

Abstract
Solute Carrier (SLC) transporters are membrane proteins that transport solutes, such as ions, metabolites, peptides, and drugs, across biological membranes, using diverse energy coupling mechanisms. In human, there are 386 SLC transporters, many of which contribute to the absorption, distribution, metabolism, and excretion of drugs and/or can be targeted directly by therapeutics. Recent atomic structures of SLC transporters determined by X-ray crystallography and NMR spectroscopy have significantly expanded the applicability of structure-based prediction of SLC transporter ligands, by enabling both comparative modeling of additional SLC transporters and virtual screening of small molecules libraries against experimental structures as well as comparative models. In this review, we begin by describing computational tools, including sequence analysis, comparative modeling, and virtual screening, that are used to predict the structures and functions of membrane proteins such as SLC transporters. We then illustrate the applications of these tools to predicting ligand specificities of select SLC transporters, followed by experimental validation using uptake kinetic measurements and other assays. We conclude by discussing future directions in the discovery of the SLC transporter ligands.
Keywords: Membrane transporter, comparative modeling, ligand docking, protein function prediction, structure-based ligand discovery
Solute Carrier transporters
Transporters are membrane proteins that control the uptake and efflux of various solutes, including metabolites, ions, toxins, and drugs. They include solute carrier (SLC) transporters as well as ion dependent pumps and ATP-binding cassette (ABC) transporters. The majority of the SLC transporters are secondary active transporters, such as exchangers, symporters, and antiporters, for which transport is driven by various energy coupling mechanisms [1-7].
Here, we focus on the human SLC transporters, which include 386 members grouped into 52 families based on their sequences, number of transmembrane α-helices (TMHs) (typically 10-14 TMHs), and biological functions [1, 2, 4]. The SLC members play an important role in a variety of cellular functions, often cooperatively with other protein families, including receptors, enzymes, and other transporters, as illustrated by the following examples.
First, members of the SLC families SLC1 (glutamate and neutral amino acid transporter), SLC6 (Na+- and Cl--dependent neurotransmitter transporters), SLC17 (vesicular glutamate transporters), SLC18 (vesicular amine transporters), and SLC32 (vesicular inhibitory amino acid transporters) control the concentration of neurotransmitters such as glutamate and serotonin in the synapses, thereby regulating downstream neurosignaling pathways that are activated or inhibited via membrane receptors [8]. Several members of these SLC families are important drug targets. For instance, the norepinephrine transporter (NET/SLC6A2) transports norepinephrine from synaptic spaces into presynaptic neurons to regulate downstream adrenergic pathways associated with behavioral traits. Thus, several drugs, such as the attention-deficit hyperactivity disorder (ADHD) drug methylphenidate (Ritalin), inhibit NET activity to increase adrenergic signaling [9, 10].
Second, members of several SLC families, such as SLC21 (organic anion transporters), SLC22 (organic cation/anion/zwitterion transporters), and SLC47 (multidrug and toxin extrusion (MATE) transporters), are highly abundant in the liver, kidney, and blood-brain-barrier (BBB) where they regulate drug absorption, distribution, metabolism, and elimination (ADME) [11-13]. For example, the organic ion transporter 1 (OCT1/SLC22A1) transports anti-cancer and anti-viral drugs into the liver and kidney and may, therefore, mediate drug-drug interactions [14, 15].
Third, mutations in SLC members can lead to differential drug response among individuals (pharmacogenetics) [16, 17]. For instance, the intracellular concentrations of the anti-diabetic drug metformin are affected by genetic variations in OCT1 [18-21].
Sequences, structures, and mechanisms of SLC transporters
By definition, members of each of the 52 SLC transporter families share sequence identity of 20% or more to at least one other family member, and do not exhibit any significant sequence similarity to members of other families [1]. Several additional classifications of the SLC members based on their amino-acid sequences have also been suggested [3, 22]. The Transporter Classification (TC) system classifies transporters from all organisms, based on their sequence similarity and cellular functions (http://www.tcdb.org/) [3, 4]. The TC is a hierarchical classification consisting of five components, including “class” (eg, a carrier), “subclass” (eg, secondary-active) and “superfamily” (eg, the Major Facilitative Superfamily (MFS)). Due to the diversity of the human SLC members, they occur in a number of TC groups [4]. For example, the SLC6 and the SLC8 families are assigned to the Neurotransmitter:Sodium Symporter (NSS; 2.A.22) and the Ca2+:Cation Antiporter (CaCA; 2.A.19) superfamilies, respectively. Furthermore, a phylogenetic analysis of the SLC transporters using Hidden Markov Models (HMMs) refined the current classification, grouping 15 of the 52 SLC families into four distinct clusters [22].
Currently, the only human SLC transporter with an experimentally determined atomic structure is the Rhesus glycoprotein ammonium transporter (RhCG, SLC42A3) [23]. In addition, there are several high-resolution structures of proteins from prokaryotic and other eukaryotic organisms that are similar to the human SLC members, sharing at least 25% sequence identity with the human homolog [24-29]. These structures revealed that the human SLC transporters are likely to be highly diverse in structure (Fig. 1A). For example, the structure of the proton glutamate symporter Glp from Pyrococcus horikoshii (an SLC1 homolog) is dissimilar to the structure of the leucine transporter LeuT from Aquifex aeolicus (an SLC6 homolog). In addition, the structures of prokaryotic SLC homologs revealed that some SLC families share similar structural features despite weak sequence relationships (less than 15% sequence identity). For example, the prokaryotic homologs of the SLC5 (vSGLT) [24], SLC6 (LeuT) [25], SLC7 (Arginine/agmatine antiporter (AdiC) [26, 27] and ApcT transporter [28]) families, as well as other prokaryotic transporters, including the uracil transporter UraA [29], the sodium-hydantoin transporter Mhp1 [30], the Na+/betaine symporter BetP [31], the glutamate/gamma-aminobutyrate antiporter [32], and the L-carnitine/gamma-butyrobetaine antiporter CaiT [33], are classified into the same structural family [34] that is often referred to as the LeuT-like or NSS fold.
Fig. 1. Structures of SLC transporters.
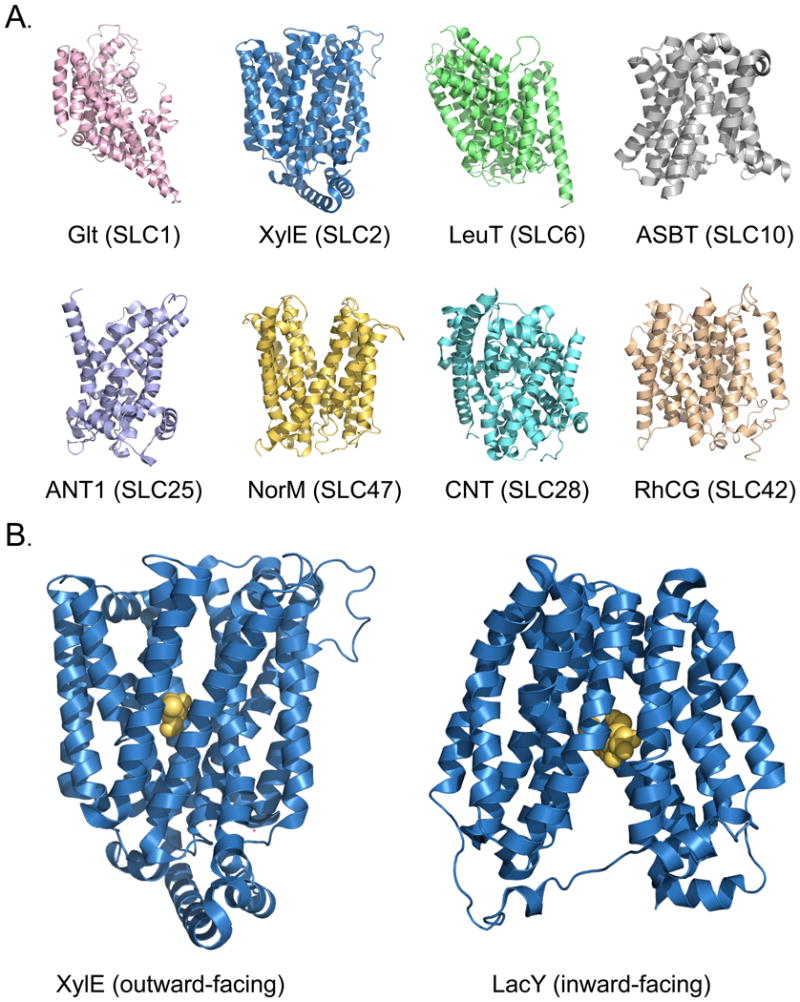
(A) Structures of SLC transporters representing select known structural classes and related human families. The structures are of the aspartate transporter Glt [153], the xylose transporter XylE [53], the leucine transporter LeuT [154], the sodium / bile acid symporter ASBT [155], the ADP/ATP translocase 1 ANT1 [156], the drug and toxin transporter NorM [142], the concentrative nucleoside transporter CNT [157], and the human rhesus glycoprotein RhCG ammonium transporter [23]. (B) The alternating access mechanism for structures with the MFS fold. The structures are of XylE in a ligand-bound outward-facing conformation (left) and the lactose permease LacY in a ligand-bound inward-facing conformation [54] (right). All structures are visualized with PyMol [158].
The majority of the SLC structures contain evolutionary-related inverted structural repeats that are highly divergent in sequence [3, 6, 35-37]. For example, the LeuT fold repeats typically include five transmembrane helices that are related to each other via a two-fold pseudo-symmetry axis parallel to the membrane plane; the first helix of each repeat is often involved in ligand binding [6, 35]. Although the intricate details of the transport mechanisms differ among the SLC members of the different folds (or even within the same fold), their structures support a common ‘alternating access’ transport mechanism, in which the transporter alternately exposes its primary binding site at either side of the membrane [5, 7, 25, 35, 36, 38-40] (Fig. 1B). For example, LeuT structures have been determined in four conformations representing distinct snapshots of a putative transport cycle [40-42]. Recent LeuT structures in complex with various amino acid substrates and inhibitors suggested a competitive inhibition mechanism in which larger and more hydrophobic ligands, such as tryptophan, stabilize an inactive outward-facing conformation [42]. An additional putative second substrate-binding site (‘S2’) has also been shown to be located on the LeuT surface [43-45], although its affinity for substrates remains uncertain [41]. However, a number of inhibitors were shown to stabilize different conformations of LeuT via sites overlapping with the S2 site [41, 46].
