

Tyrosine kinase 2 is required for release of IL-27(p28) by TLR4-activated macrophages; disruption of this pathway is protective during in vivo models of sepsis.
Keywords: inflammation, macrophages, lipopolysaccharide, shock, interferon
Abstract
The aim of this study was to test the hypothesis that gene expression and release of IL-27 may be modulated by Tyk2. Macrophages derived from the peritoneum or bone marrow of C57BL/10SnJ (WT) mice produced abundant amounts of IL-27(p28) following TLR4 activation by LPS. In contrast, production of IL-27(p28), but not EBI3, was reduced by ∼50% in TLR4-activated macrophages derived from mice with genetic deficiency of Tyk2 compared with WT macrophages. Frequencies of IL-27(p28)+F4/80+CD11b+ cells were lower in TLR4-activated macrophages derived from Tyk2−/− mice. Mechanistically, Tyk2−/− resulted in disruption of a type I IFN-dependent mechanism for production of IL-27(p28), which was induced by type I IFNs, and release of IL-27 was defective in macrophages from IFN-β−/− and IFNAR1−/− mice. In contrast, Tyk2 was not required to mediate the effects of IL-27 on target gene expression in CD4+ T cells. In vivo, we observed that Tyk2−/− mice have improved survival following endotoxic shock or polymicrobial sepsis induced by CLP. Plasma levels of IL-27(p28) during endotoxic shock or polymicrobial sepsis were markedly reduced in Tyk2−/− mice compared with WT mice. Disruption of IL-27 signaling using IL-27RA−/− mice was protective against sepsis-associated mortality. These data suggest that Tyk2 may mediate adverse outcomes of SIRS by promoting the production of IL-27. In conclusion, this report identifies Tyk2 as a prerequisite factor in the molecular networks that are involved in generation of IL-27.
Introduction
Severe sepsis and septic shock are life-threatening complications of bacterial and fungal infections [1]. SIRS is frequently observed in response to noninfectious trauma and displays similar clinical features as pathogen-induced sepsis. It has been estimated that the worldwide incidence of sepsis may reach 18 million cases/year, with mortality rates of 20–50% [2]. The pathophysiology of severe sepsis and SIRS is not understood completely [3]. The current consensus is that a dysbalance in the acute inflammatory response plays a central role during sepsis/SIRS [4]. The molecular mechanisms of acute inflammation include, but are not limited to, the activation of intracellular kinase signaling cascades. The activity of kinases regulates the downstream release of multiple cytokines, which may have protective or adverse effects during sepsis/SIRS.
Tyk2 is a member of the JAK family of intracellular receptor-associated kinases, which plays a critical role during LPS-induced endotoxin shock (an experimental model of SIRS) [5]. Expression of Tyk2 occurs ubiquitously, including various immune cell types, such as macrophages, DCs, NK cells, mast cells, T cells, and B cells [6]. Tyk2 associates with several cytokine receptors, including IFNAR1, IL-12Rβ, IL-10R2, gp130, and IL-13Rα [6]. Human T cells with a genetic defect in Tyk2 manifested failure of cytokine-induced signaling [6–8]. Whereas Tyk2−/− mice are protected during endotoxic shock [5], Tyk2−/− mice display reduced survival rates following infection with Toxoplasma gondii or Listeria monocytogenes [9, 10]. Tyk2 is required for the production of IFN-α and IFN-β (type I IFNs) in response to TLR4 activation by LPS in macrophages and during endotoxic shock [5]. For instance, mRNA for IFN-β and IFN-α4 is diminished in LPS-activated macrophages derived from Tyk2−/− mice, whereas generation of several other proinflammatory cytokines and NO remains intact in Tyk2−/− mice following endotoxic shock [5].
IL-27 is another factor that appears to be involved in the outcome of experimental sepsis (IL-27) [11]. IL-27 is a heterodimeric cytokine assembled by noncovalent interactions of the subunit proteins, IL-27(p28) and EBI3 [12, 13]. IL-27 binds with high affinity to the IL-27RA receptor (also known as WSX-1), transducing signaling via gp130 [14]. Signaling via the IL-27RA/gp130R complex initiates phosphorylation of STAT1 and STAT3 and downstream modulation of target genes in T lymphocytes [15, 16]. The biological effects of IL-27 vary greatly in being dependent on the immunological context. IL-27 may promote inflammation in models of chronic colitis, bacterial infection, and arthritis [11, 17, 18]. On the other hand, IL-27 may silence inflammation, as seen in protozoan infections and in autoimmune encephalitis [19–21].
Production of IL-27 has been noted in the course of viral, bacterial, fungal, and protozoan infections [11, 19, 22]. Major cellular sources of IL-27 are DCs and macrophages that have been activated by microbial products [13, 23]. Binding of LPS to the TLR4 receptor results in abundant production of IL-27 [23]. For gene expression of IL-27, MyD88 and TRIF adaptor proteins are required [24, 25]. MyD88 mediates activation of NF-κB (c-Rel) for induction of gene expression of IL-27 [25]. TRIF mediates the nuclear translocation of IRFs, such as IRF-1, IRF-3, and IRF-9, which bind to the promoter region of IL-27(p28) to promote transcriptional activation [24, 26]. In addition, we have reported recently that Akt/PI3K signaling antagonized TLR4-dependent, de novo synthesis of IL-27(p28) in cultures of peritoneal macrophages and during endotoxic shock [23].
In this study, we have investigated the molecular mechanisms of gene expression and release of IL-27(p28) in macrophages and during sepsis. As IL-27(p28) and Tyk2 have been described as central players of endotoxic shock, we hypothesized a potential cross-talk of IL-27(p28) and Tyk2. We uncover a novel role of Tyk2 as a regulating factor for production of IL-27(p28) during sepsis. Tyk2 promoted the release of IL-27(p28) by activating the endogenous production of type I IFNs. Our findings suggest that deficiency of Tyk2 protects against shock-associated mortality by disruption of the generation of adverse IL-27(p28).
MATERIALS AND METHODS
Research animals
All procedures were performed in accordance with the U.S. National Institutes of Health guidelines, the University Committee on Use and Care of Animals of the University of Michigan, the State Investigation Office of Rhineland-Palatinate, the Federation of European Laboratory Animal Science Associations guidelines, and Directive 2010/63/EU of the European Parliament and of the Council of the European Union. IL-27RA−/− (B6N.129P2-Il27ratm1Mak v/J) mice were bred at the University Medical Center Mainz. IFNAR1−/− (B6.129P2-Ifnar1tm1Agt) and IFN-β−/− (B6.129P2-Ifnbtm1TI) mice were bred at the University of Veterinary Medicine Vienna. TRIF−/− mice were bred at the University of Michigan. Male mice of strains TLR4−/− (B6.B10ScN-Tlr4lps-del/JthJ), MyD88−/− [B6.129P2(SJL)-Myd88tm1.1Defr/J], C57BL/10SnJ, and Tyk2−/− (B10.D1-H2q/SgJ, a naturally occurring mis-sense mutation in Tyk2, resulting in a loss of function), were from The Jackson Laboratory (Bar Harbor, ME, USA). Tyk2−/− mice were on a C57BL/10SnJ background, and all other strains were on a C57BL/6 background. For the data shown in Fig. 5, we used Tyk2−/− mice (B6N.129P2-Tyk2tm1Biat) on a C57BL/6N background, as described previously [27]. All animals were housed under specific pathogen-free conditions.
