

The regulation of Trex1 expression by TLRs, type I IFNs, and virus, implicates a novel role for Trex1 in DC immune activation.
Keywords: viral infection, immune activation, autoimmunity
Abstract
Mutations in the Trex1 are associated with a spectrum of type I IFN-dependent autoimmune diseases. Trex1 plays an essential role in preventing accumulation of excessive cytoplasmic DNA, avoiding cell-intrinsic innate DNA sensor activation and suppressing activation of type I IFN-stimulated and -independent antiviral genes. Trex1 also helps HIV to escape cytoplasmic detection by DNA sensors. However, regulation of Trex1 in innate immune cells remains elusive. We report that murine cDCs have high constitutive expression of Trex1 in vitro and in vivo in the spleen. In resting bone marrow-derived cDCs, type I IFNs up-regulate Trex1 expression via the IFNAR-mediated signaling pathway (STAT1- and STAT2-dependent). DC activation induced by TLR3, -4, -7, and -9 ligands also augments Trex1 expression through autocrine IFN-β production and triggering of the IFN signaling pathway, whereas TLR4 ligand LPS also stimulates an early expression of Trex1 through IFN-independent NF-κB-dependent signaling pathway. Furthermore, retroviral infection also induces Trex1 up-regulation in cDCs, as we found that a gene therapy HIV-1-based lentiviral vector induces significant Trex1 expression, suggesting that Trex1 may affect local and systemic administration of gene-therapy vehicles. Our data indicate that Trex1 is induced in cDCs during activation upon IFN and TLR stimulation through the canonical IFN signaling pathway and suggest that Trex1 may play a role in DC activation during infection and autoimmunity. Finally, these results suggest that HIV-like viruses may up-regulate Trex1 to increase their ability to escape immunosurveillance.
Introduction
DCs are key innate immune cells that orchestrate the adaptive immune response. Upon encountering danger signals, resting DCs become activated/mature [1]. Type I IFNs are endogenous danger signals that trigger DC activation [2] and also mediate in an autocrine fashion the activation stimulated by TLR ligands [3, 4]. TLRs are important PRRs required to sense the presence of the PAMPs and DAMPs that are associated with infections [5, 6] that activate DCs and initiate innate and adaptive immune responses against viruses and bacteria [7–9]. If dysregulated, activated DCs are a key source of type I IFNs, contributing to the pathogenesis of autoimmune diseases, such as SLE [10–12]. Therefore, a better understanding of DC activation by TLRs and type I IFNs may provide insights into the pathogenesis of IFN-dependent autoimmunity, as well as the mechanisms of host defense to viruses.
Trex1 (formerly DNase III) is the most abundant DNA exonuclease in mammalian cells and predominantly associated with the ER [13–15]. It has been identified recently as a checkpoint in the innate DNA recognition pathway [15, 16]. Trex1 degrades 3′-unpaired termini—ssDNA and dsDNA species—including reverse-transcribed DNA, whereas oxidized DNA is less susceptible to Trex1 degradation [17]. Trex1−/− murine embryonic fibroblasts accumulate endogenous retroviral DNA in the cytosol that induces excessive type I IFN production [15, 16]. Mice lacking Trex1 exhibit elevated IFN-α levels in many organs and develop inflammatory myocarditis in the absence of viral infection [18, 19]. This illness bears a remarkable similarity to Aicardi-Goutières syndrome patients who have Trex1 loss-of-function mutations [20, 21]. Although rare, Trex1 mutations are present in autoimmune diseases, such as familial chilblain lupus [21, 22] and SLE [23, 24]. It has been proposed that Trex1 is an essential DNA scavenger that purges cytoplasmic DNA of endogenous retroviruses, therefore preventing the activation of the intracellular nucleic acid-sensing machinery [6, 25, 26]. This DNase activity of Trex1 is hijacked by the retrovirus HIV to evade the DNA sensors, avoid autointegration, and reach the nucleus without triggering the IFN-dependent innate response [27, 28].
Trex1 also contributes to innate immunity by regulating lysosomal biogenesis and by suppressing the expression of IFN-independent antiviral genes upon viral infection in macrophages [29]. These results highlight Trex1 as a negative regulator of the antiviral response that is triggered through the STING-TBK1-IRF3 signaling pathway [19, 29]. Nevertheless, little is known about the regulation of Trex1 expression and function in innate immune cells. In one study, the authors showed that IFN-γ induces Trex1 expression in murine macrophages and identified an ISRE site on the Trex1 promoter, suggesting that Trex1 can be regulated by type I IFNs [30]. More recently, Trex1 mRNA expression has been found to be increased by IFN-α in macrophages [31, 32], and overexpression of Trex1 can reduce the type I IFN response to Mycobacterium tuberculosis in murine macrophages [33]. However, little is known about the expression and regulation of Trex1 in DCs and the possible association of Trex1 to the major inducers of type I IFNs—the TLRs—during the activation/maturation of DCs.
Here, we demonstrate that Trex1 is highly expressed in DCs in vitro and in vivo. Stimulation of TLR3, -4, -7, and -9 induced Trex1 up-regulation via autocrine IFN-β production and the canonical type I IFNR signaling pathway, and in the case of TLR4 ligand LPS, also through IFN-independent NF-κB activation. Consistently, in vivo results confirmed that TLR ligands up-regulate Trex1 in cDCs, as well as in pDCs. Moreover, we found that cDCs up-regulate Trex1 expression upon infection with a HIV-based lentivirus. Overall, these results indicate that Trex1 is an IFN- and TLR-inducible gene and that its up-regulation is associated with the kind of DC activation that occurs during viral infections, such as in response to type I IFNs and TLR ligands. The up-regulation of Trex1 by these stimuli may be a strategy of DCs to restrict exogenous and endogenous retroviral DNA species in the cytoplasm and possibly regulate the DC activation status.
MATERIALS AND METHODS
Mice
B6, from The Jackson Laboratory (Bar Harbor, ME, USA), and IFNAR1KO, 129, STAT1KO, and STAT2KO mice were bred and maintained in our colonies in accordance with the guidelines of the Institutional Animal Care and Use Committees of Temple University, a member of the American Association for the Accreditation of Laboratory Animal Care accredited facilities. Female mice were used between 6 and 12 weeks of age.
In vitro bone marrow-derived cDC cultures
As a model of cDCs, bone marrow-derived DCs were generated as described previously [12]. Briefly, bone marrow precursors were flushed from femurs and tibias of mice and then seeded at 5 × 105/ml in complete IMDM (10% FBS, penicillin/streptomycin, gentamicin, and 2-ME), enriched with 3.3 ng/ml GM-CSF (BD Biosciences, San Jose, CA, USA) in 48-well plates. Medium (0.5 ml) was added on Day 2, and 0.5 ml medium was replaced from Day 5 and subsequently, each day until the culture was used. Resting cDC cultures were stimulated on Day 6 or 7 of culture with the following stimuli: 1500 U/ml IFN-α (HyCult Biotechnology, PB Uden, the Netherlands), 100 ng/ml LPS (Sigma-Aldrich, St. Louis, MO, USA), 10 μg/ml CpG-B 1826 (IDT Biotechnologies, Coralville, IA, USA), 1 μg/ml R848 (Invivogen, San Diego, CA, USA), and 200 ng/ml PolyI:C (Alexis Biochemicals, San Diego, CA, USA). To block NF-κB activity, cDCs were treated with 10−6 M BAY11 (Sigma-Aldrich) for 15 min and then stimulated with TLR ligands. cDCs were harvested after the indicated time-points for RNA analysis and resuspended with complete lysis buffer for Western blotting and in FACS buffer for immunostaining and FACS analysis of surface activation markers. For viral infection, Days 6–7 cDCs (5×105 cells/ml) were infected for 6 h with lentivirus at a MOI of one or five (5×105 or 2.5×106 pfu/well) and then harvested for RNA analysis.