Furthermore, various conformations of SLC transporters that are adopted during the transport cycle have been revealed using other experimental techniques, such as single molecule fluorescence resonance energy transfer (smFRET) [43-46] as well as crystallographic characterizations of various LeuT-like structures (eg, Mhp1) [30, 47, 48]. Recently, alternate states of various SLC transporters were modeled by relying on the internal symmetry of their structures in other states [25, 49] and Molecular Dynamics (MD) simulations [50]. The mechanisms of transport and inhibition, even for such an extensively studied model transporter as LeuT, are far from being fully described.
Comparison and clustering of SLC transporters
We recently performed a comprehensive analysis of SLC transporters that included topology prediction, profile-profile sequence alignment, sequence-based clustering to create similarity networks, and clustering analysis [51]. A node of a sequence similarity network corresponds to a protein sequence; edges connect those pairs of sequences that are related above a defined similarity threshold. A variety of different functional annotations, including substrate type, transport mode, organism conservation, and tissue specificity, were mapped on the similarity networks. The goal was to identify relationships among human SLC transporters and extend their classification.
SLC families of homologs with similar structures generally clustered together, despite exhibiting relatively weak sequence similarities that do not traditionally suffice for a reliable transfer of structural and functional annotation (∼10% sequence identity) [52]. For example, the SLC5, SLC6, and SLC7, which exhibit significant sequence similarity to prokaryotic proteins with LeuT-like fold, were interconnected. Our analysis identified additional families, such as the SLC32 (the vesicular inhibitory amino acid transporter family), SLC36 (the proton-coupled amino acid transporter family), and SLC38 (System A and System N sodium-coupled neutral amino acid transporter family) as putative LeuT-like fold members.
The recent structure of the D-xylose-proton symporter (an SLC2 homolog) [53] and the structure of the lactose permease LacY (an SLC37 homolog) [54], both of which adopt the major facilitator superfamily (MFS) fold, confirmed our prediction that the human SLC2 and SLC37 families are related, increasing our confidence in the analysis (Fig. 2). However, some structural relationships were not detected using the SLC similarity network, indicating that better sequence comparison methods are still needed for membrane proteins. For instance, PepTSO, a homolog of the human peptide transporter family (PEPT/SLC15), also adopts the MFS fold, but is not connected to any other family in our similarity network [55] (Fig. 2). The SLC15 family members appear to be more divergent from other previously characterized MFS transporters (eg, they have additional transmembrane helical regions and large insertions in the loops between the transmembrane helices) [55].
Fig. 2. Sequence similarity network of human SLC families predicted to have the MFS fold.
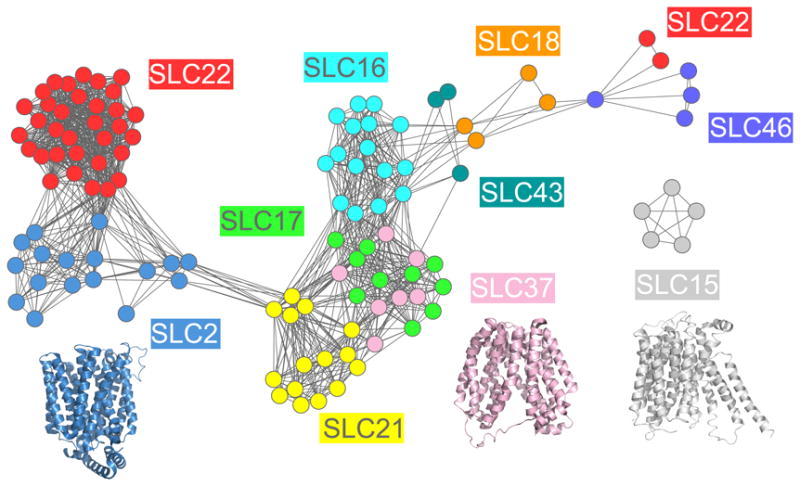
(A) The relationships between SLC sequences are visualized using Cytoscape 2.6.1 [51, 159]. (A) The nodes represent SLC sequences, including splice variants, that are similar to each other or to sequences of proteins with known MFS structures; the colors of the nodes indicate the SLC family [1]. The edges between the nodes correspond to a pairwise alignment with sequence identity of at least 10% and an E-value of less than 1 [51, 160]. The structures of XylE (blue) [53], LacY (pink) [54], and PEPTSO (grey) [55] are visualized using PyMol [158] and their colors are based on the color of the most similar human family in the similarity network.
The similarity network also shows that some families with chemically similar substrates cluster together in the sequence space. For instance, the organic ion transporter families SLC22 and SLC21, both of which are important for drug ADME, are grouped together, suggesting that the SLC similarity network can guide functional annotation of uncharacterized SLC members based on their characterized aligned homologs. Particularly, the SLC22 family was also highly inter-connected to the glucose transporter family (GLUTs/SLC2), which includes members that are associated with cancer and diabetes (Fig. 2). The predicted functional overlap between the SLC2 and SLC22 families was confirmed experimentally by kinetic measurements of representative members of these families (eg, the fructose transporter SLC2A9 and the urate transporter SLC22A12), which were found to have a common substrate (ie, uric acid) [51]. In summary, the comprehensive sequence-based comparison was shown to capture many functional and structural relationships among SLC transporters. It thus served as a guide for structural modeling of selected SLC members, as described in the next section.
Comparative modeling of SLC transporter structures
To construct a comparative model of a human SLC transporter (target), most similar known structures (templates) need to be first identified and aligned with the target sequence. Because SLC families (eg, the SLC5, SLC6, and SLC7) can be only distantly related to proteins of known structure, state-of-the-art fold assignment and alignment techniques need to be applied judiciously. The majority of the SLC families share a similar topology (10-14 transmembrane helices). Thus, a key step toward aligning their sequences involves identifying these transmembrane helical segments, which is fortunately relatively accurate [56-59]; for example, a support vector machine program MEMSAT-SVM achieves ∼90% accuracy on a testing set of experimentally determined membrane protein structures [60].
Efforts to model membrane transporters have also relied on profile-profile alignment (eg, HHpred [61]) and fold recognition methods that rely on structural information (eg, Promals3D [62]), as well as combinations of these methods and other techniques [63, 64]. For example, the alignment program AlignMe is based on a dynamic programming algorithm that takes as input the hydropathy plot of a protein in addition to its sequence, thereby improving its alignment accuracy [37]. AlignMe was shown to be accurate in identifying proteins of the LeuT fold and also captured sequence similarities among the LeuT inverted structural repeats that were not identified by other means. In addition, sequence similarity networks have been shown to be a useful tool in functional annotation and structural modeling of various protein families, including GPCRs, kinases, and enzymes families [60, 65, 66].
Once an alignment between the target sequence and one or more known structures is obtained, comparative model building can be performed [67-72]. For example, the program MODELLER computes an atomic model based on satisfaction of spatial restraints that are derived from the target-template alignment, atomic statistical potentials, and molecular mechanics [73]. Finally, comparative models can be assessed using a variety of computational criteria [69, 74-78].
Structure-based discovery of SLC transporter ligands
In structure-based ligand discovery, a large virtual library of small organic molecules is computationally screened against an atomic structure of a protein target (‘receptor’) [79, 80]. For a protein without an experimentally determined structure, virtual screening can be performed against a comparative protein structure model [81]. The small molecules are docked and scored based on their complementarity to the receptor's binding site. For example, in the program DOCK, the score is a sum of van der Waals, Poisson–Boltzmann electrostatic, and ligand desolvation penalty terms [82, 83]. The top-scoring molecules are then often analyzed manually and selected for experimental testing. Although binding affinities of ligands cannot yet be predicted accurately, the ability of docking programs to prioritize molecules from large databases, such that one needs to consider only the top few hundred molecules instead of the whole library, is its key strength [80, 84].
The accuracy of virtual screening can be estimated by the enrichment for known ligands relative to decoy compounds, calculated by docking against the tested model (‘enrichment calculations’) [85-87]. Thus, by selecting models based on their enrichment scores, comparative models can be optimized for their utility in predicting additional ligands [88-91]. Recent studies have successfully combined modeling and docking approaches with experimental validation of predicted ligands for proteins representing a wide range of functions, including GPCRs [89, 90, 92, 93], nuclear hormone receptors [94], and various enzymes [95-97].
Because of limited high-resolution structural information, most previous discovery efforts targeting SLC transporters have used ligand-based methodologies (eg, pharmacophore modeling) [98-101]. However, a recent increase in the number of atomic structures of several prokaryotic and eukaryotic homologs expanded our ability to apply structure-based ligand discovery to the human SLC members. Nevertheless, constructing comparative models that are sufficiently accurate for virtual screening remains challenging, for several reasons: First, it can be difficult, and sometimes even impossible, to align the sequences of the human SLC proteins to their prokaryotic homologs of known structure, because they can be highly divergent in sequence (sequence identity lower than 30%) [51]. Second, modeling and docking programs have not been optimized specifically for membrane proteins, as opposed to globular proteins [57, 102]. Third, the quality of the available membrane protein structures is, on average, lower than those for globular proteins. Fourth, comparative modeling and ligand docking against highly dynamic transporters, using the X-ray structure of a single and often unbound conformation, is challenging and sometimes inaccurate [86].
Next, we discuss five case studies in which computational modeling, docking, and experimental follow-up were used to describe specificity determinants of select SLC transporters.
Case study 1: the norepinephrine transporter (NET, SLC6A2)
NET transports norepinephrine from the synapse into presynaptic neurons, thereby regulating signaling pathways that are linked to behavioral traits and cardiovascular effects [103, 104]. NET is a known target for a variety of prescription drugs, including antidepressants and psychostimulants that inhibit its activity to increase neurotransmitter levels in the synapse.
NET is predicted to have one domain containing 12 transmembrane helices and is related to LeuT (sequence identity of ∼25%). The ligands transported by LeuT (amino acids) are chemically different from those of NET (positively charged monoamines), suggesting that the LeuT structure is not well suited for virtual screening for NET ligands and that the NET structure needs to be modeled. Thus, we constructed a model for NET based on the LeuT structure in an outward-occluded substrate-bound conformation [41], which likely represents an outward-occluded active conformation of LeuT [42], and computationally evaluated the model using enrichment calculations [91].