Figure 5. IL-27-mediated induction of target genes does not require Tyk2 in CD4+ T cells.
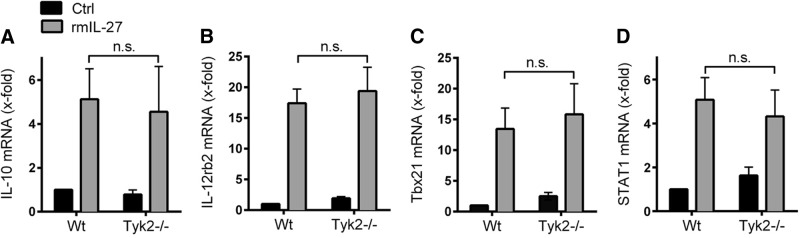
CD4+ T cells from 8- to 12-week-old WT mice and Tyk2−/− mice were stimulated with rmIL-27 (10 ng/ml) under neutral conditions or left without IL-27 (Ctrl) for 48 h. Total RNA was isolated, and mRNA expression for IL-10 (A), IL-12rb2 (B), Tbx21 (C), and STAT1 (D) was determined by RT-PCR. Data were normalized to the housekeeping gene Ube2d2 and calculated relative to untreated WT cells (Ctrl). Data are pooled from three independent experiments. One-way ANOVA; mean ± sem; n.s., not significant.
Cultures of macrophages
BMDM were obtained from femurs and tibias and incubated for 7 days in RPMI-1640 medium (25 mM HEPES, 100 U/ml penicillin-streptomycin, 20% FCS, 30% L cell conditioned media). For experiments, BMDM were plated at 5 × 105 cells/ml in RPMI 1640 (25 mM HEPES, 100 U/ml penicillin-streptomycin, 0.1% BSA). For cell activation, LPS (Escherichia coli, 0111:B4; Sigma-Aldrich, St. Louis, MO, USA) or rmIFN-α (R&D Systems, Minneapolis, MN, USA) was used. For peritoneal-elicited macrophages, mice were injected i.p. with 1 ml thioglycollate (2.4%) and cells harvested by peritoneal lavage, 4 days later, as described elsewhere [23].
Cultures of T cells
Primary CD4+ T cells were purified from splenocytes by MACS with a mouse CD4+ T Cell Isolation Kit II (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer's instructions. Cells were seeded in 24-well plates at a density of 1.5 × 106 cells/ml/well in IMDM, supplemented with 10% FCS, 100 μg/ml penicillin, 100 U/ml streptomycin, 2 mM L-glutamine, and 50 μM β-ME. Anti-mCD3ϵ (5 μg/ml) and anti-mCD28 (1 μg/ml) antibodies were added, and cells were grown under neutral condition, i.e., in the presence of anti-IL-12 (5 μg/ml), anti-IFN-γ (5 μg/ml), and anti-IL-4 (1 μg/ml), and incubated at 37°C for 48 h in the presence or absence of rmIL-27. All antibodies and cytokines were from eBioscience (San Diego, CA, USA).
Endotoxic shock
Body weight was measured for each individual mouse before injection with LPS (10 mg/kg body weight i.p.; E. coli 0111:B4; Sigma-Aldrich). Plasma was collected in EDTA (5–10 mM) and survival monitored at least twice daily for 10 days [23]. Body-surface temperature was measured using a YSI4600/YSI427 thermometer.
CLP
Polymicrobial sepsis was induced by CLP, as described elsewhere [28]. Briefly, mice were anesthetized using ketamine/xylazine before a 1-cm midline incision in the abdomen. The distal part of the cecum (5–7 mm) was ligated, punctured through-and-through with a 21-G needle, and a small portion of feces extruded to ensure patency. After wound closure, the mice received 1 ml sterile NaCl 0.9% s.c. for fluid resuscitation. The surgeon was blinded regarding the age/sex-matched groups of WT or transgenic mice. Monitoring of survival, EDTA plasma collection, and measurements of body-surface temperature was done as described above.
ELISA
ELISA kits (R&D Systems) for mouse IL-27(p28) and IFN-β were performed according to the manufacturer's instructions [23].
Analysis of mRNA by PCR
From experiments using macrophages, the total RNA was extracted [RNeasy Kit; Qiagen, Valencia, CA, USA; or TriFAST (peqGOLD peqlab)], reverse-transcribed (TaqMan; Applied Biosystems, Life Technologies, Carlsbad, CA, USA) and amplified (SYBR Green Mastermix; Applied Biosystems) as described elsewhere [23]. Results were calculated by the 2-ΔΔ comparative threshold relative quantification method and normalized to GAPDH. The following mouse primer sequences were used (Invitrogen, Carlsbad, CA, USA): p28 (forward) 5′-GGCCATGAGGCTGGATCTC-3′, p28 (reverse) 5′-AACATTTGAATCCTGCAGCCA-3′; EBI3 (forward) 5′-GGCTGAGCGAATCATCAA-3′, EBI3 (reverse) 5′-GAGAGAGAAGATGTCCGGGAA-3′; GAPDH (forward) 5′-TACCCCCAATGTGTCCGTCGTG-3′, GAPDH (reverse) 5′-CCTTCAGTGGGCCCTCAGATGC-3′.
For the work with CD4+ T cells, the total RNA was isolated with TriFAST (peqGOLD peqlab), converted to cDNA (iScript cDNA synthesis kit; Bio-Rad, Hercules, CA, USA), and PCRs performed in duplicates on a Stratagene Mx3000P machine. Expression levels were normalized to the housekeeping gene Ube2d2. Primers and probes for Ube2d2 and IL-10 are available through the Real-Time Primer and Probe Database (http://medgen.ugent.be/rtprimerdb/index.php; IDs 3377 and 3892, respectively). Primers for Tbx21 (QT00129822), Gata3 (QT00170828), and ll12rβ2 (QT00136416) were purchased from Qiagen (QuantiTect). Primers (Sigma-Aldrich) and probes (Metabion, Martinsried, Germany) for STAT1 were as follows: STAT1 (forward) 5′-GATCAGCTGCAAACGTGGTTC-3′, STAT1 (reverse) 5′-GCTTTTTAAGCTGCTGACGGA-3′, STAT1 (probe) 5′-FAM-CCATTGTTGCAGAGACCCTGCAGCA-BHQ-3′.
Flow cytometry
Macrophages were incubated with monensin (2 μM; Sigma-Aldrich) and stained using the Cytofix/Cytoperm kit (BD Biosciences, San Jose, CA, USA) and BD Fc Block. Fifty thousand events were acquired on a BD LSR II flow cytometer (BD Biosciences) and data analyzed with WinList for Win32 3.0 software (Verity Software House, Topsham, ME, USA). All antibodies used were anti-mouse, together with matched fluorochrome-labeled isotype controls (eBioscience): PE IL-27(p28) (Clone MM27-7B1), allophycocyanin F4/80 (Clone BM8), efluor450 CD11b (Clone M1/70).
Statistical analysis
In vitro experiments were performed independently, two to three times, and in vivo data were generated with the numbers of mice/group, as indicated in the figure legends. Data sets from all independent experiments were analyzed by one-way ANOVA, two-tailed Student's t-test, Mann-Whitney test, and survival data by the log-rank (Mantel-Cox) test using GraphPad Prism Version 5.04. All values are expressed as mean, error bars represent sem, and differences were considered significant when P < 0.05.
