, and historically, outcome has almost uniformly been poor. There are no prospective trials to provide guidance for the treatment of this disease, and most of the current literature consists of case reports or case review series. Chemotherapy regimens employed in other series include CHOP and CHOP-like regimen,25 alemtuzumab/cladribine,34 hyperCVAD,35 fludarabine/alemtuzumab,36 IEV (ifosfamide, epirubicin, and etoposide),37 and pentostatin.38,39 Several authors suggest superior outcome with the addition of HDT-SCT, and a number of reports include cases treated successfully with autologous and allogeneic SCT.11,12,25,33,40–47 There are only 2 reports of larger single-center experiences in the literature thus far. The first was published by Belhadj and colleagues in 2003 and includes data from 21 patients with HSTCL diagnosed between 1981 and 2001.25 In their report, most patients (90.5%) received induction treatment with CHOP or CHOP-like regimens, and 2 of 21 subjects (9.5%) were induced using platinum cytarabine-based therapy. Seven of 21 patients (33.3%) failed to respond to induction, all died within 16 months. Nine of 21 patients (43%) achieved a CR, 5 of 21 patients (24%) a good PR, the latter including patients induced with non-CHOP regimen. Nine of 21 patients (43%) went on to either allogeneic bone marrow transplantation (3 of 21 patients—2 died of treatment-related disease, 1 with POD) or autologous bone marrow transplantation (6 of 21 patients (28.6%), 4 of which died within 13–33 months). Despite response to induction treatment in two-thirds of the treated patients, long-term therapeutic results were poor with a median survival of 16 months. At the time of report only 2 of 21 patients (9.5%) were alive; both had received induction treatment with platinum/cytarabine with PR and gone on to autologous bone marrow transplant. More recently, Falchook and colleagues published their single-center experience on 14 patients with HSTCL treated at M.D. Anderson Cancer Center between 1997 and 2007.33 Despite CR rates of 50% with various induction regimens, the duration was short-lived in most cases, and median survival was 8 months. Similar to other reports, all patients treated with CHOP-like regimen (6 of 14) did poorly. The 4 patients (29%) that were alive at the time of report (11–36 months) had been induced with more intense regimen, 3 of 4 survivors had been consolidated with allogeneic SCT. From these data the authors attempted to identify possible clinical characteristics to help predict outcome. Sex was the only factor conferring a statistically different survival (median OS 25 months in female vs. 8 months in male patients). There were trends suggesting better survival in patients with TCR rearrangements in the γ chain with all 4 patients with such rearrangement achieving CR and still alive in remission, though statistically nonsignificant trends toward worse survival were seen with liver involvement at diagnosis (median OS 7.5 vs. 13 months without liver involvement) and history of prior immunocompromise (median OS 6 vs. 11 months without prior immunocompromise). No trends in OS were seen for age, presence of B-symptoms, or cytopenias at presentation, TCR status, or cytogenetics.
Our single-institution experience further supports the notion that use of non-CHOP induction chemotherapy regimens such as ICE or IVAC and early HDT-SCT consolidation might improve the outcome for patients with HSTCL compared with reported results with CHOP or CHOP-like regimens alone. At the time of this report, 7 of 14 patients (50%) remain alive, 5 of which were initially treated with non-CHOP regimen. All 7 surviving patients had preceded to HDT-SCT, 5 receiving an allogeneic, and 2 an autologous graft. Both patients treated with autologous SCT were in CR at the time of transplant, while 2 of 5 recipients of allogeneic grafts received transplant in PR. The 3 patients that did not undergo SCT as part of their management did poorly (see also Figure 1).
When applying the prognostic factors suggested by Falchook and colleagues, our data do not confirm a trend toward worse OS in patients with liver involvement at the time of diagnosis (P = .382), nor improved OS in patients with γ TCR rearrangements (P = .919) or previous history of immunosuppression (P = .455). Formally established scoring models used to assess prognosis in peripheral T-cell lymphoma include the IPI48 and the PIT.49 These systems might not be as useful for stratifying patients with HSTCL because those affected are almost universally young with stage IV disease. As noted above, we were unable to show correlation with outcome for either of the 2 prognostic models, bearing in mind the limitations of our sample size. Notably, for patients who are not estimated among the highest risk at initial presentation (PIT < 3; IPI < 3) our data show long-term survivors among those consolidated with autologous as well as allogeneic SCT.