The model of NET's primary binding site includes the following four key features that might determine its ligand-binding specificity (Figs. 3A,B): First, several polar groups are predicted to make polar interactions with the ligands. For example, the sidechain of Asp75 makes ionic interactions with the amine group of norepinephrine. Second, four residues with aromatic groups are capable of forming interactions involving π electrons (Phe72, Tyr152, Phe317, and Phe323). For example, Phe72 is predicted to form π-cation interactions with norepineprhine via its benzyl sidechain as well as a hydrogen bond via the amide oxygen of its main chain. Third, several hydrophobic residues (eg, Ala145 and Val148) are capable of interacting with ligands via van der Waals interactions and the hydrophobic effect. Fourth, the binding site is relatively small, and, hence, the size of the ligands is limited. These four features are highly conserved among other SLC6 members that transport monoamines, including the dopamine transporter (DAT, SLC6A1) and the serotonin transporter (SERT, SLC6A4)). For example, the residues corresponding to NET's Asp75 in SERT (Asp98) [105] and DAT (Asp79) [106] were proposed to have similar key roles in ligand recognition [64, 107].
Fig. 3. Binding sites and modes of ligand binding in SLC6 members.
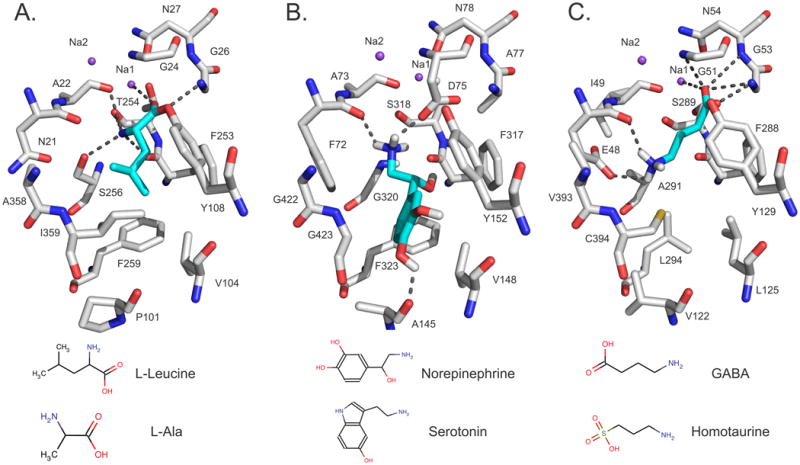
(A) The X-ray structure of LeuT as well as the comparative models of (B) NET and (C) GAT-2, in the ligand-bound occluded conformation, are visualized with PyMol [158]. Atoms are displayed as sticks, with oxygen, nitrogen, and hydrogen atoms in red, blue, and white, respectively. The sodium ions Na1 and Na2 are visualized with purple spheres. The ligands L-Leucine, norepinephrine, and GABA are illustrated in cyan sticks and their hydrogen bonds with the binding-site residues of LeuT (Ala22, Gly26, Thr254, Ser256, and Na1), GAT-2 (Glu48, Gly51, Gly53, Asn54, and Na1), and NET (Ala145, Phe72, and Asp75) are displayed as dotted gray lines. Sketches of two representative ligands of each transporter are shown at the bottom.
We computationally screened 6,436 drugs from the Kyoto Encyclopedia of Genes (KEGG DRUG) against the NET model. 18 top-scoring molecules were tested experimentally using a cis-inhibition assay in which the molecules are tested for their ability to inhibit transport of a known transporter substrate (radiolabeled or fluorescent) without distinguishing between inhibitors and substrates [87]. Ten drugs were found to be potent NET ligands; five of these were chemically novel ligands of NET (ie, Tanimoto coefficient of <0.5 against all known ligands, using the Daylight fingerprints [108]). These results may rationalize the efficacy of several sympathetic and antidepressant drugs, as well as side effects of diabetes and Alzheimer's drugs. For example, talsaclidine is a muscarinic M1 receptor agonist that was under development for the treatment of Alzheimer's disease, but failed in clinical trials due to various side effects, such as tachycardia, high blood pressure, nausea, diarrhea, excessive sweating, and palpitation [109]. It was proposed that the pharmacological effects of the drug are caused by its effect on the cholinergic system via binding to the M2 and M3 receptors as well as on the adrenergic system via an unknown mechanism [109]. The pharmacodynamic properties of talsaclidine, coupled with our uptake experiments, suggest that inhibition of the NET uptake might occur in clinically relevant concentrations [17, 87]. Thus, the positive and negative pharmacological effects of the drug can be partially rationalized by NET binding. Finally, these observations highlight the utility of virtual screening against a comparative model, even when the target and template share less than 30% sequence identity and have different ligand binding profiles in the primary binding site.
Case study 2: the GABA transporter 2 (GAT-2, SLC6A13)
GABA is a key inhibitory neurotransmitter in mammals that activates GABAergic receptors in inhibitory neurons (eg, GABAA) [110] and in peripheral tissues, such as the liver, kidney and lungs [110-112]. The GABA transporter (GAT) family consists of four transporters that regulate GABAergic signaling pathway [110, 112] and are targeted by various anticonvulsants and relaxants (eg, tiagabine). GAT-2 (SLC6A13) is primarily expressed in the liver, kidney and other peripheral tissues (eg, lungs [110-112]), and might also regulate ADME of GABAergic drugs. We modeled two different conformations of GAT-2, using the LeuT structures in occluded-outward-facing (‘occluded’) and outward-facing (‘outward’) conformations (Fig. 3C). The models were iteratively refined with molecular dynamics simulations and side-chain optimization, and were subsequently evaluated using enrichment calculations [90]. We validated the models experimentally, using site-directed mutagenesis, kinetic uptake measurements, and cis-inhibition assays.
The occluded and outward-facing GAT-2 models are similar to each other, differing only by minor backbone and side-chain rearrangements that make the putative binding site of the outward conformation slightly more accessible to a ligand. However, almost all predicted polar interactions between GABA and GAT-2 are conserved in both the occluded and the outward-facing models (eg, the carboxy group of GABA interacts with the sodium ion Na1, Gly51, and Gly53) (Fig. 3C). Furthermore, the amino group of GABA forms a key hydrogen bond with the main chain oxygen of Glu48 as well as a polar interaction with the negatively charged side-chain of the same residue. During model refinement, the conformation of the Glu48 side-chain correlated with the enrichment scores for the models, suggesting its importance for ligand recognition (Fig 3C). Site-directed mutagenesis indeed confirmed this residue as important for the function, demonstrating the utility of combining model refinement and ligand docking to predict a key functional residue. Interestingly, Glu48 and almost all other putative binding site residues are highly conserved among three of the four GAT's (GAT-2, GAT-3, and BGT1) [113], suggesting a common ligand recognition mechanism for these transporters. The GAT-1 binding site, however, is predicted to be unique and likely interacts with GABA via a different mechanism [38, 114].
A virtual screen of 594,166 drugs, metabolites, and fragment-like molecules in the ZINC database [115] against the two GAT-2 conformations retrieved two groups of chemically distinct ligands (Fig. 4). Particularly, molecules predicted to interact with GAT-2 using the outward-facing inhibitor-bound model (eg, baclofen) were larger and more hydrophobic than those predicted using the occluded substrate-bound model (eg, homotaurine), and covered a different area of the chemical space (Fig. 4). We tested 31 small organic molecules experimentally, using the cis-inhibition assay. Twelve ligands were validated, six of which were chemically novel (eg, homotaurine). The validated ligands suggest that GAT-2 is a high-selectivity/low-affinity transporter that is resistant to inhibition by typical GABAergic inhibitors. For example, even ligands that are within one heavy atom of GABA, such as GABOB, were found to inhibit GAT-2 significantly more weakly than GABA (Km of 26.2 µM for GABA versus IC50 of 402 µM for GABOB) [91]. This difference suggests that GAT-2 has a different binding profile from those of GAT-3 and BGT1 [110-112], despite their conserved primary binding sites. Because GAT-2 is highly abundant in the BBB, it is possible that it evolved to be highly selective to prevent toxins and other molecules from entering the CNS. Furthermore, the pharmacological effects of some of the discovered ligands can still be partially rationalized by GAT-2 binding. For example, the hemeprecursor 5-aminolevulinic acid (5-ALA) is involved in neurological side-effects (eg, porphyria) [116, 117] and is also used for the detection of CNS tumors [118-120]. Thus, it is plausible that GAT-2 transports 5-ALA across the BBB into the CNS.
Fig. 4. Structure-based ligand prediction for GAT-2.
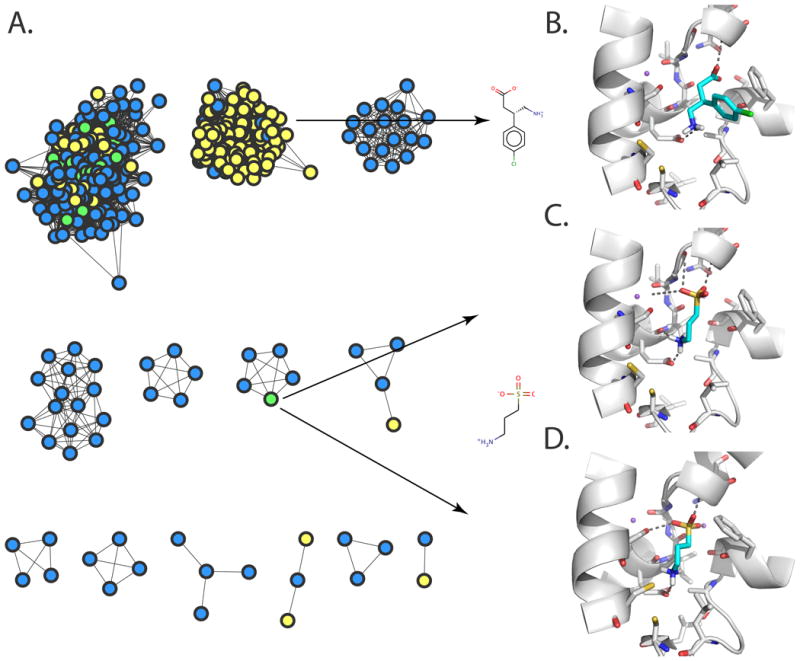
(A) A network view of predicted GAT-2 ligand drugs and their similarities [91]. The nodes represent top small molecule hits predicted to bind GAT-2, using the occluded model (blue), the outward-facing model (yellow), or both models (green). Predicted structures of GAT-2 in the outward-facing (B and C) and occluded (D) conformations, in complex with the representative experimentally confirmed hits baclofen (B) and homotaurine (C and D). Small molecules ligands are colored in cyan, with oxygen, nitrogen, sulfur, and hydrogen atoms in red, blue, yellow, and white, respectively. The sodium ions Na1 and Na2 are visualized as purple spheres. The GAT-2 TMH regions are illustrated in white ribbons. Important residues for binding are depicted as sticks; predicted hydrogen bonds between ligands and GAT-2 are displayed as dotted gray lines.