RESULTS
Tyk2 promotes the production of IL-27(p28) by TLR4-activated F4/80+CD11b+ macrophages
To study a potential cross-talk of Tyk2 with the production of IL-27, BMDM were obtained from C57BL/10SnJ WT mice or Tyk2−/− mice. BMDM were stimulated with LPS, followed by analysis of mRNA levels for IL-27(p28), using RT-PCR (Fig. 1A). As expected, TLR4 activation strongly induced gene expression of IL-27(p28) in BMDM from WT mice. The relative gene expression of IL-27(p28) was reduced by >50% in TLR4-activated BMDM from Tyk2−/− mice compared with BMDM from WT mice (Fig. 1A). When mRNA for EBI3 was quantified, a fivefold increase following TLR4 activation by LPS was observed compared with EBI3 mRNA amounts in resting BMDM (Fig. 1B). Baseline gene-expression levels of EBI3 appeared to be higher than for IL-27(p28) (data not shown). Interestingly, the amounts of mRNA for EBI3 were not significantly lower in LPS-activated BMDM from Tyk2−/− mice compared with WT mice (Fig. 1B). As EBI3 is also a subunit for IL-35, its expression is not specific for IL-27. Accordingly, we focused our subsequent studies on IL-27(p28) and Tyk2.
Figure 1. Tyk2 is required for gene expression and release of IL-27(p28) in TLR4-activated macrophages.
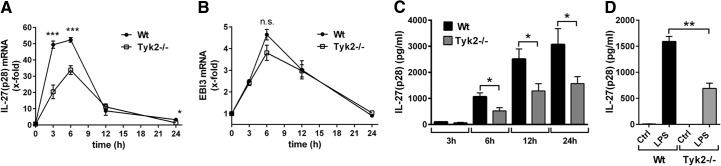
(A) RT-PCR of IL-27(p28) gene expression in BMDM from C57BL/10SnJ (WT) mice or Tyk2−/− mice after incubation with LPS (1 μg/ml) at time-points 3–24 h. (B) RT-PCR of EBI3 gene expression in LPS-activated BMDM from WT mice or Tyk2−/− mice at different time-points. Samples were from identical experiments as RT-PCR for IL-27(p28), as shown above; n.s., not significant. (C) Release of IL-27(p28) in supernatants of cultured BMDM from WT mice or Tyk2−/− mice after incubation with LPS for the indicated time-points; ELISA. (D) Quantification of IL-27(p28) in supernatants of cultured thioglycollate-elicited peritoneal macrophages from WT mice or Tyk2−/− mice left untreated (Ctrl) or activated with LPS (1 μg/ml) for 8 h; ELISA. One-way ANOVA; mean ± sem; *P < 0.05; **P < 0.01; ***P < 0.001. Graphs are aggregates from three independent experiments.
In response to LPS activation, IL-27(p28) protein accumulated in supernatants of BMDM from WT mice in a time-dependent manner (6–24 h), whereas much less IL-27(p28) was released by TLR4-activated BMDM from Tyk2−/− mice (Fig. 1C). In LPS-activated, thioglycollate-elicited peritoneal macrophages (“tissue macrophages”), the appearance of IL-27(p28) in supernatant fluids was also partly defective with genetic absence of Tyk2 (Fig. 1D). Production of IL-27(p28) was similar in studies with LPS-activated BMDM compared with peritoneal macrophages. For subsequent experiments, we used BMDM, as BMDM preparations yielded higher cell numbers and purity.
Flow cytometry staining for intracellular IL-27(p28) in cultured BMDM, derived from WT mice or Tyk2−/− mice, showed reduced intracellular staining for IL-27(p28) in F4/80+CD11b+ cells from Tyk2−/− mice after LPS (45.9% vs. 26.4%; Fig. 2A and B). Only minimal staining was observed with isotype control antibodies, and virtually all BMDM from both strains of mice were CD11b+F4/80+ double-positive (>95%; data not shown).
Figure 2. Lower frequencies of IL-27(p28)+F4/80+CD11b+ macrophages derived from Tyk2−/− mice compared with WT mice.
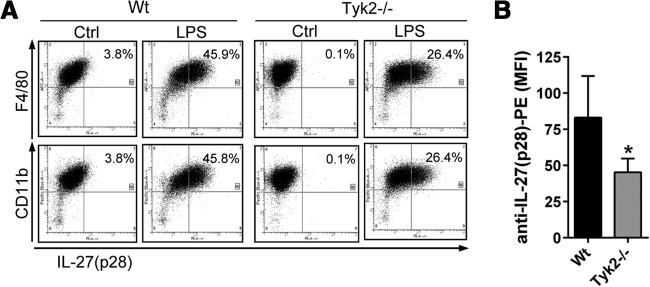
(A) Flow cytometry for intracellular IL-27(p28) and surface markers F4/80 and CD11b in TLR4-activated BMDM derived from WT mice or Tyk2−/− mice; LPS (1 μg/ml), 2 μM monensin, 12 h. Ctrl, BMDM treated with 2 μM monensin but without LPS. (B) Mean fluorescence intensity (MFI) of anti-IL-27(p28) PE-labeled antibody staining in LPS-activated BMDM from Tyk2−/− mice or WT mice. Data are shown as representatives of three independent experiments (A) or were pooled from three independent experiments (B), each done in duplicates. Student's two-tailed t-test; mean ± sem; *P < 0.05.
The production of IL-27(p28) by macrophages in response to TLR4 activation required the presence of both adaptor proteins MyD88 and TRIF at all time-points studied (Supplemental Fig. 1). As the MyD88-dependent pathways (IκB, ERK, JNK, p38) are fully functional in Tyk2−/− cells [5], we hypothesized that Tyk2 may modulate IL-27(p28) production via TRIF-dependent mechanisms, such as IFNs.
Tyk2 is part of a type I IFN-dependent mechanism to promote IL-27(p28) production by F4/80+CD11b+ macrophages
To test the hypothesis that endogenous-secreted factors may be involved in the production of LPS-induced IL-27(p28), we performed supernatant transfer experiments (Fig. 3A). BMDM, derived from WT mice, were incubated with LPS or with medium for 12 h. Thereafter, conditioned supernatants were collected and transferred to BMDM from TLR4−/− mice for another 3-h incubation, followed by RNA isolation and determination of IL-27(p28) expression by RT-PCR in BMDM (TLR4−/−). Macrophages from TLR4−/− mice were used to prevent direct activation of IL-27(p28) gene expression by the presence of LPS in the transferred supernatants. Indeed, coincubation with conditioned supernatants from LPS-treated BMDM (WT) resulted in the expression of IL-27(p28) mRNA in BMDM from TLR4−/− mice (Fig. 3B), suggesting an involvement of endogenous-secreted factors.
Figure 3. A role of endogenous factors for promoting the release of IL-27(p28) in cultures of TLR4-activated macrophages.
