

Abstract
Recent advances in sequencing technologies provide the means for identifying copy number variation (CNV) at an unprecedented resolution. A single next-generation sequencing experiment offers several features that can be used to detect CNV, yet current methods do not incorporate all available signatures into a unified model. cnvHiTSeq is an integrative probabilistic method for CNV discovery and genotyping that jointly analyzes multiple features at the population level. By combining evidence from complementary sources, cnvHiTSeq achieves high genotyping accuracy and a substantial improvement in CNV detection sensitivity over existing methods, while maintaining a low false discovery rate. cnvHiTSeq is available at http://sourceforge.net/projects/cnvhitseq
Background
Next-generation sequencing (NGS) technologies are rapidly superseding microarrays as the leading platform for identifying and cataloging genomic variation. Unlike genotyping arrays, NGS can potentially assess all forms of variation at an unprecedented resolution. While high-coverage, whole-genome sequencing (WGS) remains prohibitive in large sample sets, low-coverage sequencing at the population level has been proposed as a more efficient alternative [1].
Copy number variation (CNV) is pervasive in the human genome, and has been estimated to contribute more to genetic diversity than single nucleotide polymorphisms [2]. Moreover, rare copy number variants (CNVs) have been shown to be highly penetrant for complex diseases such as obesity [3]. As a result, there has been an increased interest in developing algorithms to identify CNVs using low-coverage WGS [4], while accurate CNV genotyping from population sequence data has received less attention.
Most NGS-based CNV detection algorithms rely on mapping sequence reads back to a reference genome in search of discrepancies that may provide evidence for different types of variants. CNV signatures of all classes (deletions, insertions and duplications) can be obtained from the different features of a single NGS experiment, with varying degrees of sensitivity. Methods that focus on read depth (RD) are better suited for determining the absolute copy number [5] but often suffer from low breakpoint resolution and lower sensitivity for small variants (<1 kb). Methods that focus on the distance (span) and orientation of read pairs (RPs) are more sensitive to CNVs caused by retrotransposable elements but often fail to detect CNVs flanked by repetitive sequence [6]. Finally, approaches based on split reads (SRs) can achieve single-base-pair resolution but depend highly on the length of the reads and are less reliable in repetitive regions. Achieving a high sensitivity across the CNV spectrum, therefore, requires taking the strengths and weaknesses of each approach into account and incorporating information from multiple data sources. While there are a few methods that analyze data from any two of these sources, they use step-wise approaches to combine the results. Notably, Genome STRiP [7] considers discordant RPs as a starting point and RD as a downstream filter. Similarly, DELLY [8] analyzes discordant RPs first and then attempts to strengthen the results with supporting SRs.
Here we present cnvHiTSeq, an integrative approach to sequencing-based CNV detection and genotyping that jointly models all available NGS features at the population level. By organically combining evidence from RD, RPs and SRs, cnvHiTSeq provides sensitive and precise discovery of all CNV classes even from low-coverage sequence data. Furthermore, the probabilistic model employed by our method allows it to pool information across individual samples and reconcile copy number differences among data sources, thus achieving a high CNV genotyping accuracy.
Results
cnvHiTSeq integrates evidence from three distinct and largely complementary sequencing data sources: RD, RPs, and SRs (Figure 1). Each data source consists of a vector of one (or more) real values measured at a user-specified resolution across a chromosome. RD corresponds to the number of reads aligning to a specific genomic position and is proportional to the underlying copy number. In our model, RD is represented by a single measurement of the average normalized read count within a given window (Figure 1a,d). RP analysis utilizes discrepancies between the observed and the expected distance of mapped paired-end reads to infer the presence of CNVs. RP is incorporated into cnvHiTSeq using a pair of measurements, the first being the number of RPs that span a given position, and the second being the average insert size of these reads (Figure 1b,e). Finally, SR analysis involves detecting single reads that happen to encompass the breakpoints of a CNV and thus appear to be 'split' between two genomic locations when mapped to the reference. SR is summarized by the count of split reads that span a given genomic position (Figure 1c).
Figure 1.

Two characteristic CNVs on chromosome 8 and their corresponding paired-end sequencing signatures. Gray reads give rise to read depth measurements, represented as black circles. Blue reads correspond to discordant read pairs and contribute to read pair measurements denoted as blue squares. Split reads and their corresponding counts are represented in green. Hybrid green/blue reads provide evidence of split reads combined with abnormal read pair distance. (a) Read depth signature for well described homozygous deletion on chromosome 8. Reads are absent inside the deleted region. (b) Read pair signature for the same deletion as (a). The distance of the pairs spanning the deletion is significantly increased compared to the expected insert size. (c) Combination of read depth, read pair and split read signatures. Two split reads that span the breakpoints of the deletion were identified. The joint analysis of all the sources improves the breakpoint detection. (d) Read depth signature for multi-copy tandem duplication on chromosome 8. Significantly more reads are present inside the duplicated region. (e) Read pair signature for the same duplication as (d). The read pair distance appears significantly decreased compared to the expected insert size. (f) Combination of read depth and read pair signatures provides improved breakpoints. The split reads detected in the region are used to rescue their orphaned pair and are thus incorporated into the read pair signature.