Case study 3: the SLC6 family
The SLC6 transporter family of Na+- and Cl-- dependent transporters can be classified into four groups based on their amino acid sequences and functions – the monoamine transporters, GABA transporters, amino acid transporters, and “orphan” transporters that may also transport amino acids [10, 103]. Interestingly, the SLC6 members are closely related in sequence despite their variability in function. Comparisons between structural models of representative members of the groups within the SLC6 family in complex with their ligands suggest key specificity determinants within this family (Fig. 3). For example, GAT-2 and NET share sequence identity of ∼45%, but bind chemically different ligands in their putative primary binding sites (GABA-like molecules and aromatic monoamines for GAT-2 and NET, respectively). The following three differences in the predicted binding sites of NET and GAT-2 likely rationalize their differences in ligand-binding selectivity (Fig. 3B,C): First, the number of aromatic residues (two in GAT-2 and four in NET). Second, the number and location of the charged groups (Glu48 and Na1 in GAT-2, and Asp75 in NET). Third, the size and shape of the binding site (ie, Ala145, Val148, Gly422, and Gly423 in NET are replaced by the larger Val122, Leu125, Val393, and Cys394 in GAT-2). These proposed ligand-binding determinants are consistent with previous studies characterizing other monoamine SLC6 transporters [38, 64], SERT [105] and DAT [106] as well as other GATs [38, 64, 113], such as GAT-3 (mouse GAT-4) [113]. In the future, characterization of SLC6 members in additional conformations and at higher resolution will enable us to further address functional variability within the SLC6 family. For example, why do GAT-2 and GAT-3 exhibit substantial differences in binding affinity to ligands, despite almost identical binding sites?
Case study 4: the large-neutral amino acid transporter (LAT-1, SLC7A5)
LAT-1 is a Na+-independent exchanger of large-neutral amino acids (eg, tyrosine), thyroid hormones (eg, triiodothyronine (T3)), and prescription drugs (eg, the anti-convulsant gabapentin) [121]. LAT-1 is found in the brain, testis, and placenta, and is highly abundant in the BBB where it is responsible for the transport of key metabolites and drugs into the CNS [122-124]. LAT-1 is also highly upregulated in a variety of cancerous tumors, such as non-small cell lung cancer and glioblastoma multiforme (GBM) [125, 126], for which it is thought to provide essential amino acids that are used as nutrients and signaling molecules for proliferation [127, 128]. LAT-1 is inactive in isolation and becomes active upon binding to the single transmembrane helix glycoprotein SLC3A2 [129]. The putative transmembrane domain (12 predicted transmembrane helices) of LAT-1 is similar in sequence to prokaryotic transporters of the acid/polyamine/organo-cation transporter (APC) family that adopt a LeuT-like fold [34, 130]. Therefore, LAT-1 was modeled based on the structures of the outward-occluded arginine-bound arginine/agmatine transporter AdiC from E. coli [48] and the inward-apo conformation of the amino acid, polyamine, and organo-cation transporter ApcT from Methanococcus jannaschii [28, 130].
The model of the LAT-1 complex with its natural ligands provides structural hypotheses for the similarities and differences in ligand binding profiles among the amino acid transporters, such as LAT-1 and AdiC. In particular, backbone atoms in residues interacting with the carboxylate and amine groups of the amino acid ligands are highly conserved between the LAT-1 model and the AdiC template structure (eg, Thr62, Ile63, and Ile64 in LAT-1 correspond to Ala22, Ile23, and Met24 in AdiC). Conversely, two key differences in their binding sites can explain why their ligands are amino acids with different physicochemical properties (large and hydrophobic amino acids for LAT-1 versus small and polar amino acids for AdiC). First, larger residues in the AdiC binding site are replaced with smaller residues in LAT-1, making the LAT-1 binding site much larger (eg, Val148, Gly255, and Ser342 in LAT-1 correspond to Met104, Ile205, and Trp293 in AdiC). Second, polar residues in AdiC correspond to residues with aromatic and hydrophobic side-chains in LAT-1 (eg, Thr361 in AdiC corresponds to Trp405 in LAT-1).
We identified four small-molecule ligands for LAT-1 (ie, 3,5-diiodo-L-tyrosine, 3-iodo-L-tyrosine, fenclonine, and acivicin) by virtual screening of 19,166 endogenous metabolites and prescription drugs from the KEGG database, followed by kinetic uptake experiments of the 12 top-scoring hits. Two of the four ligands (ie, the anti-cancer agent acivicin and the tryptophan hydroxylase inhibitor fenclonine) were also confirmed as substrates by a trans-stimulation assay; in this assay, cells overexpressing the transporter exchange the intracellular known radiolabeled substrate with the tested extracellular molecule, but only if the tested molecule is a substrate [130]. These results rationalize some of the positive and negative pharmacological effects of these ligands. For example, acivicin failed in clinical trials for cancer therapy due to CNS-related toxic side effects that resulted from active transport [131]. Our results suggest that such toxicities were facilitated by LAT-1 active transport of acivicin through the BBB.
A potential drug that is a ligand of a cancer-upregulated exchange or import transporter can act via one or both of the following mechanisms. First, a substrate can “hijack” the transporter to deliver a drug against a target in the cell (eg, a metabolic enzyme) (‘mechanism 1’). Second, a transporter inhibitor can selectively block transport activity, thus denying the cancer cell key nutrients (eg, the glucose transporter (GLUT1) and LAT-1) (‘mechanism 2’). Both of the newly discovered LAT-1 ligands, the inhibitor 3-iodo-L-tyrosine and substrate acivicin, are capable of inhibiting cancer cell proliferation in a GBM cell line, which expresses LAT-1 at high levels. This finding suggests that the two newly discovered LAT-1 ligands inhibited the GBM cell line proliferation via two distinct mechanisms, including nutrient deprivation by 3-iodo-L-tyrosine (mechanism 1) and cytotoxicity by acivicin (mechanism 2). Future structure-based ligand discovery studies will be applied simultaneously to LAT-1 and other proteins that might function together in cancer (eg GLUT1) or BBB permeability (P-gp). For instance, a comparative model of another key BBB transporter, the ATP-dependent efflux transporter P-glycoprotein (P-gp), was constructed based on the structure of its mouse homolog and then used, via molecular docking, to predict drugs that may get exported from the BBB [132]. Analyzing the commonalities and differences in the substrate specificities of LAT-1 and P-gp, including the identification of correlations in ligand properties such as size, polarity, charge, hydrogen-bonding potential, and other features, may facilitate rational design of drugs with optimal BBB permeability.
Case study 5: the multidrug and toxin extrusion transporter (MATE, SLC47) family
The multidrug and toxin extrusion transporter (MATE) family of proteins consists of two members, MATE1 and MATE2 [12, 133]. MATE proteins contribute to the excretion of diverse organic cations through the membranes of various cell types by exchanging their substrates with an oppositely directed proton gradient [133, 134]. In the human kidney, MATE1 and MATE2-K (a splice variant of MATE2) are localized to the apical membrane of the proximal and distal convoluted tubule [133]. The MATEs have ligands similar to those of key SLC22 transporters (OCT1-3), and together these transporters contribute to the tissue distribution and excretion of many drugs (eg, metformin) (Fig. 5A). Several genetic variants of MATE1 have been associated with differential response to metformin among diabetic patients [135, 136].
Fig. 5. Ligand discovery for MATE-1.
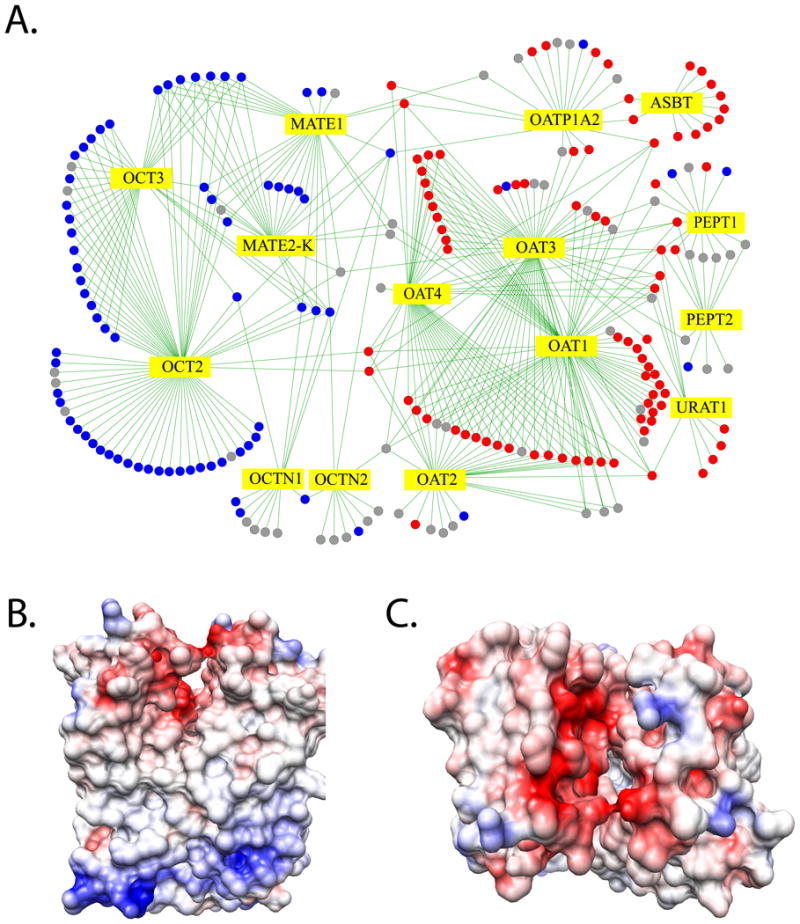
(A) An interaction network for prescription drugs and SLC transporters in kidney [140]. SLC transporters are depicted as yellow diamonds. Positively charged drugs are shown as solid blue circles, negatively charged drugs as solid red circles, and uncharged drugs as gray circles. Edges between transporters and drugs correspond to known transporter-drug interactions. (B) A preliminary human MATE-1 comparative model based on the structure of its prokaryotic homolog NorM. The model is visualized using the Coulombic Surface Coloring in UCSF Chimera [161]. Negative and positive electrostatic potentials are illustrated in red and blue, respectively.