Lentiviral production
HIV-1-based lentiviruses were produced as described previously [34]. Briefly, the GFP reporter, under the transcriptional control of the CMV promoter, was inserted into the lentiviral transfer plasmid described previously [34]. The self-inactivating HIV-1-based vectors, pseudotyped with the vesicular stomatitis–G protein envelop, were generated by three-plasmid cotransfection in human embryonic kidney 293–T cells [35]. Viral supernatants, concentrated by ultracentrifugation, were titrated using serial dilutions.
Lentiviral transduction in cDCs
Day 2 bone marrow cells (5×105 cells/well), grown in GM-CSF-enriched complete medium, as described above, were treated with protamine sulfate 50 μg/ml for 1 h and then transduced without or with HIV-based lentiviruses at a MOI of one, five, and ten. After 3–4 h, cells were washed with PBS once and replaced with fresh GM-CSF-enriched complete media until Day 5. Lentivirus-transduced cells were replaced with 0.5 ml fresh media on Day 5, harvested on Day 6, and assayed with intracellular staining of Trex1. CD11c+ gated cells expressing GFP were considered as successfully transduced DCs. Trex1 ΔMdFI was calculated in cells gated as GFP-positive and GFP-negative, subtracting the MdFI of each gated population stained with the secondary antibody IgG1 from the MdFI of the same gated cells stained with the anti-Trex1 antibody.
Intracellular staining of Trex1
Spleens were harvested from mice that were injected i.p. with 25 μg LPS or 100 μg GpG-B 1826 or equivalent volume of PBS (500 μl), 24 h earlier. Spleens were incubated with DNase I (Sigma-Aldrich) and Collagenase D type IV (Worthington, Lakewood, NJ, USA) for 1 h at 37°C under 5% CO2 and then passed through a 100-μm strainer. Red blood cells were lysed, and splenocytes were resuspended in FACS buffer and incubated with rat anti-mouse CD16/CD32 (Clone 2.4G2) mAb for 15 min to block FcRs. Cells were then stained with allophycocyanin-conjugated hamster anti-mouse CD11c and PE-conjugated rat anti-mouse B220 (BD Biosciences) to label cDCs and pDCs. After surface staining, splenocytes were fixed overnight at 4°C with permeabilization/fixation solution (eBioscience, San Diego, CA, USA) and then stained with mouse anti-mouse Trex1 antibody (BD Biosciences) for 30 min at 4°C, followed by secondary antibody goat anti-mouse IgG1 FITC (SouthernBiotech, Birmingham, AL, USA) for 15 min at 4°C. Stained cells were washed and analyzed on a FACSCanto cytometer (BD Biosciences). Trex1−/− cells were used as negative controls to assess the specificity of the antibody for Trex1. FlowJo (TreeStar, Ashland, OR, USA) was used for analysis.
qRT-PCR
Gene expression in cDCs was analyzed by qRT-PCR using TaqMan probes described before [12]. Briefly, RNA was extracted using the Qiagen RNeasy Plus kit, following the manufacturer's protocol (Qiagen, Valencia, CA, USA). cDNA was synthesized using the cDNA archive kit, followed by a preamplification reaction (Applied Biosystems, Foster City, CA, USA). TaqMan primers and probes for Trex1 and IFN-β were from Applied Biosystems. Cyclophilin was used as the reference gene for normalization. The Ct method of relative quantification of gene expression was used for these TaqMan PCRs (ΔΔCt) [12], and the normalized Ct values (against cyclophilin) were calibrated against the control sample (untreated B6 cDCs) in each experiment. In each independent experiment, one cDC culture generated from a KO mouse was compared with one cDC culture generated from its appropriate WT control mouse, either B6 or 129.
Western blot analysis
Total cell lysates (30–40 μg) were used for Western blotting [12, 36]. In brief, denatured protein samples were boiled for 5 min, run on 10% NuPAGE Bis-Tris gels, and then transferred to nitrocellulose membranes. Membranes were blocked for 1 h with blocking buffer (2% nonfat milk in PBS) and then incubated overnight at 4°C with the primary antibodies diluted in blocking buffer with 0.1% Tween 20. We used mouse anti-mouse Trex1 (BD Biosciences) to detect Trex1 protein and rabbit anti-mouse β-actin [(I-19)-R sc-1616-R; Santa Cruz Biotechnology, Santa Cruz, CA, USA] as a loading control. Membranes were then washed and incubated for 1 h with IRDye 800 goat anti-mouse and IRDye 680 goat anti-rabbit (LI-COR Biosciences, Lincoln, NE, USA), diluted in blocking buffer plus 0.1% Tween 20. After three washes, proteins were visualized on an Odyssey infrared imaging system (LI-COR Biosciences) in 700 nm and 800 nm channels.
Statistical analysis
Means and se were calculated, averaging results from independent experiments, performed with independent bone marrow cultures or spleens, obtained from individual mice/each experiment. Prism software (GraphPad, San Diego, CA, USA) was used for statistical analysis, and one sample t-test and two-tailed unpaired t-test for comparison between two groups and one-way ANOVA for multiple groups, followed by Newman-Keuls post hoc correction, were used as appropriate. P < 0.05 (marked in the figures as *P<0.05, **P<0.01, and ***P<0.001) was considered significant.
RESULTS
Type I IFNs increase Trex1 expression in cDCs
To investigate the regulation of Trex1 expression in DCs, we analyzed the expression of Trex1 in an in vitro model of cDCs, the DCs grown in rGM-CSF, as described previously [36]. It has been reported previously that mouse macrophages up-regulate the Trex1 transcript, along with other ISGs upon IFN-αβ and IFN-γ stimulation [31, 37]. We have shown previously that cDCs are an important source of type I IFNs, such as IFN-β, and of ISGs, which can be stimulated by TLR ligands and IFN-α [36]. In this investigation, we found that stimulation of B6 bone marrow-derived cDCs with IFN-α significantly up-regulated Trex1 RNA, up to 20-fold, as measured by qRT-PCR (Fig. 1A), with similar kinetics to the up-regulation of IFN-β RNA (Fig. 1B), an early ISG induced in DCs by IFN-α stimulation. cDCs constitutively expressed high levels of Trex1 protein that doubled in the IFN-α-stimulated cDCs, and such high expression was maintained even 48 h after stimulation (Fig. 1C). Trex1 RNA induction within 1 h after IFN-α stimulation suggests that Trex1 is an early ISG. Thus, the expression of Trex1 is constitutively high in cDCs, and it is up-regulated quickly by IFN-α, indicating that Trex1 is an early ISG in cDCs.
Figure 1. Type I IFNs induce Trex1 expression in B6 cDCs.