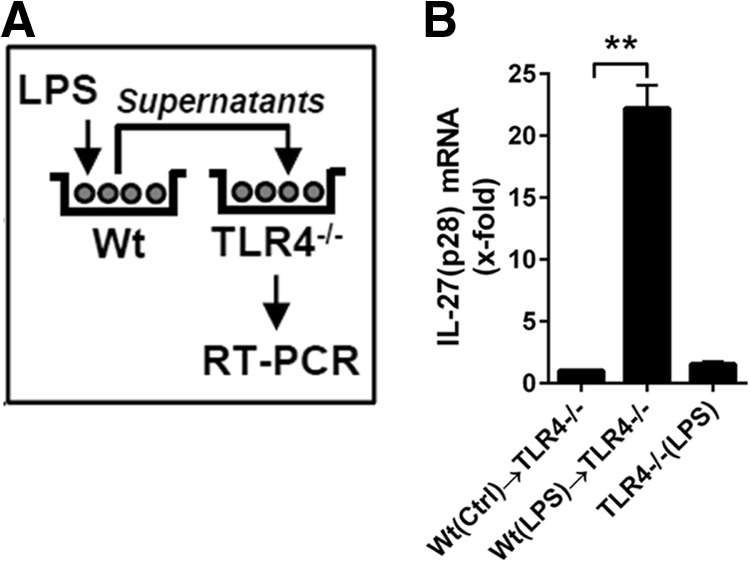
(A) Schematic overview of the experimental design of supernatant-transfer experiments. BMDM, from WT mice, were incubated with LPS (1 μg/ml) for 12 h before transfer of conditioned supernatants to BMDM derived from TLR4−/− mice, followed by RNA isolation and RT-PCR, 3 h later. TLR4−/− BMDM were used to exclude direct effects of LPS during the second incubation step. (B) RT-PCR of the relative IL-27(p28) expression normalized to GAPDH of the experiments described above. Levels of mRNA for IL-27(p28) induced by supernatants from unstimulated WT BMDM were used as onefold (Ctrl; left bar). Conditioned supernatants from LPS-activated WT BMDM strongly up-regulated mRNA for IL-27(p28) when incubated with TLR4−/− BMDM (center bar). The right bar shows negative controls demonstrating the absence of IL-27(p28) production when TLR4−/− BMDM were incubated directly with LPS in normal media (no transfer of WT BMDM conditioned supernatants). Data are representative of three independent experiments. One-way ANOVA; mean ± sem; **P < 0.01.
According to the existing literature, we favored the possibility of IFN-dependent expression of IL-27 via Tyk2 signaling [24, 26]. Genetic deficiency of Tyk2 was confirmed to result in defective type I IFN production in LPS-activated BMDM (Fig. 4A), as described before in thioglycollate-elicited peritoneal macrophages [5]. Addition of type I IFN to cultures of BMDM from WT mice induced the production of IL-27(p28) (Fig. 4B). LPS-activated BMDM from IFNAR1−/− and IFN-β−/− mice displayed a substantially defective generation of IL-27(p28) in supernatants (Fig. 4C). When intracellular IL-27(p28) was assessed, the frequencies of IL-27(p28)+F4/80+CD11b+ cells were lower in LPS-activated BMDM from IFNAR1−/− mice (26.9%) and IFN-β−/− mice (23.9%) compared with BMDM from WT mice (48.3%; Fig. 4D and E). Collectively, a partial requirement of Tyk2 for production of IL-27(p28) in LPS-activated macrophages appeared to be linked to impaired synthesis of type I IFNs in Tyk2−/− macrophages.
Figure 4. Tyk2 activates the endogenous production of type I IFNs for enhancing the release of IL-27(p28).

(A) Detection of IFN-β secretion by LPS-activated macrophages from WT mice or Tyk2−/− mice; Ctrl, unstimulated control macrophages; 8 h; ELISA. (B) Induction of IL-27(p28) release from BMDM (WT) after incubation with IFN-α (1000 U/ml); 8 h; ELISA. (C) IL-27(p28) release of BMDM from WT mice, IFNAR1−/− mice, or IFN-β−/− mice after LPS; 8 h. (D) Flow cytometry of IL-27(p28)+F4/80+CD11b+ BMDM from WT mice, IFNAR1−/− mice, or IFN-β−/− mice after LPS; 12 h; monensin (2 μM). Numbers indicate the percentage of IL-27(p28)+F4/80+CD11b+ cells. (E) Representative dot-plot images of the experiments described in D; all dot-plots were gated on CD11b+ cells. Data are pooled from three independent experiments (A–C) or done with n = 4 mice/group (D and E). One-way ANOVA (A, C, and D) or Student's two-tailed t-test (B); mean ± sem; *P < 0.05; ***P < 0.001.
Tyk2 is dispensable for mediating the effects of IL-27 on CD4+ T cells
As Tyk2 is required for gene expression of IL-27(p28) (Fig. 1), we tested whether Tyk2 is also involved in mediating the biological effects of IL-27 in T cells. For these studies, CD4+ T cells were purified from spleens of healthy WT mice and Tyk2−/− mice and incubated under neutral conditions (anti-IL-4, anti-IL-12, anti-IFN-γ). Following incubation with rmIL-27, the RNA expression levels of several target genes of IL-27 signaling were analyzed. A major biological function of IL-27 is known to be induction of gene expression of IL-10 in T cells [15, 16]. In our experiments, IL-27 promoted gene expression of IL-10 in CD4+ T cells without a significant difference observed in CD4+ T cells from WT mice compared with Tyk2−/− mice (Fig. 5A). IL-27 strongly induced mRNA for IL-12rb2 in CD4+ T cells in a similar fashion as cells from WT mice and Tyk2−/− mice (Fig. 5B). Tbx21 is a Th1 cell-specific transcription factor controlling the expression of IFN-γ in Th1 cells. rIL-27 strongly induced gene expression of Tbx21 after 48 h without requiring the presence of Tyk2 (Fig. 5C). Finally, IL-27 activated expression of STAT1 in CD4+ T cells (WT) and CD4+ T cells (Tyk2−/−; Fig. 5D). Similar observations for gene expression of IL-10, Tbx21, IL-12rb2, and STAT1 were made when CD4+ T cells from WT mice and Tyk2−/− mice were incubated with different concentrations of IL-27 (1 and 100 ng/ml; two to three independent experiments; data not shown). In conclusion, these findings suggest that Tyk2 is not essential in T cells to mediate the biological effects following ligation of IL-27 with the IL-27RA/gp130R complex.
IL-27 mediates Tyk2-associated mortality during experimental sepsis
To evaluate the in vivo relevance of Tyk2 as a regulator of IL-27(p28) production, we used LPS-induced shock as a model of SIRS. We have reported earlier on a role for Tyk2 during endotoxic shock [5]. In the current study, we first confirmed a clear survival benefit of Tyk2−/− mice (B10.D1-H2q/SgJ; naturally occurring loss-of-function mutation) during endotoxic shock (Fig. 6A). Survival of Tyk2−/− mice was 89% compared with 33% in C57BL/10SnJ WT mice receiving the same dose of LPS from E. coli (10 mg/kg body weight i.p.; Fig. 6A). Hypothermia is an early predictor of mortality during endotoxic shock in rodents and was also found to be less-severe in Tyk2−/− mice (Fig. 6B). The plasma concentrations of type I IFNs were substantially lower in Tyk2−/− mice compared with WT mice (Fig. 6C), as reported before [5].
Figure 6. Survival and release of IL-27(p28) in Tyk2−/− mice following endotoxic shock.