Serial reports of HSTCL in patients receiving immunosuppressive therapy with agents blocking TNF-α have prompted a safety alert through the US FDA, originally issued in 2009 and recently updated.50An increase in incidence has also been reported with other immunosuppressants such as azathioprine, or 6-MP.24,51–53 The contribution of the underlying autoimmune disease itself is less well established for inflammatory bowel diseases54 than for autoimmune arthritis.55 In our series, 8 of 14 patients carried a diagnosis of autoimmune disease, 6 of which were treated with systemic immunosuppressants other than corticosteroids. Only 3 had received previous TNF-α blockade, which was given sequentially or in combination with 6-MP. The other 3 patients had been treated with 6-MP alone. Two patients with a previous diagnosis of autoimmune disease had no documented history of such immunosuppression, but for 1 of them previous treatment records for juvenile arthritis were unavailable to our review.
Conclusion
Our choice of transplant approach has largely been guided by the availability of a suitable donor. Though our series is clearly too small to draw definitive conclusions our preference is for allogeneic SCT. However, our experience suggests HDT and consolidation with autologous SCT represents a reasonable alternative for those who can achieve a CR to their initial chemotherapy. Certainly for patients without a suitable allogeneic donor, this is our preferred approach.
As a case series, this study's principal limitations are the small number of subjects and its retrospective nature. It certainly cannot dissect the relative importance of the individual components of therapy (non-CHOP induction vs. HDT consolidation). In the absence of prospective trials for this very rare disease, it might however provide guidance in the treatment of these young patients with no established standard of care.
Clinical Practice Points.
Hepatosplenic T-cell lymphoma typically presents with infiltration of spleen and liver and rarely involves nodal regions.
A history of inflammatory bowel disease might predispose patients to the development of HSTCL; the use of TNF-α antagonist or 6-MP analogs might also increase this risk.
Of the 14 patients in our study, 7 remain alive; 6 received non–CHOP-based induction therapies (ICE or IVAC).
All patients who remained without evidence of disease underwent consolidative transplantation.
In our experience, consideration of a non–CHOP-based induction therapy with intent to consolidate with an allogeneic SCT in first CR appears to be a necessity considering the high propensity of primary refractory or short interval relapse associated with HSTCL.
Footnotes
Disclosures: The authors have stated that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Farcet JP, Gaulard P, Marolleau JP, et al. Hepatosplenic T-cell lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma delta. Blood. 1990;75:2213–9. [PubMed] [Google Scholar]
- 2.Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the international lymphoma Study Group. Blood. 1994;84:1361–92. [PubMed] [Google Scholar]
- 3.Harris NL, Jaffe ES, Diebold J, et al. Lymphoma classification–from controversy to consensus: the R.E.A.L. and WHO Classification of lymphoid neoplasms. Ann Oncol. 2000;11(Suppl 1):3–10. [PubMed] [Google Scholar]
- 4.Vega F, Medeiros LJ, Gaulard P. Hepatosplenic and other gammadelta T-cell lymphomas. Am J Clin Pathol. 2007;127:869–80. doi: 10.1309/LRKX8CE7GVPCR1FT. [DOI] [PubMed] [Google Scholar]
- 5.Gaulard P, Belhadj K, Reyes F. Gammadelta T-cell lymphomas. Semin Hematol. 2003;40:233–43. doi: 10.1016/s0037-1963(03)00137-9. [DOI] [PubMed] [Google Scholar]
- 6.Harris NL, Jaffe ES, Diebold J, et al. The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Report of the clinical advisory committee meeting, Airlie house, Virginia, November, 1997. Ann Oncol. 1999;10:1419–32. doi: 10.1023/a:1008375931236. [DOI] [PubMed] [Google Scholar]