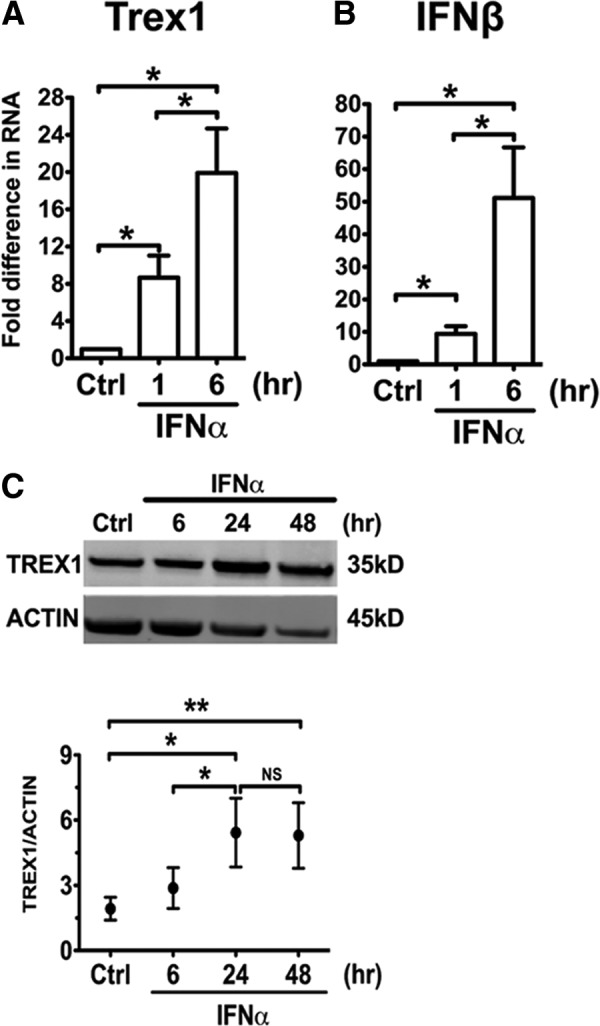
We stimulated B6 cDCs at Days 6–7 of culture, with or without IFN-α 1500 U/ml, and measured RNA expression of Trex1 (A) and IFN-β (B) by qRT-PCR after 1 or 6 h of stimulation. We normalized the results to the housekeeping gene cyclophilin and show fold differences compared with unstimulated B6 cDCs. Mean ± se are from four independent experiments. Ctrl, Control. (C) We measured by Western blotting protein levels of Trex1 in B6 cDCs after 6, 24, and 48 h of IFN-α stimulation. One representative blot is shown (upper), and Trex1 intensity values were normalized to β-actin (lower) and shown as mean ± se of three independent experiments, performed with three independent bone marrow-derived cultures, generated from three different mice.
IFN-α induces Trex1 expression through an IFNAR1-, STAT1-, and STAT2-dependent signaling pathway
To confirm that Trex1 is induced by IFN-α, we analyzed cDCs from WT and type I IFNARKO mice (Fig. 2A). The deficiency of IFNAR1 resulted in the abrogation of Trex1 RNA up-regulation by IFN-α after 1 and 6 h of stimulation (Fig. 2A) and was paralleled by the expected, deficient up-regulation of IFN-β (Fig. 2A). Western blot analysis showed that the increase in Trex1 protein levels, which in WT cDCs, started in the first 7.5 h and doubled after 24 h of stimulation with IFN-α, was absent in IFNARKO cDCs (Fig. 2B). Thus, IFNAR1 is essential for IFN-α-induced Trex1 expression in cDCs.
Figure 2. IFN-α-induced Trex1 expression is IFNAR1-, STAT1-, and STAT2-dependent.

We stimulated cDCs from IFNARKO (A and B) or STAT1KO or STAT2KO (C and D) and their appropriate WT (B6 or 129) mice with IFN-α 1500 U/ml. (A) Trex1 and IFN-β RNA were measured by qRT-PCR in WT and IFNARKO cDCs. (B) Protein expression of Trex1 in WT and IFNARKO (KO) cDCs was stimulated with IFN-α for 2.5, 7.5, and 24 h. (C) Trex1 RNA was measured in WT, STAT2KO, and STAT1KO cDCs stimulated with IFN-α for 1 and 6 h. (D) Protein expression of Trex1 was measured in WT and STAT1KO (S1KO) and STAT2KO (S2KO) cDCs stimulated with IFN-α for 8 h. qRT-PCR results were normalized to the housekeeping gene cyclophilin and shown as fold differences compared with unstimulated WT cDCs in A and C. Mean ± se are from four (A) and four (C) independent experiments in WT and STAT2KO, and results are from one experiment for STAT1KO cDCs. One representative blot and mean ± se from three independent experiments are shown in B and D. Trex1 intensity values were normalized to β-actin.
The canonical signaling pathway downstream of IFNAR1 involves engagement of STAT1 and STAT2 heterodimers and STAT1 homodimers [38]. Previously, STAT1 engagement was identified to mediate IFN-γ-induced Trex1 in macrophages [30]. To assess whether STAT1 homodimers and STAT1/STAT2 heterodimers contribute to the IFN-α-induced up-regulation of Trex1, we stimulated WT and STAT1KO or STAT2KO cDCs with IFN-α. In contrast to WT cDCs, neither STAT1KO nor STAT2KO cDCs up-regulated Trex1 expression at the RNA (Fig. 2C) or protein (Fig. 2D) levels upon exposure to IFN-α. Overall, these findings reveal that IFN-α drives Trex1 induction in cDCs via the canonical IFNAR signaling pathway through STAT1/STAT2 heterodimers rather than STAT1 homodimers.
TLR7, TLR9, and TLR3 ligands up-regulate Trex1 expression in cDCs through autocrine type I IFN production
TLR ligands are pivotal stimulators of DC activation. Mouse and human DCs respond to TLR ligands by activating their proinflammatory and APC functions and producing abundant type I IFNs [4]. To determine whether activation upon TLR stimulation induces Trex1 expression in DCs, we stimulated B6 cDCs with R848, CpG-B, and PolyI:C which are TLR7, TLR9, and TLR3 ligands, respectively, and found that these TLR stimuli did not up-regulate Trex1 at 1 h, as instead, IFN-α stimulation did (compare Fig. 1A with Fig. 3A). However, TLR7, -9, and -3 stimulation increased Trex1 RNA transcription significantly, by 20- to 40-fold compared with untreated B6 cDCs, at 6 h after stimulation (Fig. 3A). IFN-β expression was increased at an early time-point at 1 h by TLR ligands (Fig. 3A). Considering the fact that TLR-induced activation of DCs can be mediated by autocrine type I IFNs [3, 4, 39] and by IFN-independent mediators (NF-κB and MAPK, etc.), a possible explanation for the different kinetics in Trex1 up-regulation between TLR and IFN-α stimulation would be that TLR-induced Trex1 up-regulation is mediated by autocrine type I IFNs. To test this hypothesis, we stimulated IFNARKO and WT cDCs with R848, CpG-B, and PolyI:C and determined Trex1 expression. There was minimal Trex1 up-regulation in WT and IFNARKO cDCs at 1 h of stimulation; however, after 6 h, we found increased Trex1 RNA expression in WT that was absent in IFNARKO cDCs (Fig. 3B). Consistently, reduction in IFN-β RNA levels at 6 h paralleled these results (Fig. 3B). To determine further whether the signaling pathway downstream of IFNAR plays a role in Trex1 expression, we studied STAT1KO or STAT2KO and WT cDCs stimulated with TLR ligands. As observed in IFNARKO cDCs, there was little up-regulation by TLR stimuli at 1 h in WT, STAT1KO, and STAT2KO cDCs (Fig. 3C). STAT1 or STAT2 deficiency prevented TLR-induced up-regulation of Trex1 RNA at 6 h (Fig. 3C), paralleled with a greatly diminished expression of IFN-β (data not shown) and also, a suppressed up-regulation of Trex1 protein at 8 h after stimulation with R848, CpG-B, and PolyI:C (Fig. 3D and E). Thus, our findings show that TLR7, -9, and -3 ligands induce Trex1 expression in cDCs and that an intact IFNAR1, STAT1, and STAT2 signaling pathway is required for this induction. These results indicate that TLR7, -9, and -3 activation leads to autocrine type I IFN production, which up-regulates Trex1 expression. Furthermore, as Trex1 is up-regulated by four major DC stimuli, it could be important in DC activation/maturation, and therefore, we aimed to determine whether it is induced by DC activators other than TLR7, TLR9, and TLR3 in an IFN-independent manner.