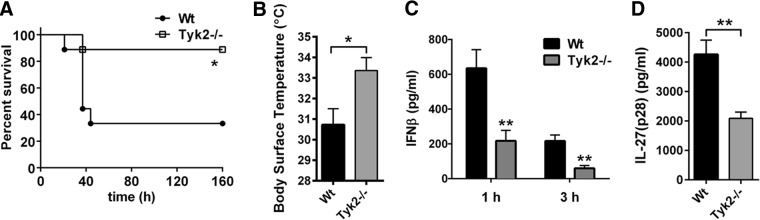
(A) Survival of WT mice (n=9) and Tyk2−/− mice (n=9) during endotoxic shock following injection with LPS (10 mg/kg body weight i.p.). (B) Body-surface temperature of WT mice and Tyk2−/− mice, 24 h after LPS injection of the experiment described above. (C) Plasma concentrations of IFN-β following endotoxic shock (1 and 3 h) in WT mice and Tyk2−/− mice (n=6/group); ELISA. (D) Detection of IL-27(p28) in plasma during endotoxic shock in WT mice (n=7) and Tyk2−/− mice (n=8); 6 h; ELISA. Survival curves were analyzed using the log-rank (Mantel-Cox) test, and frames (B–D) were analyzed using the nonparametric Mann-Whitney test. *P < 0.05; **P < 0.01.
Concentrations of IL-27(p28) in plasma were highly elevated in WT mice during endotoxic shock, whereas IL-27(p28) plasma levels were reduced substantially in Tyk2−/− mice (Fig. 6D). IL-27(p28) was not detectable in plasma of healthy, untreated WT or Tyk2−/− mice (data not shown).
Next, we used the model of polymicrobial sepsis following CLP to expand our findings on the cross-talk between Tyk2 and IL-27(p28). Genetic deficiency of Tyk2 was protective at time-points >60 h after CLP with 7-day survival rates of 53% in Tyk2−/− mice versus 6% in WT mice (Fig. 7A). Early measurements of body-surface temperature after CLP were already indicative of a beneficial outcome of polymicrobial sepsis in Tyk2−/− mice (Fig. 7B). The detection of IL-27(p28) in plasma revealed a >50% reduction of IL-27(p28) after CLP in Tyk2−/− mice (Fig. 7C), similar to the observations for endotoxic shock (Fig. 6D).
Figure 7. Survival and release of IL-27(p28) in Tyk2−/− mice during polymicrobial sepsis following CLP.
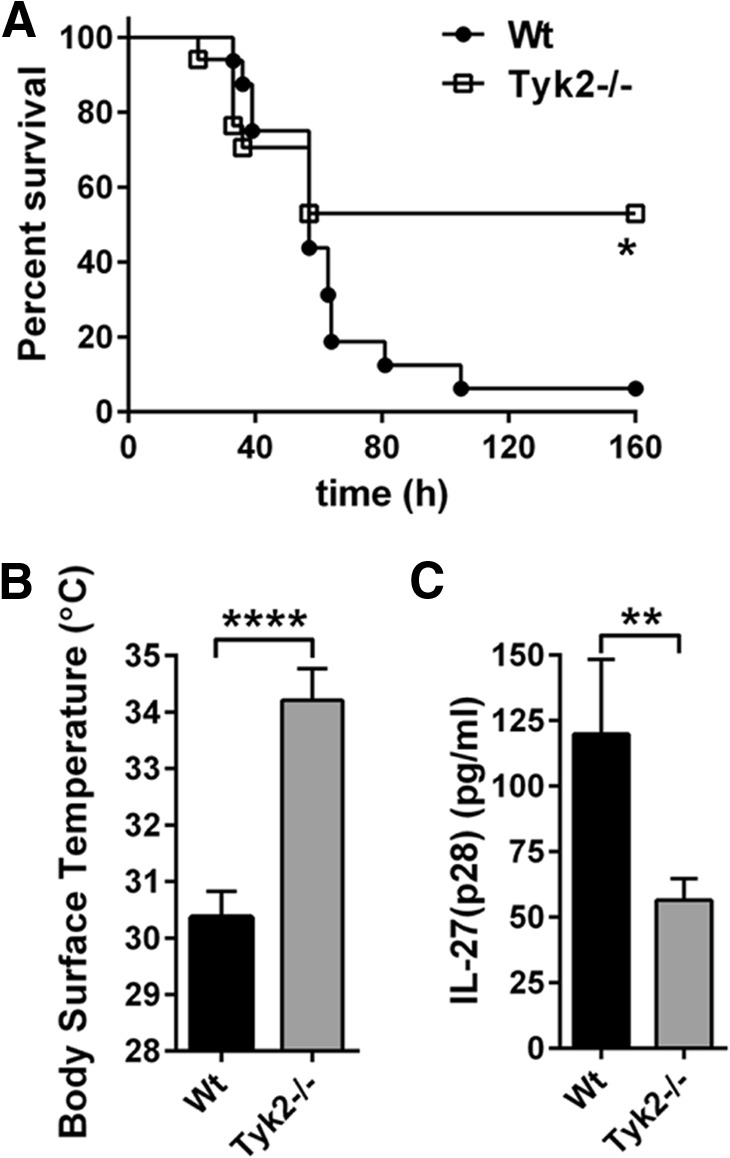
(A) Survival of WT mice (n=16) and Tyk2−/− mice (n=17) in polymicrobial sepsis following CLP (“high-grade”). (B) Body-surface temperature of WT mice (n=16) and Tyk2−/− mice (n=16), 24 h after CLP from the same experiment described in A. (C) Plasma concentrations of IL-27(p28), 10 h after CLP in WT mice (n=7) and Tyk2−/− mice (n=8). Survival curves were analyzed using the log-rank (Mantel-Cox) test, and frames (B and C) were analyzed using the nonparametric Mann-Whitney test. **P < 0.01; ****P < 0.0001.
To evaluate the relevance of IL-27 for the outcome of LPS-induced shock, mice with targeted genetic deletion of IL-27RA (IL-27RA−/−) were used. Whereas five out of n = 11 C57BL/6 mice in the control group had a lethal outcome of endotoxic shock, all IL-27RA−/− mice (n=10) survived (Fig. 8). However, IL-27RA−/− mice showed clinical symptoms of shock following LPS injections (lethargy, scuffing, piloerection), suggesting that protection of the IL-27RA−/− strain is not complete. The differences in survival rates of WT mice in Figs. 6 and 8 may be explained by using C57BL/6 mice in Fig. 6 and C57BL/10SnJ in Fig. 8 as the appropriate WT control strains. In conclusion, IL-27RA−/− mice were greatly protected against LPS-induced mortality compared with C57BL/6 WT mice (Fig. 8).
Figure 8. Protection of IL-27RA−/− mice from shock-associated mortality.
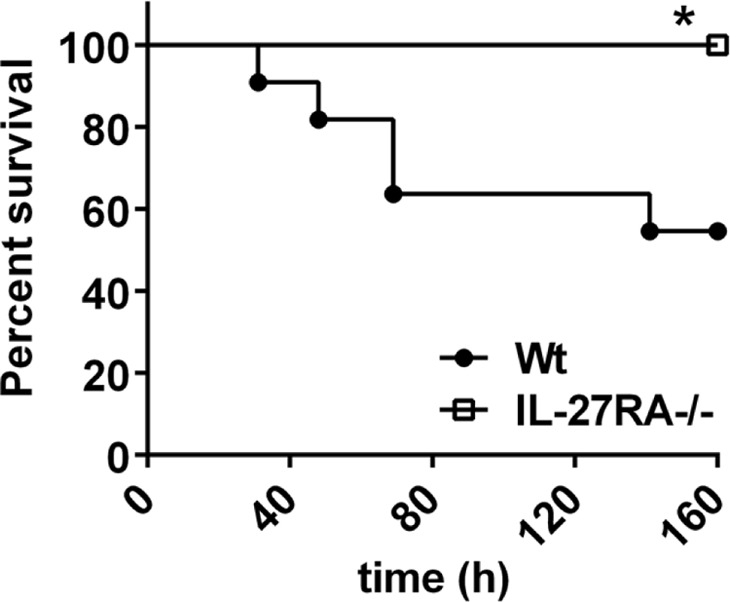
Survival curves of IL-27RA−/− mice (n=10) compared with C57BL/6 (WT) mice (n=10) following endotoxic shock (LPS 10 mg/kg body weight i.p.). Data were acquired with the numbers of mice/group as indicated. Log-rank (Mantel-Cox) test; *P < 0.05.