- 7.Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. A progress report. Am J Clin Pathol. 1999;111(Suppl 1):S8–12. [PubMed] [Google Scholar]
- 8.Gaulard P, Bourquelot P, Kanavaros P, et al. Expression of the alpha/beta and gamma/delta T-cell receptors in 57 cases of peripheral T-cell lymphomas. Identification of a subset of gamma/delta T-cell lymphomas. Am J Pathol. 1990;137:617–28. [PMC free article] [PubMed] [Google Scholar]
- 9.Wong KF, Chan JK, Matutes E, et al. Hepatosplenic gamma delta T-cell lymphoma. A distinctive aggressive lymphoma type. Am J Surg Pathol. 1995;19:718–26. doi: 10.1097/00000478-199506000-00013. [DOI] [PubMed] [Google Scholar]
- 10.Wei SZ, Liu TH, Wang DT, et al. Hepatosplenic gammadelta T-cell lymphoma. World J Gastroenterol. 2005;11:3729–34. doi: 10.3748/wjg.v11.i24.3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooke CB, Krenacs L, Stetler-Stevenson M, et al. Hepatosplenic T-cell lymphoma: a distinct clinicopathologic entity of cytotoxic gamma delta T-cell origin. Blood. 1996;88:4265–74. [PubMed] [Google Scholar]
- 12.Weidmann E. Hepatosplenic T cell lymphoma. A review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990. Leukemia. 2000;14:991–7. doi: 10.1038/sj.leu.2401784. [DOI] [PubMed] [Google Scholar]
- 13.Wu H, Wasik MA, Przybylski G, et al. Hepatosplenic gamma-delta T-cell lymphoma as a late-onset posttransplant lymphoproliferative disorder in renal transplant recipients. Am J Clin Pathol. 2000;113:487–96. doi: 10.1309/YTTC-F55W-K9CP-EPX5. [DOI] [PubMed] [Google Scholar]
- 14.Khan WA, Yu L, Eisenbrey AB, et al. Hepatosplenic gamma/delta T-cell lymphoma in immunocompromised patients. Report of two cases and review of literature. Am J Clin Pathol. 2001;116:41–50. doi: 10.1309/TC9U-FAV7-0QBW-6DFC. [DOI] [PubMed] [Google Scholar]
- 15.Steurer M, Stauder R, Grünewald K, et al. Hepatosplenic gammadelta-T-cell lymphoma with leukemic course after renal transplantation. Hum Pathol. 2002;33:253–8. doi: 10.1053/hupa.2002.31301. [DOI] [PubMed] [Google Scholar]
- 16.Hassan R, Franco SA, Stefanoff CG, et al. Hepatosplenic gammadelta T-cell lymphoma following seven malaria infections. Pathol Int. 2006;56:668–73. doi: 10.1111/j.1440-1827.2006.02027.x. [DOI] [PubMed] [Google Scholar]
- 17.François A, Lesesve JF, Stamatoullas A, et al. Hepatosplenic gamma/delta T-cell lymphoma: a report of two cases in immunocompromised patients, associated with isochromosome 7q. Am J Surg Pathol. 1997;21:781–90. doi: 10.1097/00000478-199707000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Colombel JF, Loftus EV, Jr, Tremaine WJ, et al. The safety profile of infliximab in patients with Crohn's disease: the Mayo Clinic experience in 500 patients. Gastroenterology. 2004;126:19–31. doi: 10.1053/j.gastro.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 19.Peyrin-Biroulet L, Deltenre P, de Suray N, et al. Efficacy and safety of tumor necrosis factor antagonists in Crohn's disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol. 2008;6:644–53. doi: 10.1016/j.cgh.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Zeidan A, Sham R, Shapiro J, et al. Hepatosplenic T-cell lymphoma in a patient with Crohn's disease who received infliximab therapy. Leuk Lymphoma. 2007;48:1410–3. doi: 10.1080/10428190701345433. [DOI] [PubMed] [Google Scholar]
- 21.Mackey AC, Green L, Liang LC, et al. Hepatosplenic T cell lymphoma associated with infliximab use in young patients treated for inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2007;44:265–7. doi: 10.1097/MPG.0b013e31802f6424. [DOI] [PubMed] [Google Scholar]
- 22.Rosh JR, Gross T, Mamula P, et al. Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn's disease: a cautionary tale? Inflamm Bowel Dis. 2007;13:1024–30. doi: 10.1002/ibd.20169. [DOI] [PubMed] [Google Scholar]
- 23.Rosh JR, Oliva-Hemker M. Infliximab use and hepatosplenic T cell lymphoma: questions to be asked and lessons learned. J Pediatr Gastroenterol Nutr. 2007;44:165–7. doi: 10.1097/MPG.0b013e318031d61a. [DOI] [PubMed] [Google Scholar]