Figure 3. TLR7, -9, and -3 ligands up-regulate Trex1 expression in cDCs in a STAT1-, STAT2-, and IFNAR1-mediated manner.

We stimulated cDCs from B6 (A), IFNARKO (B), STAT2KO and STAT1KO (C), and appropriate WT mice with R848 1 μg/ml, CpG 10 μg/ml, and PolyI:C (PI:C) 200 ng/ml for 1 and 6 h and analyzed Trex1 and IFN-β RNA by qRT-PCR. Results of qRT-PCR were normalized to cyclophilin, and mean ± se of three (A), four (B), and three (C) independent experiments are shown. One representative experiment is shown for STAT1KO in C. (D and E) Protein expression of Trex1 in WT, STAT1KO, and STAT2KO cDCs stimulated with the same TLR ligands. Western blotting results were normalized to β-actin, and one representative blot and mean ± se from three independent experiments are shown in D and E.
LPS induces Trex1 expression using type I IFN- and NF-κB-dependent signaling pathways
We assessed whether LPS, a TLR4 ligand and potent DC activator, also affects Trex1 expression in cDCs. LPS started to up-regulate Trex1 RNA after 1 h of stimulation and continued to induce robust production after 6 h, which paralleled the early-elevated IFN-β expression (Fig. 4A). Moreover, IFNAR1 and STAT2 deficiencies dramatically abolished LPS-induced up-regulation of Trex1 RNA at 6 h but not at 1 h poststimulation (Fig. 4B and C). Similarly, STAT1KO and STAT2KO cDCs stimulated with LPS for 8 h displayed a moderate induction in Trex1 protein levels compared with unstimulated controls, although not to the same extent observed in LPS-stimulated WT cDCs (Fig. 4D). These results reveal that type I IFN-dependent and -independent pathways contribute to early Trex1 stimulation by LPS. In contrast, later Trex1 induction by LPS in WT cDCs is mostly dependent on the type I IFNAR-mediated signaling, as it was greatly diminished in IFNAR1, STAT1KO, and STAT2KO cDCs.
Figure 4. LPS induces Trex1 expression via a STAT1, STAT2, and IFNAR signaling pathway, as well as a NF-κB-dependent pathway.

We stimulated cDCs from B6 (A), IFNARKO (B), and STAT2KO (C) and WT mice with LPS 100 ng/ml and measured Trex1 and IFN-β RNA by qRT-PCR after 1 or 6 h of stimulation. Mean ± se are from five (A), four (B), and four (C) independent experiments. (D) Western blot analysis of Trex1 in WT and STAT1KO and STAT2KO cDCs, stimulated with LPS for 8 h. One representative blot and mean ± se of three independent experiments are shown. (E) Trex1 RNA of WT, STAT1KO, and STAT2KO cDCs, pretreated with NF-κB inhibitor BAY11 (10−6 M) for 15 min and then stimulated with LPS for 1 h. Mean ± se of three independent experiments of WT and STAT2KO and one representative experiment for STAT1KO are shown in E. (F) Trex1 protein levels of WT cDCs, pretreated with BAY11 and stimulated with LPS for 6 and 24 h. RAW 264.7 cell lysates were loaded as positive (Pos.) control, as they express high Trex1 proteins. One representative blot is shown. Protein levels of Trex1 were normalized to β-actin in D and F, and mean ± se from three independent experiments are shown.
To determine the potential mechanism leading to the early IFN-independent Trex1 induction by LPS, we studied NF-κB activation because of its pivotal role in mediating signaling downstream of TLRs [40, 41]. We examined the effect of IκB kinase inhibitor BAY11 [42]: we pretreated WT, STAT1KO, and STAT2KO cDCs, with or without BAY11, for 15 min and then stimulated them with LPS. BAY11 at 10−6 M did not cause cell death for up to 24 h (data not shown) and did not induce Trex1 expression by itself (Fig. 4E and F). BAY11 treatment reduced the early Trex1 RNA expression in response to 1 h of LPS stimulation in WT cDCs and greatly inhibited it in STAT1KO and STAT2KO cDCs at the same time-point (Fig. 4E). Trex1 protein level was also inhibited by BAY11 in WT cDCs at an early time-point (6 h), although the differences did not reach statistical significance because of variations between experiments in the amounts of activation by LPS (Fig. 4F). The inhibitory effect of BAY11 on Trex1 expression was lost at 24 h (Fig. 4F), although the inhibitory effects on other readouts, such as costimulatory molecules in DCs, were persistent for up to 24 h (data not shown). These results indicate that the blockade of NF-κB signaling by BAY11 affects only early Trex1 up-regulation. These results suggest that LPS uses NF-κB to up-regulate early Trex1 expression in cDCs. This conclusion is consistent with the identification of a NF-κB box in the Trex1 promoter [30]. Although NF-κB mediates only early Trex1 induction, this is the first evidence that the NF-κB signaling plays a role in the regulation of Trex1 expression. These results indicate that Trex1 can be induced in DCs via signaling pathways other than type I IFNs and suggest that Trex1 may play a role in DC activation in general.
Trex1 is up-regulated by TLRs in DCs in vivo
We next explored whether TLR ligands increase Trex1 expression in DCs in vivo. We i.p.-injected WT mice with the TLR ligands LPS and CpG-B or PBS, and then we determined Trex1 expression in splenic DC subsets after 24 h. We found that constitutive expression of Trex1 was significantly higher in cDCs and pDCs than in other splenocytes (Fig. 5A and B), indicating that high Trex1 expression is intrinsic in DC subsets. In response to LPS or CpG-B, cDCs appeared as a single population that increased Trex1 expression significantly compared with PBS controls (Fig. 5A and B). These in vivo results confirm our in vitro data that cDCs up-regulate Trex1 upon TLR stimulation. PDCs showed two distinct levels of Trex1 expression (low and high) in PBS controls, up-regulated in LPS- or CpG-B-injected mice (Fig. 5A). Previous studies indicated that mouse and human pDCs express TLR4 [43, 44], and it is possible that the observed up-regulation of Trex1 in pDCs is a result of the direct response to LPS upon triggering of TLR4. Although pDCs do not induce much IFN-α production upon CpG-B stimuli, they are able to up-regulate activation markers [45]. Taken together, our in vivo results suggest that Trex1 up-regulation in pDCs may be a result of the direct response of pDCs to LPS and CpG-B; moreover, we cannot exclude the possibility that this up-regulation in pDCs is a result of the paracrine production of type I IFNs by cDCs upon LPS or CpG-B and therefore, that type I IFNs may also induce Trex1 in pDCs. Overall, in DC populations, Trex1 regulation may be a general response to infections and equivalent sources of danger signals.