DISCUSSION
In this report, we found Tyk2 to be required for the release of IL-27(p28) by macrophages and during in vivo models of sepsis/SIRS. The lower amounts of IL-27(p28) detected in Tyk2−/− mice may contribute to the enhanced resistance of Tyk2−/− mice during sepsis/SIRS, which was previously linked to the effects of type I IFNs [5]. Our data suggest that type I IFNs during sepsis/SIRS may act via IL-27, given the protection of IL-27RA−/− mice during endotoxic shock (Fig. 8). In this context, Tyk2 may modulate TRIF-dependent rather than MyD88-dependent production of IL-27(p28), as signaling pathways downstream of MyD88 have been reported to remain functional in Tyk2−/− macrophages [5]. Other reports have also suggested the involvement of endogenous IFN production as contributing factors for gene expression of IL-27 [26]. We have expanded these previous observations by specific quantification of the p28 subunit in supernatants and using intracellular staining techniques in F4/80+CD11b+ macrophages. The resistance of IL-27RA−/− mice in LPS-induced shock is in line with a previous study, demonstrating up-regulation of mRNA for EBI3 and p28 during polymicrobial sepsis following CLP [11]. The use of EBI3−/− mice or administration of a IL-27RA-Fc fusion protein to disrupt IL-27 functions during polymicrobial sepsis results in improved survival rates [11]. EBI3−/− mice show enhanced clearance of bacteria in the peritoneal cavity and blood, most likely as a result of a better recruitment of leukocytes to the site of infection [11]. In addition, leukocytes from EBI3−/− mice may have better bactericidal activity because of higher amounts of ROS produced following activation [11]. In a model of severe CLP (“overwhelming sepsis”), a trend for better survival of Tyk2−/− mice (20% Tyk2−/− mice vs. 0% WT mice) correlated with lower concentrations of IL-6 and CXCL10 [29]. Therefore, we suggest that several cytokines, in addition to IL-27, may cooperate to confer protection of Tyk2−/− mice during sepsis.
In support of our findings, earlier reports have associated Tyk2 gene disruption with partial defects in cytokine responses and diminished LPS-induced NO production in macrophages [27]. Genetic deficiency of Tyk2 results in changes in basal gene expression and proteome patterns [30, 31]. The production of type I IFNs and IFN-inducible gene expression is, in particular, dependent on the presence of Tyk2 [5, 27, 31, 32]. In addition to regulation of IL-27 as reported here, Tyk2 is involved in expression of IL-12 and IL-23 following TLR4 activation, at least in DCs [33].
Although phosphorylation of Tyk2 in response to IL-27 has been demonstrated in keratinocytes and naive CD4+ T cells [34, 35], it has not yet been tested whether Tyk2 is nonredundant for IL-27 signaling. In our experiments, following IL-27 stimulation of cultures of CD4+ T cells from WT mice or Tyk2−/− mice, we did not find differences in the expression of target genes, such as IL-10, STAT1, IL-12rb2, and Tbx21 (Fig. 5). This suggests that association of Tyk2 with gp130 may not be required in CD4+ T cells for IL-27 signaling. This is in analogy to the situation with IL-6 and LIF in murine cells. IL-6 and LIF use the gp130R chain and phosphorylate Tyk2 [36], whereas Tyk2 signaling is not essential for IL-6 and LIF signaling [27, 37, 38].
Heterodimeric IL-27 is formed by the subunit proteins IL-27(p28) and EBI3. We focused our studies on detection of IL-27(p28), as EBI3 is also incorporated into IL-35, and EBI3 appeared not to be modulated by Tyk2 (Fig. 1B). Additional binding partners of IL-27(p28), such as CLF-1, have been reported [39]. In our preliminary experiments, gene expression of CLF-1 was not inducible by LPS in macrophages (data not shown). In addition, the potential existence of IL-27(p28) monomers or homodimers cannot be ruled out. In the current absence of specific quantification methods for IL-27(p28) monomers or homodimers, this is difficult to study. Interestingly, in a model of transgenic overexpression, IL-27(p28) was found to antagonize gp130 signaling and heterodimeric IL-27 [40].
SIRS and severe sepsis are major causes of morbidity and mortality [41, 42]. The release of cytokines is a central factor during the pathogenesis of SIRS and sepsis, contributing to early exuberant inflammation and adverse outcomes [43]. A change in paradigms has also acknowledged that during the later phases of sepsis, profound immunosuppression, including apoptosis of immune cells, can occur [44, 45]. IL-27 can exert biphasic effects by promoting acute inflammation, while down-modulating chronic inflammation via IL-10 [13]. Thereby, the functional characteristics of IL-27 are in parallel to the natural course of sepsis (early hyperinflammation and later immunosuppression) leading to mortality. For these reasons, interception of IL-27 may be an appealing prospective strategy for therapeutic reversal of the dysbalanced immune responses of SIRS and sepsis. Furthermore, IL-27 has been identified recently as a diagnostic biomarker for sepsis in critically ill children [46]. In adult patients with sepsis, measurements of IL-27 alone were inferior to testing of procalcitonin, whereas combined analysis of IL-27 and procalcitonin improved diagnostic discrimination in patients with nonlung sources of infection [47].
In conclusion, it has been suggested that IL-27 is a critical factor balancing acute and chronic inflammation, but how production of IL-27 is regulated remains insufficiently understood. In this report, we describe for the first time that Tyk2 is required for full release of IL-27(p28) from LPS-activated macrophages and during sepsis. Our studies suggest the existence of a detrimental “Tyk2/IL-27 axis” during SIRS/sepsis.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by grants GM-29507 and GM-61656 (to P.A.W.) and Community Office for Resource Efficiency (CORE) Grant P30 CA21765 (to P.J.M.) from the U.S. National Institutes of Health, along with the Deutsche Forschungsgemeinschaft (BO3482/1-1 and BO3482/3-1; to M.B.), the Federal Ministry of Education and Research (01EO1003; to M.B.), a Marie Curie Career Integration Grant of the European Union (Project 334486; to M.B.), the Mainzer Forschungsförderungsprogramm (MAIFOR Program) of the University Medical Center Mainz (to M.B.), the Hartwell Foundation (to P.J.M.), the American Lebanese Syrian Associated Charities (to P.J.M.), and the Austrian Science Fund (FWF-SFB-F28, to B.S. and M.M.; and FWF-P25642-B22, to B.S.).