- 24.Kandiel A, Fraser AG, Korelitz BI, et al. Increased risk of lymphoma among inflammatory bowel disease patients treated with azathioprine and 6-mercaptopurine. Gut. 2005;54:1121–5. doi: 10.1136/gut.2004.049460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belhadj K, Reyes F, Farcet JP, et al. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood. 2003;102:4261–9. doi: 10.1182/blood-2003-05-1675. [DOI] [PubMed] [Google Scholar]
- 26.Vega F, Medeiros LJ, Bueso-Ramos C, et al. Hepatosplenic gamma/delta T-cell lymphoma in bone marrow. A sinusoidal neoplasm with blastic cytologic features. Am J Clin Pathol. 2001;116:410–9. doi: 10.1309/BM40-YM6J-9T3X-MH8H. [DOI] [PubMed] [Google Scholar]
- 27.Macon WR, Levy NB, Kurtin PJ, et al. Hepatosplenic alphabeta T-cell lymphomas: a report of 14 cases and comparison with hepatosplenic gammadelta T-cell lymphomas. Am J Surg Pathol. 2001;25:285–96. doi: 10.1097/00000478-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Lai R, Larratt LM, Etches W, et al. Hepatosplenic T-cell lymphoma of alphabeta lineage in a 16-year-old boy presenting with hemolytic anemia and thrombocytopenia. Am J Surg Pathol. 2000;24:459–63. doi: 10.1097/00000478-200003000-00016. [DOI] [PubMed] [Google Scholar]
- 29.Alonsozana EL, Stamberg J, Kumar D, et al. Isochromosome 7q: the primary cytogenetic abnormality in hepatosplenic gammadelta T cell lymphoma. Leukemia. 1997;11:1367–72. doi: 10.1038/sj.leu.2400742. [DOI] [PubMed] [Google Scholar]
- 30.Jonveaux P, Daniel MT, Martel V, et al. Isochromosome 7q and trisomy 8 are consistent primary, non-random chromosomal abnormalities associated with hepatosplenic T gamma/delta lymphoma. Leukemia. 1996;10:1453–5. [PubMed] [Google Scholar]
- 31.Rossbach HC, Chamizo W, Dumont DP, et al. Hepatosplenic gamma/delta T-cell lymphoma with isochromosome 7q, translocation t(7;21), and tetrasomy 8 in a 9-year-old girl. J Pediatr Hematol Oncol. 2002;24:154–7. doi: 10.1097/00043426-200202000-00020. [DOI] [PubMed] [Google Scholar]
- 32.Wlodarska I, Martin-Garcia N, Achten R, et al. Fluorescence in situ hybridization study of chromosome 7 aberrations in hepatosplenic T-cell lymphoma: isochromosome 7q as a common abnormality accumulating in forms with features of cytologic progression. Genes Chromosomes Cancer. 2002;33:243–51. doi: 10.1002/gcc.10021. [DOI] [PubMed] [Google Scholar]
- 33.Falchook GS, Vega F, Dang NH, et al. Hepatosplenic gamma-delta T-cell lymphoma: clinicopathological features and treatment. Ann Oncol. 2009;20:1080–5. doi: 10.1093/annonc/mdn751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaeger G, Bauer F, Brezinschek R, et al. Hepatosplenic gammadelta T-cell lymphoma successfully treated with a combination of alemtuzumab and cladribine. Ann Oncol. 2008;19:1025–6. doi: 10.1093/annonc/mdn119. [DOI] [PubMed] [Google Scholar]
- 35.Tey SK, Marlton PV, Hawley CM, et al. Post-transplant hepatosplenic T-cell lymphoma successfully treated with HyperCVAD regimen. Am J Hematol. 2008;83:330–3. doi: 10.1002/ajh.21062. [DOI] [PubMed] [Google Scholar]
- 36.Mittal S, Milner BJ, Johnston PW, et al. A case of hepatosplenic gamma-delta T-cell lymphoma with a transient response to fludarabine and alemtuzumab. Eur J Haematol. 2006 Jun;76:531–4. doi: 10.1111/j.1600-0609.2006.00646.x. [DOI] [PubMed] [Google Scholar]
- 37.Moleti ML, Testi AM, Giona F, et al. Gamma-delta hepatosplenic T-cell lymphoma. Description of a case with immunophenotypic and molecular follow-up successfully treated with chemotherapy alone. Leuk Lymphoma. 2006;47:333–6. doi: 10.1080/10428190500293321. [DOI] [PubMed] [Google Scholar]
- 38.Corazzelli G, Capobianco G, Russo F, et al. Pentostatin (2′-deoxycoformycin) for the treatment of hepatosplenic gammadelta T-cell lymphomas. Haematologica. 2005;90:ECR14. [PubMed] [Google Scholar]