Different methodologies, including quantitative structure-activity relationship (QSAR) modeling, have been previously employed to find common and selective substrates and inhibitors of MATE and OCT proteins [98, 137-139]. We recently identified 84 MATE1 ligands using iterative ligand-based computational modeling and experimental testing by the cis-inhibition experiments. We first performed an experimental high-throughput screen (HTS) for 910 compounds from 124 therapeutic classes, including antidepressants, antibacterials, antivirals, and antihypertensives [140]. This experimental data was then used to develop a computational QSAR model of ligand binding, using the Random Forest algorithm. The QSAR model performed significantly better in identifying true ligands than random selection (as judged by the area under the receiver operating curve (AUC) of 0.7, p-value < 0.0001). Next, selected positive and negative predictions were validated by low-throughput experiments. For example, eight inhibitors (eg, a histamine H2 receptor antagonist ranitidine) did not show inhibition in the original high-throughput screen, but were predicted as inhibitors by the QSAR model, and were later confirmed by low-throughput experiments. Thus, the QSAR model was used to save false negatives from the high-throughput screen. Likewise, several false positives in the high-throughput screen, such as the painkiller phenacetin and the vitamin niacin, were also corrected using the computational model and additional focused testing. We then refined the QSAR model based on the additional compounds identified by the low-throughput experiments, significantly improving its performance (ie, AUC=0.78). Next, the refined QSAR model was used to screen 6,122 compounds from the DrugBank library [141]. Finally, five of the eight chemically novel predicted ligands that were selected for experimental testing were confirmed experimentally as MATE1 inhibitors, including maraviroc, an antiretroviral drug in the CCR5 receptor antagonist class that is used in the treatment of HIV-inhibited MATE1 (IC50 of 17.3 µM).
With the aid of the QSAR model, we also identified several features that are important for MATE1 inhibition. MATE1 inhibitors are bulky as evidenced by a higher molecular weight, a higher number of bonds, and a higher number of rings among MATE1 inhibitors versus non-inhibitors. We found that lipophilicity is the key determinant of the MATE1 ligands. Interestingly, while charge is clearly important for the MATE1 ligands, about half of the inhibitors are acids or uncharged at the physiological pH.
The MATEs are predicted to have 13 transmembrane helices and are related to the transporter NorM from Vibrio cholerae, whose structure was determined at atomic resolution and exhibits a 12-TMH topology [142]. A recent comparative model of the rabbit MATE1 based on the NorM structure, followed by experimental validation, confirmed that a ‘core’ domain containing the N-terminal 12 transmembrane helices is responsible for transport and suggested that TMH 13 likely influences transporter turnover [143, 144]. We constructed a comparative model of MATE-1 that consists of a large pore with negative electrostatic potential and hydrophobic patches. This preliminary comparative model rationalizes the results obtained with our ligand-based QSAR models. For example, the negative electrostatic potential on the surface of MATE1 pore complements the cationic nature of the MATE1 ligands (Fig. 5B).
A model of MATE-2K was recently constructed based on the NorM structure and used to rationalize the effects of four non-synonymous protein coding single nucleotide polymorphisms (nsSNPs), including Lys64Asn, Pro162Leu, Gly211Val, and Gly393Arg [145, 146]. Lys64 is predicted to be in an extracellular loop near the membrane surface; thus, mutation of the positively charged lysine to an uncharged asparagine residue might have an effect on the interaction of MATE-2K with the membrane phospholipid head groups. Pro162 and Gly211 are located in transmembrane helices 4 and 6, respectively; therefore, mutations of these residues to leucine and valine, respectively, might reduce the main chain rigidity (Pro) or flexibility (Gly) as well as disrupt helix–helix packing. Finally, Gly393 is predicted to be located in the channel close to the putative proton-binding site. Mutation of Gly393 to a positively charged residue might, therefore, perturb the interaction of the transporter with the cationic ligands and protons.
Conclusions and future outlook
In this review, we first outlined the cellular and pharmacological functions of the human SLC transporters, as well as their sequences, structures and mechanisms of transport (Fig. 1). We then described various sequence- and structure-based tools that can be used, in combination with experiment, to characterize interactions between the SLC transporters and their small-molecule ligands (Fig. 2). We illustrated the utility of computational strategies, including structure-based (NET, GAT-2, and LAT-1) and ligand-based (MATE-1) modeling, to predict ligands for biomedically important SLC transporters (Figs. 3-5). Finally, we demonstrated how a comparative model can be used to rationalize the effect of a nsSNP on pharmacogenetics of the antidiabetic drug metformin, a substrate for the kidney and liver drug transporter MATE-2K. We now present our outlook on future directions in the field of structure-based ligand discovery for human SLC transporters.
Virtual screening against comparative models of different transporter conformations was found useful for identifying chemically novel transporter ligands (Fig. 3) [91]. To identify additional unexplored classes of molecules targeting SLC transporters, virtual screening should be performed against structures representing additional states in the conformational cycle of the transporter, characterized experimentally or computationally. Applications of structural symmetry [6] and MD simulations [50] have been helpful to model experimentally undefined conformations. However, because SLC transporters are exceptionally diverse in their structures and mechanisms (including transport of ligands on widely different time scales), new approaches to characterizing their dynamics with sufficient accuracy for structure-based ligand discovery are still needed.
The applicability of structure-based ligand discovery tools to human SLC transporters has been steadily increasing, for two reasons. First, recent experimental determinations of high-resolution transporter structures have been instrumental in extending the structural coverage of human SLC transporters via comparative modeling. The number of membrane transporter structures representing uncharacterized conformations and unexplored families is expected to grow considerably in the next few years, as a result of both traditional structural biology and structural genomics efforts [147-149]. Second, the accuracy and automation of computational tools for structure-based ligand discovery is improving in general [86, 87, 90, 91, 150, 151].
It is now appreciated that SLC transporters function in a coordinated manner with each other as well as other proteins, including receptors and enzymes, in various major biological and pharmacological processes. For example, several SLC members directly mediate drug-drug interactions in the liver, kidney, and BBB, which can affect clinical outcome [17, 152]. This led the FDA to strongly recommend that drug candidates should be tested for drug interactions with seven drug transporters, five of which are SLC members (eg, OAT1) [17]; this number is expected to grow [152]. Furthermore, SLC transporters such as LAT-1 and GLUT1 can function cooperatively in reprogrammed cancer metabolic pathways, by providing nutrients and signaling molecules to the transforming cancer cells; they may thus be targets for cancer therapy [127, 128].
The increasing applicability of structure-based ligand discovery enables us to characterize interactions between small molecules and multiple protein targets that make up a ‘system’. Such systems can be a biological pathway associated with cancer (eg, the mTOR pathway) or an organ important for drug clearance (eg, kidney). Therefore, future discovery efforts should aim at describing interactions of small molecules with multiple proteins as well as appropriately integrating these interactions with additional ‘omics’ data, such as protein-protein interactions and gene expression data, to better describe these systems and to ultimately relate them to clinical observations.
Acknowledgments
We are grateful to Robert Stroud, Andrew Waight, Bjørn Pedersen, Matthias Wittwer, Ethan Geier, Sook Wah Yee, Kari Morrison, Ligong Chen, and Arik Zur (all UCSF), as well as Ron Kaback (UCLA), Greg Madej (UCLA), Christoph Grewer (Bingahmton University), Amber Dahlin (Brigham and Women's Hospital), Lucy Forrest (Max Planck Institute for Biophysics), David Smith (University of Michigan), and Seok-Yong Lee (Duke) for discussions about SLC transporters, and Matt Jacobson (UCSF) for comments on the manuscript. The project was supported by grants from National Institutes of Health (R01 GM54762, U54 GM074929, P01 GM71790 to ASali, U01 GM61390 to ASali and KMG). This review is based in part on our papers [51, 87, 91, 130, 140, 146].