Figure 5. TLRs up-regulate Trex1 in vivo.
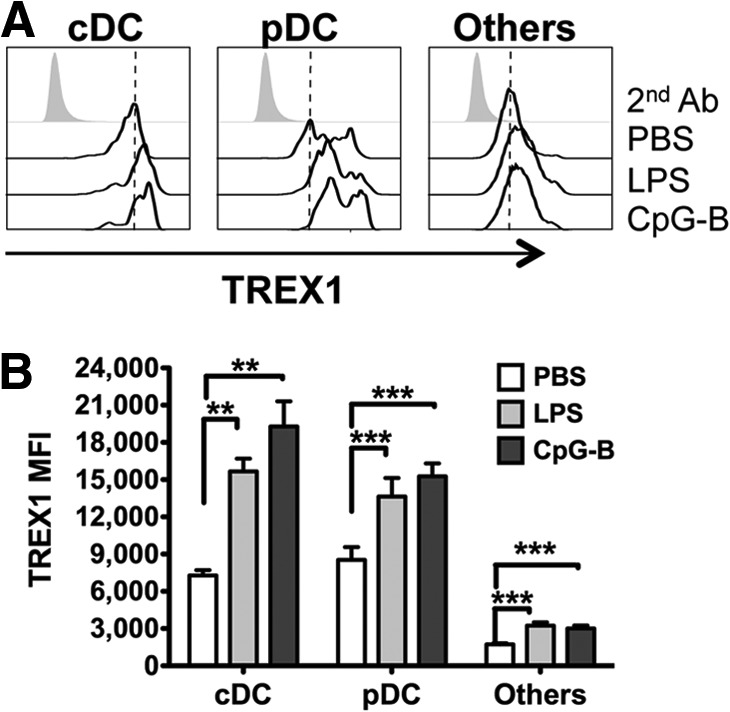
Intracellular staining of Trex1 in spleens from B6 mice injected i.p. with LPS, CpG-B, or PBS, 24 h earlier. (A) Representative histograms of Trex1 expression in different populations: cDC (CD11chighB220−), pDC (CD11clowB220+), and other cells (CD11c−B220−). 2nd Ab, Secondary antibody. (B) Mean ± se of Trex1 MFI (n=3 mice/group) in cDC, pDC, and other cells.
Lentivirus induces Trex1 in cDCs
After finding that rIFN-α and potent inducers of type I IFNs, such as the TLR ligands, stimulate Trex1 expression in DCs in vitro and in vivo, we next sought to determine whether Trex1 expression is up-regulated during the natural circumstance in which cDCs are normally exposed to type I IFNs and TLR ligands, i.e., viral infections. Because of the pathogenic role of Trex1 in HIV infection [28], we chose a HIV-derived replication-defective lentivirus to determine whether viral exposure can induce Trex1 expression. This lentivirus has been developed as a vector for gene therapy [35] and tested for expression in vitro and in vivo [34]. We and others have found that it transduces cDCs as well in vitro (data not shown) [46]. We infected B6 cDCs with the lentivirus at a MOI of one and five, and after 6 h, we measured the RNA levels of Trex1 and other ISGs. We found that the lentivirus induced Trex1 expression (Fig. 6A), along with the expression of IFN-β (Fig. 6B) and the type I IFN responsive gene Isg15 (Fig. 6C). To determine whether Trex1 up-regulation requires direct viral infection or whether it occurs in response to the type I IFNs produced upon the infection, we measured Trex1 protein levels in transduced DCs (GFP+CD11c+ cells) versus nontransduced DCs (GFP−CD11c+ cells) in vitro. We took advantage of the fact that the transduction efficiency of this lentivirus in cDCs is 30–50% to study Trex1 expression in transduced and nontransduced cells in the same culture wells. We infected bone marrow cells at Day 2 of culture with a GFP-expressing lentivirus at a MOI of one, five, and ten, and after 4 days, we stained intracellularly for Trex1 protein. We chose this time-point, because in our experience, this is when the expression of the lentivirus, as measured by GFP percent and MFI, reaches its peak. We found that transduced cDCs up-regulated Trex1 expression at all tested MOIs (Fig. 6D and E). Interestingly, the nontransduced cDCs also up-regulated Trex1 but with significantly lower Trex1 expression than that observed in the transduced cDCs (Fig. 6D and E). Consistent to our previous findings, these results demonstrate that during a retroviral infection or lentiviral vector-based gene therapy, cDCs can up-regulate Trex1 expression in response to direct viral infection and upon stimulation by paracrine mediators produced upon infection, such as type I IFNs.
Figure 6. Viral infection induces Trex1 expression.

We infected B6 cDCs at Day 6 of culture with a HIV-based lentivirus at different MOIs and measured Trex1 (A), IFN-β (B), and the IFN-responsive gene, Isg15 (C) RNA by qRT-PCR after 5–6 h. We normalized the results to the housekeeping gene cyclophilin and show fold differences compared with uninfected B6 cDCs. Mean ± se are from five (A and C) and four (B) independent experiments. (D and E) We transduced Day 2 bone marrow cells without (untreated) or with HIV-based lentiviruses expressing GFP at different MOIs (1, 5, and 10) for 96 h, and on Day 6, we measured Trex1 protein levels by intracellular staining. (D) Representative histograms of Trex1 expression in the two populations: transduced cDCs (GFP+CD11c+; upper) and nontransduced cDC (GFP−CD11c+; lower) cells; the solid gray line represents the curve of Trex1 expression from untreated cDCs (lower). (E) Trex1 ΔMdFI in GFP+CD11c+ or GFP−CD11c+ cells was calculated as Trex1 MdFI minus the MdFI of the secondary antibody IgG1 expression in each population. Mean ± se are shown from three independent experiments performed in duplicate.
DISCUSSION
TLRs are critical early host-defense players against invading pathogens. Together with many other cell types, DCs express most TLRs, and once those are triggered, DCs activate and produce proinflammatory cytokines, which are pivotal to initiate the immune response [47–49]. DCs also express type I IFNRs, which amplify DC activation by stimulating the up-regulation of IFN-α/β themselves and of other ISGs that modulate biological functions in a cell-specific manner [38]. Among hundreds of ISGs, few of them have been confirmed for their role in viral infections [50]. We show here that the most abundant exonuclease Trex1 is an ISG, and its expression is induced by type I IFNs, TLR ligands, and lentiviruses in cDCs. During the revision of our manuscript, it was reported that TLR ligands stimulate in vitro Trex1 RNA expression in many immune cells, including DCs [32]. Our results support similar conclusions; furthermore, we also show that TLR ligands require autocrine type I IFNs and the canonical signaling pathway downstream of IFNAR to up-regulate Trex1 expression. Moreover, the early Trex1 expression induced by the potent proinflammatory stimulus LPS in cDCs involves IFN-dependent and -independent pathways, the latter mediated by NF-κB activation. Recently, Trex1 gene expression was shown to be augmented by IFN-α/β in macrophages [31]. Also, another group reported that IFN-γ regulates Trex1 expression transcriptionally in a STAT1-dependent manner [30], and indeed, Trex1 promoter includes IFN-γ-activated sequence elements, ISRE, AP-1, and NF-κB sites [30, 51]. All of these results suggest that Trex1 might be up-regulated by other proinflammatory stimuli, or it may be associated with the process of DC activation in general. Trex1−/− mice have been studied in few reports [19, 28, 29, 32], and we still do not fully know all of the immunological functions of Trex1. For the first time, we studied the expression of Trex1 protein in immune cells in vivo and found that Trex1 expression is higher in splenic cDCs and pDCs than in other splenocytes and is increased further by TLR stimulation, therefore suggesting that an unknown role for Trex1 in DC functions may be in the regulation of DC activation/maturation. It will be important to understand the outcomes of this up-regulation. We also found that pDCs, but not cDCs, can be divided in two subpopulations, expressing high and low levels of Trex1, both constitutively and after LPS or CpG-B injection in vivo. As pDCs are heterogeneous in their origin, deriving from a myeloid or a lymphoid precursor, in the expression of lineage markers and even in their function (immunity vs. tolerance) [52], we can speculate that the differential expression of Trex1 may be linked to one of these variables, and further studies are needed to test these hypotheses.