We thank Vinay Patel, Mikel Haggadone, and Fabien Meta for technical assistance, as well as Beverly Schumann, Sue Scott, and Robin Kunkel for excellent staff support.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- −/−
- deficient
- BMDM
- bone marrow-derived macrophage(s)
- CLF-1
- cytokine-like factor 1
- CLP
- cecal ligation and puncture
- EBI3
- EBV-induced gene 3
- IFNAR1
- IFN-αR 1
- IL-12rb2
- IL-12R, β-2 subunit
- IL-27RA
- IL-27Rα
- IRF
- IFN regulatory factor
- LIF
- leukemia inhibitory factor
- p28
- subunit of IL-27
- rm
- recombinant mouse
- SIRS
- systemic inflammatory response syndrome
- Tbx21
- T-box transcription factor 21
- TRIF
- Toll/IL-1R domain-containing adaptor-inducing IFN-β
- Tyk2
- tyrosine kinase 2
- Ube2d2
- ubiquitin-conjugating enzyme E2 D2
- WT
- Wild type
AUTHORSHIP
M.B. designed and performed experiments, analyzed data, and coordinated the study. N.K., F.P., D.R., J.J.G., and B.S. performed experiments and analyzed data. P.J.M. provided ideas and useful comments. B.S. and M.M. provided IFNAR1−/− and IFN-β−/− macrophages and helpful comments. P.A.W. designed experiments and supervised the work. M.B. wrote the manuscript, which was edited by P.A.W., P.J.M., B.S., and M.M.
REFERENCES
- 1. Hotchkiss R. S., Karl I. E. (2003) The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348, 138–150 [DOI] [PubMed] [Google Scholar]
- 2. Slade E., Tamber P. S., Vincent J. L. (2003) The surviving sepsis campaign: raising awareness to reduce mortality. Crit. Care 7, 1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ward P. A., Bosmann M. (2012) A historical perspective on sepsis. Am. J. Pathol. 181, 2–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bosmann M., Ward P. A. (2013) The inflammatory response in sepsis. Trends Immunol. 34, 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karaghiosoff M., Steinborn R., Kovarik P., Kriegshauser G., Baccarini M., Donabauer B., Reichart U., Kolbe T., Bogdan C., Leanderson T., Levy D., Decker T., Muller M. (2003) Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol. 4, 471–477 [DOI] [PubMed] [Google Scholar]
- 6. Strobl B., Stoiber D., Sexl V., Mueller M. (2011) Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front. Biosci. 17, 3214–3232 [DOI] [PubMed] [Google Scholar]
- 7. Minegishi Y., Saito M., Morio T., Watanabe K., Agematsu K., Tsuchiya S., Takada H., Hara T., Kawamura N., Ariga T., Kaneko H., Kondo N., Tsuge I., Yachie A., Sakiyama Y., Iwata T., Bessho F., Ohishi T., Joh K., Imai K., Kogawa K., Shinohara M., Fujieda M., Wakiguchi H., Pasic S., Abinun M., Ochs H. D., Renner E. D., Jansson A., Belohradsky B. H., Metin A., Shimizu N., Mizutani S., Miyawaki T., Nonoyama S., Karasuyama H. (2006) Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 25, 745–755 [DOI] [PubMed] [Google Scholar]
- 8. O'Shea J. J., Holland S. M., Staudt L. M. (2013) JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 368, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aizu K., Li W., Yajima T., Arai T., Shimoda K., Nimura Y., Yoshikai Y. (2006) An important role of Tyk2 in APC function of dendritic cells for priming CD8+ T cells producing IFN-γ. Eur. J. Immunol. 36, 3060–3070 [DOI] [PubMed] [Google Scholar]
- 10. Yap G. S., Ortmann R., Shevach E., Sher A. (2001) A heritable defect in IL-12 signaling in B10.Q/J mice. II. Effect on acute resistance to Toxoplasma gondii and rescue by IL-18 treatment. J. Immunol. 166, 5720–5725 [DOI] [PubMed] [Google Scholar]
- 11. Wirtz S., Tubbe I., Galle P. R., Schild H. J., Birkenbach M., Blumberg R. S., Neurath M. F. (2006) Protection from lethal septic peritonitis by neutralizing the biological function of interleukin 27. J. Exp. Med. 203, 1875–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pflanz S., Timans J. C., Cheung J., Rosales R., Kanzler H., Gilbert J., Hibbert L., Churakova T., Travis M., Vaisberg E., Blumenschein W. M., Mattson J. D., Wagner J. L., To W., Zurawski S., McClanahan T. K., Gorman D. M., Bazan J. F., de Waal Malefyt R., Rennick D., Kastelein R. A. (2002) IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity 16, 779–790 [DOI] [PubMed] [Google Scholar]
- 13. Bosmann M., Ward P. A. (2013) Modulation of inflammation by interleukin-27. J. Leukoc. Biol. 94, 1159–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vignali D. A., Kuchroo V. K. (2012) IL-12 family cytokines: immunological playmakers. Nat. Immunol. 13, 722–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Batten M., Kljavin N. M., Li J., Walter M. J., de Sauvage F. J., Ghilardi N. (2008) Cutting edge: IL-27 is a potent inducer of IL-10 but not FoxP3 in murine T cells. J. Immunol. 180, 2752–2756 [DOI] [PubMed] [Google Scholar]
- 16. Stumhofer J. S., Silver J. S., Laurence A., Porrett P. M., Harris T. H., Turka L. A., Ernst M., Saris C. J., O'Shea J. J., Hunter C. A. (2007) Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat. Immunol. 8, 1363–1371 [DOI] [PubMed] [Google Scholar]
- 17. Cox J. H., Kljavin N. M., Ramamoorthi N., Diehl L., Batten M., Ghilardi N. (2011) IL-27 promotes T cell-dependent colitis through multiple mechanisms. J. Exp. Med. 208, 115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cao Y., Doodes P. D., Glant T. T., Finnegan A. (2008) IL-27 induces a Th1 immune response and susceptibility to experimental arthritis. J. Immunol. 180, 922–930 [DOI] [PubMed] [Google Scholar]
- 19. Villarino A., Hibbert L., Lieberman L., Wilson E., Mak T., Yoshida H., Kastelein R. A., Saris C., Hunter C. A. (2003) The IL-27R (WSX-1) is required to suppress T cell hyperactivity during infection. Immunity 19, 645–655 [DOI] [PubMed] [Google Scholar]
- 20. Stumhofer J. S., Laurence A., Wilson E. H., Huang E., Tato C. M., Johnson L. M., Villarino A. V., Huang Q., Yoshimura A., Sehy D., Saris C. J., O'Shea J. J., Hennighausen L., Ernst M., Hunter C. A. (2006) Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat. Immunol. 7, 937–945 [DOI] [PubMed] [Google Scholar]
- 21. Batten M., Li J., Yi S., Kljavin N. M., Danilenko D. M., Lucas S., Lee J., de Sauvage F. J., Ghilardi N. (2006) Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat. Immunol. 7, 929–936 [DOI] [PubMed] [Google Scholar]
- 22. Gafa V., Lande R., Gagliardi M. C., Severa M., Giacomini E., Remoli M. E., Nisini R., Ramoni C., Di Francesco P., Aldebert D., Grillot R., Coccia E. M. (2006) Human dendritic cells following Aspergillus fumigatus infection express the CCR7 receptor and a differential pattern of interleukin-12 (IL-12), IL-23, and IL-27 cytokines, which lead to a Th1 response. Infect. Immun. 74, 1480–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bosmann M., Haggadone M. D., Hemmila M. R., Zetoune F. S., Sarma J. V., Ward P. A. (2012) Complement activation product C5a is a selective suppressor of TLR4-induced, but not TLR3-induced, production of IL-27(p28) from macrophages. J. Immunol. 188, 5086–5093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Molle C., Nguyen M., Flamand V., Renneson J., Trottein F., De Wit D., Willems F., Goldman M., Goriely S. (2007) IL-27 synthesis induced by TLR ligation critically depends on IFN regulatory factor 3. J. Immunol. 178, 7607–7615 [DOI] [PubMed] [Google Scholar]