References
- 1.Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pflugers Arch. 2004;447(5):465–468. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- 2.Povey S, Lovering R, Bruford E, Wright M, Lush M, Wain H. The HUGO Gene Nomenclature Committee (HGNC) Hum Genet. 2001;109(6):678–680. doi: 10.1007/s00439-001-0615-0. [DOI] [PubMed] [Google Scholar]
- 3.Saier MH., Jr A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol Mol Biol Rev. 2000;64(2):354–411. doi: 10.1128/mmbr.64.2.354-411.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saier MH, Jr, Yen MR, Noto K, Tamang DG, Elkan C. The Transporter Classification Database: recent advances. Nucleic Acids Res. 2009;37(Database issue):D274–278. doi: 10.1093/nar/gkn862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forrest LR, Rudnick G. The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology (Bethesda) 2009;24:377–386. doi: 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forrest LR, Kramer R, Ziegler C. The structural basis of secondary active transport mechanisms. Biochim Biophys Acta. 2011;1807(2):167–188. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 7.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211(5052):969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 8.Gether U, Andersen PH, Larsson OM, Schousboe A. Neurotransmitter transporters: molecular function of important drug targets. Trends Pharmacol Sci. 2006;27(7):375–383. doi: 10.1016/j.tips.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Blakely RD, DeFelice LJ. All aglow about presynaptic receptor regulation of neurotransmitter transporters. Mol Pharmacol. 2007;71(5):1206–1208. doi: 10.1124/mol.107.035493. [DOI] [PubMed] [Google Scholar]
- 10.Chen NH, Reith ME, Quick MW. Synaptic uptake and beyond: the sodium- and chloride-dependent neurotransmitter transporter family SLC6. Pflugers Arch. 2004;447(5):519–531. doi: 10.1007/s00424-003-1064-5. [DOI] [PubMed] [Google Scholar]
- 11.You G, Morris ME, Wang B. Drug Transporters: Molecular Characterization and Role in Drug Disposition. Wiley; Hoboken: 2007. [Google Scholar]
- 12.Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci U S A. 2005;102(50):17923–17928. doi: 10.1073/pnas.0506483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyer zu Schwabedissen HE, Verstuyft C, Kroemer HK, Becquemont L, Kim RB. Human multidrug and toxin extrusion 1 (MATE1/SLC47A1) transporter: functional characterization, interaction with OCT2 (SLC22A2), and single nucleotide polymorphisms. American journal of physiology Renal physiology. 2010;298(4):F997–F1005. doi: 10.1152/ajprenal.00431.2009. [DOI] [PubMed] [Google Scholar]
- 14.Leabman MK, Huang CC, DeYoung J, Carlson EJ, Taylor TR, de la Cruz M, Johns SJ, Stryke D, Kawamoto M, Urban TJ, Kroetz DL, Ferrin TE, Clark AG, Risch N, Herskowitz I, Giacomini KM. Natural variation in human membrane transporter genes reveals evolutionary and functional constraints. Proc Natl Acad Sci U S A. 2003;100(10):5896–5901. doi: 10.1073/pnas.0730857100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koepsell H, Schmitt BM, Gorboulev V. Organic cation transporters. Rev Physiol Biochem Pharmacol. 2003;150:36–90. doi: 10.1007/s10254-003-0017-x. [DOI] [PubMed] [Google Scholar]
- 16.Cropp CD, Yee SW, Giacomini KM. Genetic variation in drug transporters in ethnic populations. Clin Pharmacol Ther. 2008;84(3):412–416. doi: 10.1038/clpt.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. Membrane transporters in drug development. Nature reviews Drug discovery. 2010;9(3):215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, Sheardown SA, Yue L, Burchard EG, Brett CM, Giacomini KM. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83(2):273–280. doi: 10.1038/sj.clpt.6100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, Brett CM, Giacomini KM. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117(5):1422–1431. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shu Y, Leabman MK, Feng B, Mangravite LM, Huang CC, Stryke D, Kawamoto M, Johns SJ, DeYoung J, Carlson E, Ferrin TE, Herskowitz I, Giacomini KM. Evolutionary conservation predicts function of variants of the human organic cation transporter, OCT1. Proc Natl Acad Sci U S A. 2003;100(10):5902–5907. doi: 10.1073/pnas.0730858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dresser MJ, Gray AT, Giacomini KM. Kinetic and selectivity differences between rodent, rabbit, and human organic cation transporters (OCT1) J Pharmacol Exp Ther. 2000;292(3):1146–1152. [PubMed] [Google Scholar]
- 22.Fredriksson R, Nordstrom KJ, Stephansson O, Hagglund MG, Schioth HB. The solute carrier (SLC) complement of the human genome: phylogenetic classification reveals four major families. FEBS Lett. 2008;582(27):3811–3816. doi: 10.1016/j.febslet.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 23.Gruswitz F, Chaudhary S, Ho JD, Schlessinger A, Pezeshki B, Ho CM, Sali A, Westhoff CM, Stroud RM. Function of human Rh based on structure of RhCG at 2.1 A. Proc Natl Acad Sci U S A. 2010;107(21):9638–9643. doi: 10.1073/pnas.1003587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karpowich NK, Wang DN. Structural biology. Symmetric transporters for asymmetric transport. Science. 2008;321(5890):781–782. doi: 10.1126/science.1161495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc Natl Acad Sci U S A. 2008;105(30):10338–10343. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao X, Lu F, Zhou L, Dang S, Sun L, Li X, Wang J, Shi Y. Structure and mechanism of an amino acid antiporter. Science. 2009;324(5934):1565–1568. doi: 10.1126/science.1173654. [DOI] [PubMed] [Google Scholar]
- 27.Fang Y, Jayaram H, Shane T, Kolmakova-Partensky L, Wu F, Williams C, Xiong Y, Miller C. Structure of a prokaryotic virtual proton pump at 3.2 A resolution. Nature. 2009;460(7258):1040–1043. doi: 10.1038/nature08201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. Structure and mechanism of a na+-independent amino Acid transporter. Science. 2009;325(5943):1010–1014. doi: 10.1126/science.1176088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu F, Li S, Jiang Y, Jiang J, Fan H, Lu G, Deng D, Dang S, Zhang X, Wang J, Yan N. Structure and mechanism of the uracil transporter UraA. Nature. 2011;472(7342):243–246. doi: 10.1038/nature09885. [DOI] [PubMed] [Google Scholar]
- 30.Weyand S, Shimamura T, Yajima S, Suzuki S, Mirza O, Krusong K, Carpenter EP, Rutherford NG, Hadden JM, O'Reilly J, Ma P, Saidijam M, Patching SG, Hope RJ, Norbertczak HT, Roach PC, Iwata S, Henderson PJ, Cameron AD. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science. 2008;322(5902):709–713. doi: 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ressl S, Terwisscha van Scheltinga AC, Vonrhein C, Ott V, Ziegler C. Molecular basis of transport and regulation in the Na(+)/betaine symporter BetP. Nature. 2009;458(7234):47–52. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- 32.Ma D, Lu P, Yan C, Fan C, Yin P, Wang J, Shi Y. Structure and mechanism of a glutamate-GABA antiporter. Nature. 2012;483(7391):632–636. doi: 10.1038/nature10917. [DOI] [PubMed] [Google Scholar]
- 33.Schulze S, Koster S, Geldmacher U, Terwisscha van Scheltinga AC, Kuhlbrandt W. Structural basis of Na(+)-independent and cooperative substrate/product antiport in CaiT. Nature. 2010;467(7312):233–236. doi: 10.1038/nature09310. [DOI] [PubMed] [Google Scholar]
- 34.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22(5):623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 35.Krishnamurthy H, Piscitelli CL, Gouaux E. Unlocking the molecular secrets of sodium-coupled transporters. Nature. 2009;459(7245):347–355. doi: 10.1038/nature08143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abramson J, Wright EM. Structure and function of Na(+)-symporters with inverted repeats. Curr Opin Struct Biol. 2009;19(4):425–432. doi: 10.1016/j.sbi.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khafizov K, Staritzbichler R, Stamm M, Forrest LR. A study of the evolution of inverted-topology repeats from LeuT-fold transporters using AlignMe. Biochemistry. 2010;49(50):10702–10713. doi: 10.1021/bi101256x. [DOI] [PubMed] [Google Scholar]
- 38.Kanner BI, Zomot E. Sodium-coupled neurotransmitter transporters. Chem Rev. 2008;108(5):1654–1668. doi: 10.1021/cr078246a. [DOI] [PubMed] [Google Scholar]
- 39.Guan L, Kaback HR. Lessons from lactose permease. Annu Rev Biophys Biomol Struct. 2006;35:67–91. doi: 10.1146/annurev.biophys.35.040405.102005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481(7382):469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nyola A, Karpowich NK, Zhen J, Marden J, Reith ME, Wang DN. Substrate and drug binding sites in LeuT. Curr Opin Struct Biol. 2010;20(4):415–422. doi: 10.1016/j.sbi.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322(5908):1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30(6):667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch JA. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature. 2010;465(7295):188–193. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, Javitch JA. Substrate-modulated gating dynamics in a Na(+)-coupled neurotransmitter transporter homologue. Nature. 2011 doi: 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Claxton DP, Quick M, Shi L, de Carvalho FD, Weinstein H, Javitch JA, McHaourab HS. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nat Struct Mol Biol. 2010;17(7):822–829. doi: 10.1038/nsmb.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimamura T, Weyand S, Beckstein O, Rutherford NG, Hadden JM, Sharples D, Sansom MS, Iwata S, Henderson PJ, Cameron AD. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science. 2010;328(5977):470–473. doi: 10.1126/science.1186303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao X, Zhou L, Jiao X, Lu F, Yan C, Zeng X, Wang J, Shi Y. Mechanism of substrate recognition and transport by an amino acid antiporter. Nature. 2010;463(7282):828–832. doi: 10.1038/nature08741. [DOI] [PubMed] [Google Scholar]
- 49.Radestock S, Forrest LR. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J Mol Biol. 2011;407(5):698–715. doi: 10.1016/j.jmb.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Faraldo-Gomez JD, Forrest LR. Modeling and simulation of ion-coupled and ATP-driven membrane proteins. Curr Opin Struct Biol. 2011;21(2):173–179. doi: 10.1016/j.sbi.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 51.Schlessinger A, Matsson P, Shima JE, Pieper U, Yee SW, Kelly L, Apeltsin L, Stroud RM, Ferrin TE, Giacomini KM, Sali A. Comparison of human solute carriers. Protein science : a publication of the Protein Society. 2010;19(3):412–428. doi: 10.1002/pro.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rost B, Liu J, Nair R, Wrzeszczynski KO, Ofran Y. Automatic prediction of protein function. Cellular and molecular life sciences : CMLS. 2003;60(12):2637–2650. doi: 10.1007/s00018-003-3114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature. 2012;490(7420):361–366. doi: 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- 54.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301(5633):610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 55.Newstead S, Drew D, Cameron AD, Postis VL, Xia X, Fowler PW, Ingram JC, Carpenter EP, Sansom MS, McPherson MJ, Baldwin SA, Iwata S. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. The EMBO journal. 2011;30(2):417–426. doi: 10.1038/emboj.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kernytsky A, Rost B. Static benchmarking of membrane helix predictions. Nucleic Acids Res. 2003;31(13):3642–3644. doi: 10.1093/nar/gkg532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elofsson A, von Heijne G. Membrane protein structure: prediction versus reality. Annu Rev Biochem. 2007;76:125–140. doi: 10.1146/annurev.biochem.76.052705.163539. [DOI] [PubMed] [Google Scholar]