Type I IFNs and many more proinflammatory cytokines induce suppressor of cytokine signaling, which negatively regulate IFN-dependent antiviral responses by inhibiting the interaction of IFNAR1 with tyrosine kinase 2 [53–55]. Likewise, TLRs and type I IFNs could induce Trex1 to regulate negatively their own signaling or contain an ongoing antiviral response. The observation that the IFNAR1 deficiency rescues mice from the tissue damage and mortality caused by the absence of Trex1 [15, 19] suggests that type I IFNs are the main cytokines inhibited by Trex1, which can induce pathology, but it does not exclude that Trex1 may participate in regulating other less-damaging gene products.
Trex1 was previously found to degrade DNA species from endogenous retroelements and retrotranscribed HIV-1 DNA in the cytoplasm, which both would trigger type I IFN production [15, 16]. As exposure to viral DNA, as well as to type I IFNs and nucleic acids, such as CpGs, dsRNA (PolyI:C), and ssRNA (R848), induces Trex1 up-regulation in DCs, it would be intriguing to propose that Trex1 is an IFN-dependent, antiviral strategy triggered in DCs by nucleic acid sensors to control cytoplasmic DNA and render DCs more resistant to viruses. Previous evidence indicates that this process is hijacked by reverse-transcribed HIV [28]. During the process, Trex1 acts with the ER-associated complex, inhibits HIV autointegration [27], and negatively regulates the type I IFN response that is triggered by HIV DNA through the STING-TBK1-IRF3 axis [28]. Surprisingly, Trex1−/− cells displayed antiviral resistance to RNA viruses, besides DNA viruses, characterized by the activation of several IFN-independent antiviral genes [29]. Therefore, Trex1 does not seem to benefit the host but rather, the viruses. Nevertheless, in one large-scale ISG screen against YFV, Trex1 was able to inhibit viral replication by 50% compared with control [56], suggesting an unidentified antiviral role of Trex1 in the YFV infection cycle and possibly in other viral infections yet to be determined.
As we used, as a model of viral infection, a HIV-based lentivirus that was generated and is currently tested as a gene therapy vector [35], the fact that this vector induced Trex1 expression in cDCs suggests that Trex1 may play a role in controlling the persistence and expression of genes carried by lentiviral vectors. We have shown that Trex1 expression is not only up-regulated in lentivirus-transduced cDCs but also in nontransduced cDCs, although with lower expression, suggesting that transduced and nontransduced DCs may up-regulate Trex1 expression in two different ways: transduced DCs may increase Trex1 expression directly upon recognition of the virus, possibly through TLRs, whereas nontransduced DCs may increase Trex1 expression because of paracrine-type I IFN produced by the transduced DCs. Therefore, our results indicate that it is important to determine the impact of Trex1 in the success of gene therapy protocols, and we are presently performing these studies in WT and Trex1−/− mice.
Trex1 may also have the housekeeping function to establish the threshold for type I IFNs to activate DCs. By degrading excessive endogenous DNA species, Trex1 may restrain the amount of type I IFNs that is induced by DNA species through STING and other unknown DNA sensors and lower DC exposure to autocrine and paracrine type I IFNs; thus, Trex1 may contribute to balancing beneficial activated DCs and detrimental overactivated DCs. Very recently, Gehrke et al. [17] showed that oxidized DNA, derived from phagocytosis of adenovirus DNA, is highly resistant to Trex1 degradation and can enhance the type I IFN response, serving as a DAMP to myeloid DCs. This hypothesis may provide a mechanism to explain why Trex1 is linked to autoimmunity [15]. Trex1 is one of the few genes whose mutations lead to monogenic lupus [57], and excessive levels of IFN-α in the serum [58] and a type I IFN signature in PBMCs are associated with lupus [59–61]. Lupus-prone mice, genetically resistant to type I IFNs, develop a milder disease [62, 63], and DCs from lupus-prone mice have enhanced activation [64] and antigen presentation [64, 65] and express a type I IFN signature before disease onset [12]. Considering the pathogenic role of the TLR-IFN axis in autoimmunity [66], the absence of Trex1 may increase the strength of the response to TLRs and participate in the unregulated type I IFN signature and the abnormal DC activation and dysregulated immune response during autoimmunity.
In summary, our results show that the constitutive expression of Trex1 is high in cDCs and pDCs and higher than in other immune cells in vivo. We propose a novel model in which Trex1 is an early ISG that is up-regulated by type I IFNs and TLR ligands in cDCs via the canonical type I IFN signaling pathway and also via NF-κB during early LPS stimulation. Our data suggest that Trex1 may have a role in antiviral responses. Alternatively, Trex1 could be involved in functions for which there are experimental data, such as apoptosis [14]. Furthermore, Trex1 could regulate DC activation, and this latter regulatory mechanism could explain the pathogenic link between Trex1 loss-of-function mutations and autoimmunity.
ACKNOWLEDGMENTS
This study was supported by the U.S. National Institutes of Health, National Institute of Allergy and Infectious Diseases, grant RO1-AI076423 (to S.G.).
We thank Drs. Roberto Caricchio, Philip L. Cohen, Michael F. Denny, Doina Ganea, and Weidong Xiao and the members of the Lab of DC Biology (Marita Chakhtoura, Paul Gallo, Robert Chain, and Dr. Uma Sriram) for reading the manuscript. We thank Linda Varghese for the outstanding care of our mouse colony.
Footnotes
- 129
- 129/SvJ
- B6
- C57BL/6
- BAY11
- BAY11-7082
- cDC
- conventional DC
- Ct
- comparative threshold
- DAMP
- damage-associated molecule pattern
- IFNAR1
- IFN-αR 1
- IFNARKO
- IFN-αR knockout
- IRF3
- IFN regulatory factor 3
- ISG
- IFN-stimulated gene
- ISRE
- IFN-stimulated response element
- KO
- knockout
- MdFI
- median fluorescence intensity
- MFI
- mean fluorescence intensity
- MOI
- multiplicity of infection
- pDC
- plasmacytoid DC
- PolyI:C
- polyinosinic:polycytidylic acid
- qRT-PCR
- quantitative real-time RT-PCR
- R848
- resiquimod
- SLE
- systemic lupus erythematosus
- STAT1/2KO
- STAT1/2 knockout
- STING
- stimulator of IFN genes
- TBK1
- TRAF-associated NF-κB activator-binding kinase 1
- Trex1
- three repair exonuclease 1
- Trex1−/−
- three repair exonuclease 1-deficient
- YFV
- yellow fever virus
AUTHORSHIP
J.X. performed the studies, analyzed data, and drafted the manuscript. P.W.Z. generated the lentiviruses. A.M.G. contributed to the discussion. P.W.Z. and A.M.G. reviewed the manuscript. S.G. designed and supervised the study and wrote the manuscript.