- 25. Liu J. G., Guan X. Q., Ma X. J. (2007) Regulation of IL-27 p28 gene expression in macrophages through MyD88- and interferon-γ-mediated pathways. J. Exp. Med. 204, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Molle C., Goldman M., Goriely S. (2010) Critical role of the IFN-stimulated gene factor 3 complex in TLR-mediated IL-27p28 gene expression revealing a two-step activation process. J. Immunol. 184, 1784–1792 [DOI] [PubMed] [Google Scholar]
- 27. Karaghiosoff M., Neubauer H., Lassnig C., Kovarik P., Schindler H., Pircher H., McCoy B., Bogdan C., Decker T., Brem G., Pfeffer K., Muller M. (2000) Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity 13, 549–560 [DOI] [PubMed] [Google Scholar]
- 28. Bosmann M., Russkamp N. F., Patel V. R., Zetoune F. S., Sarma J. V., Ward P. A. (2011) The outcome of polymicrobial sepsis is independent of T and B cells. Shock 36, 396–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Herzig D., Fang G., Toliver-Kinsky T. E., Guo Y., Bohannon J., Sherwood E. R. (2012) STAT1-deficient mice are resistant to cecal ligation and puncture-induced septic shock. Shock 38, 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Radwan M., Miller I., Grunert T., Marchetti-Deschmann M., Vogl C., O'Donoghue N., Dunn M. J., Kolbe T., Allmaier G., Gemeiner M., Muller M., Strobl B. (2008) The impact of tyrosine kinase 2 (Tyk2) on the proteome of murine macrophages and their response to lipopolysaccharide (LPS). Proteomics 8, 3469–3485 [DOI] [PubMed] [Google Scholar]
- 31. Vogl C., Flatt T., Fuhrmann B., Hofmann E., Wallner B., Stiefvater R., Kovarik P., Strobl B., Muller M. (2010) Transcriptome analysis reveals a major impact of JAK protein tyrosine kinase 2 (Tyk2) on the expression of interferon-responsive and metabolic genes. BMC Genomics 11, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Strobl B., Bubic I., Bruns U., Steinborn R., Lajko R., Kolbe T., Karaghiosoff M., Kalinke U., Jonjic S., Muller M. (2005) Novel functions of tyrosine kinase 2 in the antiviral defense against murine cytomegalovirus. J. Immunol. 175, 4000–4008 [DOI] [PubMed] [Google Scholar]
- 33. Tokumasa N., Suto A., Kagami S., Furuta S., Hirose K., Watanabe N., Saito Y., Shimoda K., Iwamoto I., Nakajima H. (2007) Expression of Tyk2 in dendritic cells is required for IL-12, IL-23, and IFN-γ production and the induction of Th1 cell differentiation. Blood 110, 553–560 [DOI] [PubMed] [Google Scholar]
- 34. Kanda N., Watanabe S. (2008) IL-12, IL-23, and IL-27 enhance human β-defensin-2 production in human keratinocytes. Eur. J. Immunol. 38, 1287–1296 [DOI] [PubMed] [Google Scholar]
- 35. Kamiya S., Owaki T., Morishima N., Fukai F., Mizuguchi J., Yoshimoto T. (2004) An indispensable role for STAT1 in IL-27-induced T-bet expression but not proliferation of naive CD4+ T cells. J. Immunol. 173, 3871–3877 [DOI] [PubMed] [Google Scholar]
- 36. Stahl N., Boulton T. G., Farruggella T., Ip N. Y., Davis S., Witthuhn B. A., Quelle F. W., Silvennoinen O., Barbieri G., Pellegrini S., et al. (1994) Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 β receptor components. Science 263, 92–95 [DOI] [PubMed] [Google Scholar]
- 37. Shimoda K., Kato K., Aoki K., Matsuda T., Miyamoto A., Shibamori M., Yamashita M., Numata A., Takase K., Kobayashi S., Shibata S., Asano Y., Gondo H., Sekiguchi K., Nakayama K., Nakayama T., Okamura T., Okamura S., Niho Y. (2000) Tyk2 plays a restricted role in IFN α signaling, although it is required for IL-12-mediated T cell function. Immunity 13, 561–571 [DOI] [PubMed] [Google Scholar]
- 38. Chung B. M., Kang H. C., Han S. Y., Heo H. S., Lee J. J., Jeon J., Lim J. Y., Shin I., Hong S. H., Cho Y. S., Kim C. G. (2006) Jak2 and Tyk2 are necessary for lineage-specific differentiation, but not for the maintenance of self-renewal of mouse embryonic stem cells. Biochem. Biophys. Res. Commun. 351, 682–688 [DOI] [PubMed] [Google Scholar]
- 39. Crabe S., Guay-Giroux A., Tormo A. J., Duluc D., Lissilaa R., Guilhot F., Mavoungou-Bigouagou U., Lefouili F., Cognet I., Ferlin W., Elson G., Jeannin P., Gauchat J. F. (2009) The IL-27 p28 subunit binds cytokine-like factor 1 to form a cytokine regulating NK and T cell activities requiring IL-6R for signaling. J. Immunol. 183, 7692–7702 [DOI] [PubMed] [Google Scholar]
- 40. Stumhofer J. S., Tait E. D., Quinn W. J., III, Hosken N., Spudy B., Goenka R., Fielding C. A., O'Hara A. C., Chen Y., Jones M. L., Saris C. J., Rose-John S., Cua D. J., Jones S. A., Elloso M. M., Grotzinger J., Cancro M. P., Levin S. D., Hunter C. A. (2010) A role for IL-27p28 as an antagonist of gp130-mediated signaling. Nat. Immunol. 11, 1119–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adhikari N. K., Fowler R. A., Bhagwanjee S., Rubenfeld G. D. (2010) Critical care and the global burden of critical illness in adults. Lancet 376, 1339–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vincent J. L., Sakr Y., Sprung C. L., Ranieri V. M., Reinhart K., Gerlach H., Moreno R., Carlet J., Le Gall J. R., Payen D. (2006) Sepsis in European intensive care units: results of the SOAP study. Crit. Care Med. 34, 344–353 [DOI] [PubMed] [Google Scholar]
- 43. Rittirsch D., Flierl M. A., Ward P. A. (2008) Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 8, 776–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boomer J. S., To K., Chang K. C., Takasu O., Osborne D. F., Walton A. H., Bricker T. L., Jarman S. D., II, Kreisel D., Krupnick A. S., Srivastava A., Swanson P. E., Green J. M., Hotchkiss R. S. (2011) Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306, 2594–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hotchkiss R. S., Coopersmith C. M., McDunn J. E., Ferguson T. A. (2009) The sepsis seesaw: tilting toward immunosuppression. Nat. Med. 15, 496–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wong H. R., Cvijanovich N. Z., Hall M., Allen G. L., Thomas N. J., Freishtat R. J., Anas N., Meyer K., Checchia P. A., Lin R., Bigham M. T., Sen A., Nowak J., Quasney M., Henricksen J. W., Chopra A., Banschbach S., Beckman E., Harmon K., Lahni P., Shanley T. P. (2012) Interleukin-27 is a novel candidate diagnostic biomarker for bacterial infection in critically ill children. Crit. Care 16, R213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wong H. R., Lindsell C. J., Lahni P., Hart K. W., Gibot S. (2013) Interleukin 27 as a sepsis diagnostic biomarker in critically ill adults. Shock 40, 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.