- 58.Punta M, Forrest LR, Bigelow H, Kernytsky A, Liu J, Rost B. Membrane protein prediction methods. Methods. 2007;41(4):460–474. doi: 10.1016/j.ymeth.2006.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nugent T, Jones DT. Membrane protein structural bioinformatics. J Struct Biol. 2012;179(3):327–337. doi: 10.1016/j.jsb.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 60.Nugent T, Jones DT. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics. 2009;10:159. doi: 10.1186/1471-2105-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33(Web Server issue):W244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pei J, Kim BH, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36(7):2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Albers T, Marsiglia W, Thomas T, Gameiro A, Grewer C. Defining substrate and blocker activity of alanine-serine-cysteine transporter 2 (ASCT2) Ligands with Novel Serine Analogs. Mol Pharmacol. 2012;81(3):356–365. doi: 10.1124/mol.111.075648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharmacol. 2006;70(5):1630–1642. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- 65.Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One. 2009;4(2):e4345. doi: 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brown SD, Babbitt PC. Inference of functional properties from large-scale analysis of enzyme superfamilies. The Journal of biological chemistry. 2012;287(1):35–42. doi: 10.1074/jbc.R111.283408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 68.Petrey D, Honig B. Protein structure prediction: inroads to biology. Mol Cell. 2005;20(6):811–819. doi: 10.1016/j.molcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 69.Schmidt T, Haas J, Gallo Cassarino T, Schwede T. Assessment of ligand-binding residue predictions in CASP9. Proteins. 2011;79 Suppl 10:126–136. doi: 10.1002/prot.23174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Larsson P, Wallner B, Lindahl E, Elofsson A. Using multiple templates to improve quality of homology models in automated homology modeling. Protein science : a publication of the Protein Society. 2008;17(6):990–1002. doi: 10.1110/ps.073344908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ginalski K. Comparative modeling for protein structure prediction. Curr Opin Struct Biol. 2006;16(2):172–177. doi: 10.1016/j.sbi.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 72.Fiser A. Template-based protein structure modeling. Methods Mol Biol. 2010;673:73–94. doi: 10.1007/978-1-60761-842-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 74.Eramian D, Eswar N, Shen MY, Sali A. How well can the accuracy of comparative protein structure models be predicted? Protein Sci. 2008;17(11):1881–1893. doi: 10.1110/ps.036061.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eramian D, Shen MY, Devos D, Melo F, Sali A, Marti-Renom MA. A composite score for predicting errors in protein structure models. Protein Sci. 2006;15(7):1653–1666. doi: 10.1110/ps.062095806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schwede T, Sali A, Honig B, Levitt M, Berman HM, Jones D, Brenner SE, Burley SK, Das R, Dokholyan NV, Dunbrack RL, Jr, Fidelis K, Fiser A, Godzik A, Huang YJ, Humblet C, Jacobson MP, Joachimiak A, Krystek SR, Krystek SR, Jr, Kortemme T, Kryshtafovych A, Montelione GT, Moult J, Murray D, Sanchez R, Sosnick TR, Standley DM, Stouch T, Vajda S, Vasquez M, Westbrook JD, Wilson IA. Outcome of a workshop on applications of protein models in biomedical research. Structure. 2009;17(2):151–159. doi: 10.1016/j.str.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wallner B, Elofsson A. All are not equal: a benchmark of different homology modeling programs. Protein science : a publication of the Protein Society. 2005;14(5):1315–1327. doi: 10.1110/ps.041253405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fasnacht M, Zhu J, Honig B. Local quality assessment in homology models using statistical potentials and support vector machines. Protein science : a publication of the Protein Society. 2007;16(8):1557–1568. doi: 10.1110/ps.072856307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shoichet BK, McGovern SL, Wei B, Irwin JJ. Lead discovery using molecular docking. Curr Opin Chem Biol. 2002;6(4):439–446. doi: 10.1016/s1367-5931(02)00339-3. [DOI] [PubMed] [Google Scholar]
- 80.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432(7019):862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jacobson M, Sali A. Annual Reports in Medicinal Chemistry. Vol. 39. Academic Press; 2004. pp. 259–276. [Google Scholar]
- 82.Shoichet BK, Stroud RM, Santi DV, Kuntz ID, Perry KM. Structure-based discovery of inhibitors of thymidylate synthase. Science. 1993;259(5100):1445–1450. doi: 10.1126/science.8451640. [DOI] [PubMed] [Google Scholar]
- 83.Mysinger MM, Shoichet BK. Rapid context-dependent ligand desolvation in molecular docking. Journal of chemical information and modeling. 2010;50(9):1561–1573. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- 84.Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257(5073):1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 85.Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49(23):6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fan H, Irwin JJ, Webb BM, Klebe G, Shoichet BK, Sali A. Molecular docking screens using comparative models of proteins. J Chem Inf Model. 2009;49(11):2512–2527. doi: 10.1021/ci9003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schlessinger A, Geier E, Fan H, Irwin JJ, Shoichet BK, Giacomini KM, Sali A. Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc Natl Acad Sci U S A. 2011;108(38):15810–15815. doi: 10.1073/pnas.1106030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Evers A, Gohlke H, Klebe G. Ligand-supported homology modelling of protein binding-sites using knowledge-based potentials. J Mol Biol. 2003;334(2):327–345. doi: 10.1016/j.jmb.2003.09.032. [DOI] [PubMed] [Google Scholar]
- 89.Cavasotto CN, Orry AJ, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O'Neill KA, Hine H, Burton MS, Voigt JH, Abagyan RA, Bayne ML, Monsma FJ., Jr Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J Med Chem. 2008;51(3):581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 90.Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol. 2011;7(11):769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schlessinger A, Wittwer MB, Dahlin A, Khuri N, Bonomi M, Fan H, Giacomini KM, Sali A. High Selectivity of the gamma-Aminobutyric Acid Transporter 2 (GAT-2, SLC6A13) Revealed by Structure-based Approach. The Journal of biological chemistry. 2012;287(45):37745–37756. doi: 10.1074/jbc.M112.388157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol Sci. 2012;33(5):268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure. 2011;19(8):1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. J Med Chem. 2003;46(14):3045–3059. doi: 10.1021/jm0300173. [DOI] [PubMed] [Google Scholar]
- 95.Diller DJ, Li R. Kinases, homology models, and high throughput docking. J Med Chem. 2003;46(22):4638–4647. doi: 10.1021/jm020503a. [DOI] [PubMed] [Google Scholar]
- 96.Song L, Kalyanaraman C, Fedorov AA, Fedorov EV, Glasner ME, Brown S, Imker HJ, Babbitt PC, Almo SC, Jacobson MP, Gerlt JA. Prediction and assignment of function for a divergent N-succinyl amino acid racemase. Nat Chem Biol. 2007;3(8):486–491. doi: 10.1038/nchembio.2007.11. [DOI] [PubMed] [Google Scholar]
- 97.Kalyanaraman C, Imker HJ, Fedorov AA, Fedorov EV, Glasner ME, Babbitt PC, Almo SC, Gerlt JA, Jacobson MP. Discovery of a dipeptide epimerase enzymatic function guided by homology modeling and virtual screening. Structure. 2008;16(11):1668–1677. doi: 10.1016/j.str.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Astorga B, Ekins S, Morales M, Wright SH. Molecular determinants of ligand selectivity for the human multidrug and toxin extruder proteins MATE1 and MATE2-K. The Journal of pharmacology and experimental therapeutics. 2012;341(3):743–755. doi: 10.1124/jpet.112.191577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Andersen J, Kristensen AS, Bang-Andersen B, Stromgaard K. Recent advances in the understanding of the interaction of antidepressant drugs with serotonin and norepinephrine transporters. Chem Commun (Camb) 2009;(25):3677–3692. doi: 10.1039/b903035m. [DOI] [PubMed] [Google Scholar]
- 100.Ekins S, Ecker GF, Chiba P, Swaan PW. Future directions for drug transporter modelling. Xenobiotica; the fate of foreign compounds in biological systems. 2007;37(10-11):1152–1170. doi: 10.1080/00498250701646341. [DOI] [PubMed] [Google Scholar]
- 101.Chang C, Swaan PW. Computational approaches to modeling drug transporters. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2006;27(5):411–424. doi: 10.1016/j.ejps.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 102.Eyre TA, Partridge L, Thornton JM. Computational analysis of alpha-helical membrane protein structure: implications for the prediction of 3D structural models. Protein engineering, design & selection : PEDS. 2004;17(8):613–624. doi: 10.1093/protein/gzh072. [DOI] [PubMed] [Google Scholar]
- 103.Hahn MK, Blakely RD. The functional impact of SLC6 transporter genetic variation. Annu Rev Pharmacol Toxicol. 2007;47:401–441. doi: 10.1146/annurev.pharmtox.47.120505.105242. [DOI] [PubMed] [Google Scholar]
- 104.Pacholczyk T, Blakely RD, Amara SG. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature. 1991;350(6316):350–354. doi: 10.1038/350350a0. [DOI] [PubMed] [Google Scholar]
- 105.Celik L, Sinning S, Severinsen K, Hansen CG, Moller MS, Bols M, Wiborg O, Schiott B. Binding of serotonin to the human serotonin transporter. Molecular modeling and experimental validation. J Am Chem Soc. 2008;130(12):3853–3865. doi: 10.1021/ja076403h. [DOI] [PubMed] [Google Scholar]
- 106.Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11(7):780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Severinsen K, Kraft JF, Koldso H, Vinberg KA, Rothman RB, Partilla JS, Wiborg O, Blough B, Schiott B, Sinning S. Binding of the Amphetamine-like 1-Phenylpiperazine to Monoamine Transporters. ACS Chem Neurosci. 2012;3(9):693–705. doi: 10.1021/cn300040f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Daylight Chemical Information Systems, Inc. Laguna Niguel; CA: p. 92677. [Google Scholar]
- 109.Adamus WS, Leonard JP, Troger W. Phase I clinical trials with WAL 2014, a new muscarinic agonist for the treatment of Alzheimer's disease. Life Sci. 1995;56(11-12):883–890. doi: 10.1016/0024-3205(95)00024-z. [DOI] [PubMed] [Google Scholar]
- 110.Madsen KK, White HS, Schousboe A. Neuronal and non-neuronal GABA transporters as targets for antiepileptic drugs. Pharmacol Ther. 2010;125(3):394–401. doi: 10.1016/j.pharmthera.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 111.Christiansen B, Meinild AK, Jensen AA, Brauner-Osborne H. Cloning and characterization of a functional human gamma-aminobutyric acid (GABA) transporter, human GAT-2. The Journal of biological chemistry. 2007;282(27):19331–19341. doi: 10.1074/jbc.M702111200. [DOI] [PubMed] [Google Scholar]