REFERENCES
- 1. Gallo P. M., Gallucci S. (2013) The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front. Immunol. 4, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gallucci S., Lolkema M., Matzinger P. (1999) Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5, 1249–1255 [DOI] [PubMed] [Google Scholar]
- 3. Hoshino K., Kaisho T., Iwabe T., Takeuchi O., Akira S. (2002) Differential involvement of IFN-β in Toll-like receptor-stimulated dendritic cell activation. Int. Immunol. 14, 1225–1231 [DOI] [PubMed] [Google Scholar]
- 4. Baccala R., Hoebe K., Kono D. H., Beutler B., Theofilopoulos A. N. (2007) TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat. Med. 13, 543–551 [DOI] [PubMed] [Google Scholar]
- 5. Medzhitov R. (2001) Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1, 135–145 [DOI] [PubMed] [Google Scholar]
- 6. Pichlmair A., Reis e Sousa C. (2007) Innate recognition of viruses. Immunity 27, 370–383 [DOI] [PubMed] [Google Scholar]
- 7. Trinchieri G. (2003) Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3, 133–146 [DOI] [PubMed] [Google Scholar]
- 8. Schnare M., Barton G. M., Holt A. C., Takeda K., Akira S., Medzhitov R. (2001) Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2, 947–950 [DOI] [PubMed] [Google Scholar]
- 9. Pulendran B., Kumar P., Cutler C. W., Mohamadzadeh M., Van Dyke T., Banchereau J. (2001) Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J. Immunol. 167, 5067–5076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jorgensen T. N., Roper E., Thurman J. M., Marrack P., Kotzin B. L. (2007) Type I interferon signaling is involved in the spontaneous development of lupus-like disease in B6.Nba2 and (B6.Nba2 x NZW)F-1 mice. Genes Immunity 8, 653–662 [DOI] [PubMed] [Google Scholar]
- 11. Elkon K. B., Stone V. V. (2011) Type I interferon and systemic lupus erythematosus. J. Interferon Cytokine Res. 31, 803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sriram U., Varghese L., Bennett H. L., Jog N. R., Shivers D. K., Ning Y., Behrens E. M., Caricchio R., Gallucci S. (2012) Myeloid dendritic cells from B6.NZM Sle1/Sle2/Sle3 lupus-prone mice express an IFN signature that precedes disease onset. J. Immunol. 189, 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mazur D. J., Perrino F. W. (1999) Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′→5′ exonucleases. J. Biol. Chem. 274, 19655–19660 [DOI] [PubMed] [Google Scholar]
- 14. Chowdhury D., Beresford P. J., Zhu P., Zhang D., Sung J. S., Demple B., Perrino F. W., Lieberman J. (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 23, 133–142 [DOI] [PubMed] [Google Scholar]
- 15. Stetson D. B., Ko J. S., Heidmann T., Medzhitov R. (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang Y. G., Lindahl T., Barnes D. E. (2007) Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886 [DOI] [PubMed] [Google Scholar]
- 17. Gehrke N., Mertens C., Zillinger T., Wenzel J., Bald T., Zahn S., Tuting T., Hartmann G., Barchet W. (2013) Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39, 482–495 [DOI] [PubMed] [Google Scholar]
- 18. Morita M., Stamp G., Robins P., Dulic A., Rosewell I., Hrivnak G., Daly G., Lindahl T., Barnes D. E. (2004) Gene-targeted mice lacking the Trex1 (DNase III) 3′-5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 24, 6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gall A., Treuting P., Elkon K. B., Loo Y. M., Gale M., Jr., Barber G. N., Stetson D. B. (2012) Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crow Y. J., Hayward B. E., Parmar R., Robins P., Leitch A., Ali M., Black D. N., van Bokhoven H., Brunner H. G., Hamel B. C., Corry P. C., Cowan F. M., Frints S. G., Klepper J., Livingston J. H., Lynch S. A., Massey R. F., Meritet J. F., Michaud J. L., Ponsot G., Voit T., Lebon P., Bonthron D. T., Jackson A. P., Barnes D. E., Lindahl T. (2006) Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 38, 917–920 [DOI] [PubMed] [Google Scholar]
- 21. Lee-Kirsch M. A., Chowdhury D., Harvey S., Gong M., Senenko L., Engel K., Pfeiffer C., Hollis T., Gahr M., Perrino F. W., Lieberman J., Hubner N. (2007) A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J. Mol. Med. (Berl). 85, 531–537 [DOI] [PubMed] [Google Scholar]
- 22. Gunther C., Meurer M., Stein A., Viehweg A., Lee-Kirsch M. A. (2009) Familial chilblain lupus—a monogenic form of cutaneous lupus erythematosus due to a heterozygous mutation in TREX1. Dermatology 219, 162–166 [DOI] [PubMed] [Google Scholar]
- 23. Hur J-W., Sung Y-K., Shin H. D., Park B. L., Cheong H. S., Bae S-C. (2007) TREX1 polymorphisms associated with autoantibodies in patients with systemic lupus erythematosus. Rheumatol. Int. 28, 783–789 [DOI] [PubMed] [Google Scholar]
- 24. Lee-Kirsch M. A., Gong M., Chowdhury D., Senenko L., Engel K., Lee Y-A., de Silva U., Bailey S. L., Witte T., Vyse T. J., Kere J., Pfeiffer C., Harvey S., Wong A., Koskenmies S., Hummel O., Rohde K., Schmidt R. E., Dominiczak A. F., Gahr M., Hollis T., Perrino F. W., Lieberman J., Hübner N. (2007) Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 39, 1065–1067 [DOI] [PubMed] [Google Scholar]
- 25. Atianand M. K., Fitzgerald K. A. (2013) Molecular basis of DNA recognition in the immune system. J. Immunol. 190, 1911–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thompson M. R., Kaminski J. J., Kurt-Jones E. A., Fitzgerald K. A. (2011) Pattern recognition receptors and the innate immune response to viral infection. Viruses 3, 920–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yan N., Cherepanov P., Daigle J. E., Engelman A., Lieberman J. (2009) The SET complex acts as a barrier to autointegration of HIV-1. PLoS Pathog. 5, e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan N., Regalado-Magdos A. D., Stiggelbout B., Lee-Kirsch M. A., Lieberman J. (2010) The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11, 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hasan M., Koch J., Rakheja D., Pattnaik A. K., Brugarolas J., Dozmorov I., Levine B., Wakeland E. K., Lee-Kirsch M. A., Yan N. (2013) Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat. Immunol. 14, 61–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Serra M., Forcales S. V., Pereira-Lopes S., Lloberas J., Celada A. (2011) Characterization of Trex1 induction by IFN-γ in murine macrophages. J. Immunol. 186, 2299–2308 [DOI] [PubMed] [Google Scholar]
- 31. Cobos Jimenez V., Booiman T., de Taeye S. W., van Dort K. A., Rits M. A., Hamann J., Kootstra N. A. (2012) Differential expression of HIV-1 interfering factors in monocyte-derived macrophages stimulated with polarizing cytokines or interferons. Sci. Rep. 2, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pereira-Lopes S., Celhar T., Sans-Fons G., Serra M., Fairhurst A. M., Lloberas J., Celada A. (2013) The exonuclease Trex1 restrains macrophage proinflammatory activation. J. Immunol. 191, 6128–6135 [DOI] [PubMed] [Google Scholar]