- 39.Iannitto E, Barbera V, Quintini G, et al. Hepatosplenic gammadelta T-cell lymphoma: complete response induced by treatment with pentostatin. Br J Haematol. 2002;117:995–6. doi: 10.1046/j.1365-2141.2002.03537_3.x. [DOI] [PubMed] [Google Scholar]
- 40.Ooi J, Iseki T, Adachi D, et al. Successful allogeneic bone marrow transplantation for hepatosplenic gammadelta T cell lymphoma. Haematologica. 2001;86:E25. [PubMed] [Google Scholar]
- 41.Gassas A, Kirby M, Weitzman S, et al. Hepatosplenic gammadelta T-cell lymphoma in a 10-year-old boy successfully treated with hematopoietic stem cell transplantation. Am J Hematol. 2004;75:113–4. doi: 10.1002/ajh.10466. [DOI] [PubMed] [Google Scholar]
- 42.Domm JA, Thompson M, Kuttesch JF, et al. Allogeneic bone marrow transplantation for chemotherapy-refractory hepatosplenic gammadelta T-cell lymphoma: case report and review of the literature. J Pediatr Hematol Oncol. 2005;27:607–10. doi: 10.1097/01.mph.0000187431.37369.f5. [DOI] [PubMed] [Google Scholar]
- 43.Sakai R, Fujisawa S, Fujimaki K, et al. Long-term remission in a patient with hepatosplenic gammadelta T cell lymphoma after cord blood stem cell transplantation following autologous peripheral blood stem cell transplantation. Bone Marrow Transplant. 2006;37:537–8. doi: 10.1038/sj.bmt.1705272. [DOI] [PubMed] [Google Scholar]
- 44.Procházka V, Papajík T, Jarosová M, et al. T-cell gamma/delta hepatosplenic lymphoma - prolonged remission induced by aggressive first line treatment. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2005;149:275–6. [PubMed] [Google Scholar]
- 45.He S, Roberts A, Ritchie D, et al. Graft-versus-lymphoma effect in progressive hepatosplenic gamma/delta T-cell lymphoma. Leuk Lymphoma. 2007;48:1448–50. doi: 10.1080/10428190701400071. [DOI] [PubMed] [Google Scholar]
- 46.Chen AI, McMillan A, Negrin RS, et al. Long-term results of autologous hematopoietic cell transplantation for peripheral T cell lymphoma: the Stanford experience. Biol Blood Marrow Transplant. 2008;14:741–7. doi: 10.1016/j.bbmt.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konuma T, Ooi J, Takahashi S, et al. Allogeneic stem cell transplantation for hepatosplenic gammadelta T-cell lymphoma. Leuk Lymphoma. 2007;48:630–2. doi: 10.1080/10428190601126941. [DOI] [PubMed] [Google Scholar]
- 48.Ansell SM, Habermann TM, Kurtin PJ, et al. Predictive capacity of the international prognostic factor index in patients with peripheral T-cell lymphoma. J Clin Oncol. 1997;15:2296–301. doi: 10.1200/JCO.1997.15.6.2296. [DOI] [PubMed] [Google Scholar]
- 49.Gallamini A, Stelitano C, Calvi R, et al. Peripheral T-cell lymphoma unspecified (PTCL-U): a new prognostic model from a retrospective multicentric clinical study. Blood. 2004;103:2474–9. doi: 10.1182/blood-2003-09-3080. [DOI] [PubMed] [Google Scholar]
- 50.FDA. Tumor necrosis factor (TNF) blockers, azathioprine and/or mercaptopurine: update on reports of hepatosplenic T-cell lymphoma in adolescents and young adults. [Accessed: July 18, 2011]; Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm251443.htm.
- 51.Pozadzides JV, Pro B. Hepatosplenic T-cell lymphoma and TNF-alpha inhibitors. Expert Rev Hematol. 2009;2:611–4. doi: 10.1586/ehm.09.62. [DOI] [PubMed] [Google Scholar]
- 52.Connell WR, Kamm MA, Dickson M, et al. Long-term neoplasia risk after azathioprine treatment in inflammatory bowel disease. Lancet. 1994;343:1249–52. doi: 10.1016/s0140-6736(94)92150-4. [DOI] [PubMed] [Google Scholar]
- 53.Farrell RJ, Ang Y, Kileen P, et al. Increased incidence of non-Hodgkin's lymphoma in inflammatory bowel disease patients on immunosuppressive therapy but overall risk is low. Gut. 2000;47:514–9. doi: 10.1136/gut.47.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones JL, Loftus EV., Jr Lymphoma risk in inflammatory bowel disease: is it the disease or its treatment? Inflamm Bowel Dis. 2007;13:1299–307. doi: 10.1002/ibd.20211. [DOI] [PubMed] [Google Scholar]
- 55.Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum. 2006;54:692–701. doi: 10.1002/art.21675. [DOI] [PubMed] [Google Scholar]