- 112.Nakashita M, Sasaki K, Sakai N, Saito N. Effects of tricyclic and tetracyclic antidepressants on the three subtypes of GABA transporter. Neurosci Res. 1997;29(1):87–91. doi: 10.1016/s0168-0102(97)00072-2. [DOI] [PubMed] [Google Scholar]
- 113.Melamed N, Kanner BI. Transmembrane domains I and II of the gamma-aminobutyric acid transporter GAT-4 contain molecular determinants of substrate specificity. Mol Pharmacol. 2004;65(6):1452–1461. doi: 10.1124/mol.65.6.1452. [DOI] [PubMed] [Google Scholar]
- 114.Kanner BI. Transmembrane domain I of the gamma-aminobutyric acid transporter GAT-1 plays a crucial role in the transition between cation leak and transport modes. The Journal of biological chemistry. 2003;278(6):3705–3712. doi: 10.1074/jbc.M210525200. [DOI] [PubMed] [Google Scholar]
- 115.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Albers JW, Fink JK. Porphyric neuropathy. Muscle Nerve. 2004;30(4):410–422. doi: 10.1002/mus.20137. [DOI] [PubMed] [Google Scholar]
- 117.Lindberg RL, Martini R, Baumgartner M, Erne B, Borg J, Zielasek J, Ricker K, Steck A, Toyka KV, Meyer UA. Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria. The Journal of clinical investigation. 1999;103(8):1127–1134. doi: 10.1172/JCI5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Musiol R, Serda M, Polanski J. Prodrugs in photodynamic anticancer therapy. Curr Pharm Des. 2011;17(32):3548–3559. doi: 10.2174/138161211798194549. [DOI] [PubMed] [Google Scholar]
- 119.Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. The lancet oncology. 2006;7(5):392–401. doi: 10.1016/S1470-2045(06)70665-9. [DOI] [PubMed] [Google Scholar]
- 120.Fukuda H, Casas A, Batlle A. Aminolevulinic acid: from its unique biological function to its star role in photodynamic therapy. The international journal of biochemistry & cell biology. 2005;37(2):272–276. doi: 10.1016/j.biocel.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 121.Kanai Y, Segawa H, Miyamoto K, Uchino H, Takeda E, Endou H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98) The Journal of biological chemistry. 1998;273(37):23629–23632. doi: 10.1074/jbc.273.37.23629. [DOI] [PubMed] [Google Scholar]
- 122.Roberts LM, Black DS, Raman C, Woodford K, Zhou M, Haggerty JE, Yan AT, Cwirla SE, Grindstaff KK. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience. 2008;155(2):423–438. doi: 10.1016/j.neuroscience.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 123.Alexander GM, Schwartzman RJ, Grothusen JR, Gordon SW. Effect of plasma levels of large neutral amino acids and degree of parkinsonism on the blood-to-brain transport of levodopa in naive and MPTP parkinsonian monkeys. Neurology. 1994;44(8):1491–1499. doi: 10.1212/wnl.44.8.1491. [DOI] [PubMed] [Google Scholar]
- 124.Wang Y, Welty DF. The simultaneous estimation of the influx and efflux blood-brain barrier permeabilities of gabapentin using a microdialysis-pharmacokinetic approach. Pharm Res. 1996;13(3):398–403. doi: 10.1023/a:1016092525901. [DOI] [PubMed] [Google Scholar]
- 125.Kaira K, Oriuchi N, Imai H, Shimizu K, Yanagitani N, Sunaga N, Hisada T, Tanaka S, Ishizuka T, Kanai Y, Endou H, Nakajima T, Mori M. Prognostic significance of L-type amino acid transporter 1 expression in resectable stage I-III nonsmall cell lung cancer. Br J Cancer. 2008;98(4):742–748. doi: 10.1038/sj.bjc.6604235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kobayashi K, Ohnishi A, Promsuk J, Shimizu S, Kanai Y, Shiokawa Y, Nagane M. Enhanced tumor growth elicited by L-type amino acid transporter 1 in human malignant glioma cells. Neurosurgery. 2008;62(2):493–503. doi: 10.1227/01.neu.0000316018.51292.19. discussion 503-494. [DOI] [PubMed] [Google Scholar]
- 127.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13(6):472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 128.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136(3):521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Archiv : European journal of physiology. 2004;447(5):532–542. doi: 10.1007/s00424-003-1086-z. [DOI] [PubMed] [Google Scholar]
- 130.Geier EG, Schlessinger A, Fan H, Gable JE, Irwin JJ, Sali A, Giacomini KM. Structure-based ligand discovery for the Large-neutral Amino Acid Transporter 1, LAT-1. Proc Natl Acad Sci U S A. 2013;110(14):5480–5485. doi: 10.1073/pnas.1218165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hidalgo M, Rodriguez G, Kuhn JG, Brown T, Weiss G, MacGovren JP, Von Hoff DD, Rowinsky EK. A Phase I and pharmacological study of the glutamine antagonist acivicin with the amino acid solution aminosyn in patients with advanced solid malignancies. Clinical cancer research : an official journal of the American Association for Cancer Research. 1998;4(11):2763–2770. [PubMed] [Google Scholar]
- 132.Dolghih E, Bryant C, Renslo AR, Jacobson MP. Predicting binding to p-glycoprotein by flexible receptor docking. PLoS Comput Biol. 2011;7(6):e1002083. doi: 10.1371/journal.pcbi.1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, Ogawa O, Inui K. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. Journal of the American Society of Nephrology : JASN. 2006;17(8):2127–2135. doi: 10.1681/ASN.2006030205. [DOI] [PubMed] [Google Scholar]
- 134.Otsuka M, Yasuda M, Morita Y, Otsuka C, Tsuchiya T, Omote H, Moriyama Y. Identification of essential amino acid residues of the NorM Na+/multidrug antiporter in Vibrio parahaemolyticus. J Bacteriol. 2005;187(5):1552–1558. doi: 10.1128/JB.187.5.1552-1558.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. The pharmacogenomics journal. 2009;9(4):242–247. doi: 10.1038/tpj.2009.15. [DOI] [PubMed] [Google Scholar]
- 136.Jablonski KA, McAteer JB, de Bakker PI, Franks PW, Pollin TI, Hanson RL, Saxena R, Fowler S, Shuldiner AR, Knowler WC, Altshuler D, Florez JC. Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes. 2010;59(10):2672–2681. doi: 10.2337/db10-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011;(201):105–167. doi: 10.1007/978-3-642-14541-4_3. [DOI] [PubMed] [Google Scholar]
- 138.Damme K, Nies AT, Schaeffeler E, Schwab M. Mammalian MATE (SLC47A) transport proteins: impact on efflux of endogenous substrates and xenobiotics. Drug Metab Rev. 2011;43(4):499–523. doi: 10.3109/03602532.2011.602687. [DOI] [PubMed] [Google Scholar]
- 139.Kido Y, Matsson P, Giacomini KM. Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J Med Chem. 2011;54(13):4548–4558. doi: 10.1021/jm2001629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wittwer MB, Zur AA, Khuri N, Kido Y, Kosaka A, Zhang X, Morrissey KM, Sali A, Huang Y, Giacomini KM. Discovery of Potent, Selective Multidrug and Toxin Extrusion Transporter 1 (MATE1, SLC47A1) Inhibitors Through Prescription Drug Profiling and Computational Modeling. J Med Chem. 2013 doi: 10.1021/jm301302s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34(Database issue):D668–672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.He X, Szewczyk P, Karyakin A, Evin M, Hong WX, Zhang Q, Chang G. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature. 2010;467(7318):991–994. doi: 10.1038/nature09408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhang X, Wright SH. MATE1 has an external COOH terminus, consistent with a 13-helix topology. American journal of physiology Renal physiology. 2009;297(2):F263–271. doi: 10.1152/ajprenal.00123.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang X, He X, Baker J, Tama F, Chang G, Wright SH. Twelve transmembrane helices form the functional core of mammalian MATE1 (multidrug and toxin extruder 1) protein. The Journal of biological chemistry. 2012;287(33):27971–27982. doi: 10.1074/jbc.M112.386979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kajiwara M, Terada T, Ogasawara K, Iwano J, Katsura T, Fukatsu A, Doi T, Inui K. Identification of multidrug and toxin extrusion (MATE1 and MATE2-K) variants with complete loss of transport activity. J Hum Genet. 2009;54(1):40–46. doi: 10.1038/jhg.2008.1. [DOI] [PubMed] [Google Scholar]
- 146.Choi JH, Yee SW, Ramirez AH, Morrissey KM, Jang GH, Joski PJ, Mefford JA, Hesselson SE, Schlessinger A, Jenkins G, Castro RA, Johns SJ, Stryke D, Sali A, Ferrin TE, Witte JS, Kwok PY, Roden DM, Wilke RA, McCarty CA, Davis RL, Giacomini KM. A common 5′-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther. 2011;90(5):674–684. doi: 10.1038/clpt.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Stroud RM, Choe S, Holton J, Kaback HR, Kwiatkowski W, Minor DL, Riek R, Sali A, Stahlberg H, Harries W. 2007 Annual progress report synopsis of the Center for Structures of Membrane Proteins. J Struct Funct Genomics. 2009 doi: 10.1007/s10969-008-9058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Love J, Mancia F, Shapiro L, Punta M, Rost B, Girvin M, Wang DN, Zhou M, Hunt JF, Szyperski T, Gouaux E, MacKinnon R, McDermott A, Honig B, Inouye M, Montelione G, Hendrickson WA. The New York Consortium on Membrane Protein Structure (NYCOMPS): a high-throughput platform for structural genomics of integral membrane proteins. Journal of structural and functional genomics. 2010;11(3):191–199. doi: 10.1007/s10969-010-9094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pieper U, Schlessinger A, Kloppmann E, Chang GA, Chou JJ, Dumont ME, Fox BG, Fromme P, Hendrickson WA, Malkowski MG, Rees DC, Stokes DL, Stowell MH, Wiener MC, Rost B, Stroud RM, Stevens RC, Sali A. Coordinating the impact of structural genomics on the human alpha-helical transmembrane proteome. Nature structural & molecular biology. 2013;20(2):135–138. doi: 10.1038/nsmb.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, Heveker N, Shoichet BK, Volkman BF. Structure-based ligand discovery for the protein-protein interface of chemokine receptor CXCR4. Proc Natl Acad Sci U S A. 2012;109(14):5517–5522. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fan H, Schneidman-Duhovny D, Irwin JJ, Dong G, Shoichet BK, Sali A. Statistical potential for modeling and ranking of protein-ligand interactions. Journal of chemical information and modeling. 2011;51(12):3078–3092. doi: 10.1021/ci200377u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zamek-Gliszczynski MJ, Hoffmaster KA, Tweedie DJ, Giacomini KM, Hillgren KM. Highlights from the international transporter consortium second workshop. Clin Pharmacol Ther. 2012;92(5):553–556. doi: 10.1038/clpt.2012.126. [DOI] [PubMed] [Google Scholar]
- 153.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445(7126):387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 154.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437(7056):215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 155.Hu NJ, Iwata S, Cameron AD, Drew D. Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature. 2011;478(7369):408–411. doi: 10.1038/nature10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426(6962):39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 157.Johnson ZL, Cheong CG, Lee SY. Crystal structure of a concentrative nucleoside transporter from Vibrio cholerae at 2.4 A. Nature. 2012;483(7390):489–493. doi: 10.1038/nature10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.DeLano WL. The PyMOL Molecular Graphics System. San Carlos; CA, USA: 2002. [Google Scholar]
- 159.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Madhusudhan MS, Webb BM, Marti-Renom MA, Eswar N, Sali A. Alignment of multiple protein structures based on sequence and structure features. Protein Eng Des Sel. 2009;22(9):569–574. doi: 10.1093/protein/gzp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]