- 33. Manzanillo P. S., Shiloh M. U., Portnoy D. A., Cox J. S. (2012) Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell. Host Microbe 11, 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Endo M., Zoltick P. W., Peranteau W. H., Radu A., Muvarak N., Ito M., Yang Z., Cotsarelis G., Flake A. W. (2008) Efficient in vivo targeting of epidermal stem cells by early gestational intraamniotic injection of lentiviral vector driven by the keratin 5 promoter. Mol. Ther. 16, 131–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sena-Esteves M., Tebbets J. C., Steffens S., Crombleholme T., Flake A. W. (2004) Optimized large-scale production of high titer lentivirus vector pseudotypes. J. Virol. Methods 122, 131–139 [DOI] [PubMed] [Google Scholar]
- 36. Sriram U., Biswas C., Behrens E. M., Dinnall J. A., Shivers D. K., Monestier M., Argon Y., Gallucci S. (2007) IL-4 suppresses dendritic cell response to type I interferons. J. Immunol. 179, 6446–6455 [DOI] [PubMed] [Google Scholar]
- 37. Liu S. Y., Sanchez D. J., Aliyari R., Lu S., Cheng G. (2012) Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. USA 109, 4239–4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Platanias L. C. (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 [DOI] [PubMed] [Google Scholar]
- 39. Montoya M., Schiavoni G., Mattei F., Gresser I., Belardelli F., Borrow P., Tough D. F. (2002) Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 99, 3263–3271 [DOI] [PubMed] [Google Scholar]
- 40. Kawai T., Adachi O., Ogawa T., Takeda K., Akira S. (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11, 115–122 [DOI] [PubMed] [Google Scholar]
- 41. Alexopoulou L., Holt A. C., Medzhitov R., Flavell R. A. (2001) Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 [DOI] [PubMed] [Google Scholar]
- 42. Mori N., Yamada Y., Ikeda S., Yamasaki Y., Tsukasaki K., Tanaka Y., Tomonaga M., Yamamoto N., Fujii M. (2002) Bay 11-7082 inhibits transcription factor NF-κB and induces apoptosis of HTLV-I-infected T-cell lines and primary adult T-cell leukemia cells. Blood 100, 1828–1834 [DOI] [PubMed] [Google Scholar]
- 43. Edwards A. D., Diebold S. S., Slack E. M., Tomizawa H., Hemmi H., Kaisho T., Akira S., Reis e Sousa C. (2003) Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 α+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33, 827–833 [DOI] [PubMed] [Google Scholar]
- 44. Dai J., Megjugorac N. J., Amrute S. B., Fitzgerald-Bocarsly P. (2004) Regulation of IFN regulatory factor-7 and IFN-α production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J. Immunol. 173, 1535–1548 [DOI] [PubMed] [Google Scholar]
- 45. Guiducci C., Ott G., Chan J. H., Damon E., Calacsan C., Matray T., Lee K. D., Coffman R. L., Barrat F. J. (2006) Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J. Exp. Med. 203, 1999–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Toscano M. G., Delgado M., Kong W., Martin F., Skarica M., Ganea D. (2010) Dendritic cells transduced with lentiviral vectors expressing VIP differentiate into VIP-secreting tolerogenic-like DCs. Mol. Ther. 18, 1035–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Akira S., Takeda K., Kaisho T. (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2, 675–680 [DOI] [PubMed] [Google Scholar]
- 48. Gautier G., Humbert M., Deauvieau F., Scuiller M., Hiscott J., Bates E. E., Trinchieri G., Caux C., Garrone P. (2005) A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J. Exp. Med. 201, 1435–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Napolitani G., Rinaldi A., Bertoni F., Sallusto F., Lanzavecchia A. (2005) Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 6, 769–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schoggins J. W., Rice C. M. (2011) Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1, 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Christmann M., Tomicic M. T., Aasland D., Berdelle N., Kaina B. (2010) Three prime exonuclease I (TREX1) is Fos/AP-1 regulated by genotoxic stress and protects against ultraviolet light and benzo(a)pyrene-induced DNA damage. Nucleic Acids Res. 38, 6418–6432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reizis B., Bunin A., Ghosh H. S., Lewis K. L., Sisirak V. (2011) Plasmacytoid dendritic cells: recent progress and open questions. Annu. Rev. Immunol. 29, 163–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fenner J. E., Starr R., Cornish A. L., Zhang J. G., Metcalf D., Schreiber R. D., Sheehan K., Hilton D. J., Alexander W. S., Hertzog P. J. (2005) Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat. Immunol. 7, 33–39 [DOI] [PubMed] [Google Scholar]
- 54. Qin H., Wilson C. A., Lee S. J., Benveniste E. N. (2006) IFN-β-induced SOCS-1 negatively regulates CD40 gene expression in macrophages and microglia. FASEB J. 20, 985–987 [DOI] [PubMed] [Google Scholar]
- 55. Piganis R. A., De Weerd N. A., Gould J. A., Schindler C. W., Mansell A., Nicholson S. E., Hertzog P. J. (2011) Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon α receptor (IFNAR1)-associated tyrosine kinase Tyk2. J. Biol. Chem. 286, 33811–33818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schoggins J. W., Wilson S. J., Panis M., Murphy M. Y., Jones C. T., Bieniasz P., Rice C. M. (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Connolly J. J., Hakonarson H. (2012) Role of cytokines in systemic lupus erythematosus: recent progress from GWAS and sequencing. J. Biomed. Biotechnol. 2012, 798924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ytterberg S. R., Schnitzer T. J. (1982) Serum interferon levels in patients with systemic lupus erythematosus. Arth. Rheum. 25, 401–406 [DOI] [PubMed] [Google Scholar]
- 59. Bennett L., Palucka A. K., Arce E., Cantrell V., Borvak J., Banchereau J., Pascual V. (2003) Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197, 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Crow M. K., Kirou K. A., Wohlgemuth J. (2003) Microarray analysis of interferon-regulated genes in SLE. Autoimmunity 36, 481–490 [DOI] [PubMed] [Google Scholar]
- 61. Baechler E. C., Batliwalla F. M., Karypis G., Gaffney P. M., Ortmann W. A., Espe K. J., Shark K. B., Grande W. J., Hughes K. M., Kapur V., Gregersen P. K., Behrens T. W. (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 100, 2610–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Santiago-Raber M. L., Baccala R., Haraldsson K. M., Choubey D., Stewart T. A., Kono D. H., Theofilopoulos A. N. (2003) Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J. Exp. Med. 197, 777–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Agrawal H., Jacob N., Carreras E., Bajana S., Putterman C., Turner S., Neas B., Mathian A., Koss M. N., Stohl W., Kovats S., Jacob C. O. (2009) Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J. Immunol. 183, 6021–6029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Colonna L., Dinnall J. A., Shivers D. K., Frisoni L., Caricchio R., Gallucci S. (2006) Abnormal costimulatory phenotype and function of dendritic cells before and after the onset of severe murine lupus. Arthritis Res. Ther. 8, R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhu J., Liu X., Xie C., Yan M., Yu Y., Sobel E. S., Wakeland E. K., Mohan C. (2005) T cell hyperactivity in lupus as a consequence of hyperstimulatory antigen-presenting cells. J. Clin. Invest. 115, 1869–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Marshak-Rothstein A. (2006) Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol. 6, 823–835 [DOI] [PMC free article] [PubMed] [Google Scholar]