cnvHiTSeq utilizes the population-haplotype framework of cnvHap [9] to incorporate the diverse data sources into a single probabilistic model. A hidden Markov model (HMM) is used to capture the spatial properties of CNV across a single chromosome copy (Figure 2). This is motivated by the hypothesis that the observed sequence data are generated by discrete unobserved states corresponding to the unknown copy numbers at each genomic position. cnvHiTSeq models the probability of every data point conditional on this hidden copy number using statistical distributions tailored to each data source (Figure 2c). The HMM can then be used to calculate the likelihood of different paths (corresponding to different CNV segmentations) through the model, and thus perform an integrated analysis of the underlying sources (Figure 2d,e). The spatial smoothing provided by the HMM allows cnvHiTSeq to detect events from low coverage data at a much finer resolution than sliding window methods without concomitant loss of power or increase in false positives due to small window noise. To benefit from large sample sizes and account for sample-independent variation in the data sources (for example, due to the local sequence composition), cnvHiTSeq uses the population distribution of data measurements at each position to update the parameters of the emission distribution during the model training. cnvHiTSeq also updates a transition rate parameter at each position in order to capture variation in CNV frequency across the genome.
Figure 2.
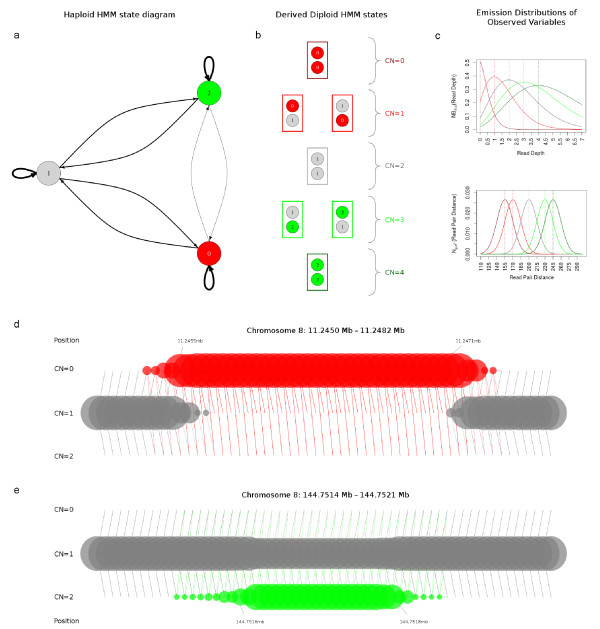
Schematic diagram of cnvHiTSeq model. (a) State diagram of the haploid HMM used by cnvHiTSeq. Vertices represent haploid copy number states (0, red; 1, gray; and 2, green) and edges represent transitions from one state to another. The thickness of the edges is proportional to the transition probabilities of the model. More haploid states can be used to model higher copy numbers. (b) By pairing two haploid copy number states cnvHiTSeq derives the final diploid model (0, dark red; 1, red; 2, gray; 3, green; 4, dark green). (c) Each observed variable is modeled using a customized emission distribution. Here, as an example, we present the emission distributions for read depth and read pair distance conditional on the copy number state. Read depth is modeled using the negative binomial distribution, while read pair distance is modeled using the normal distribution. The color scheme corresponds to the diploid copy number states presented in (b). (d) The trained haploid HMM that corresponds to the homozygous deleted region on Figure 1a-c. The rows represent copy number states and the columns represent genomic location. Positions are given in megabases (Mb). Bubble size corresponds to the probability of assignment to each state at each position. The lines between bubbles indicate possible transitions between states. The annotated positions indicate the start and the stop of the CNV call. (e) The trained haploid HMM that corresponds to the heterozygous duplicated region on Figure 1d-f.
False discovery rate estimation
We first evaluated the ability of cnvHiTSeq to detect CNV using data from the HapMap CEU trio. This trio was sequenced by the 1000 Genomes Project to high coverage (30×), but we also randomly downsampled the data to medium (10× and 20×) and low coverage (6×). We applied cnvHiTSeq separately to the child (sample NA12878) and the parents (samples NA12891 and NA12892) of the trio in order to approximate the false discovery rate (FDR) via the rate of Mendelian inconsistency in CNV prediction. This method does not utilize the population modeling capabilities of cnvHiTSeq, but provides a good genome-wide estimate of the FDR in the absence of a gold standard. Throughout our analyses we only focus on CNVs larger than 100 bp. At low coverage, cnvHiTSeq detected 2,910 deletions and 2,994 duplications in the child at a FDR of 4.3% and 9.8%, respectively. The FDR was calculated using a 1-bp overlap criterion and was found to decrease with increasing coverage (Figure 3a). When using a stricter 50% reciprocal overlap criterion between the child and the parents we report a FDR of 5.3% and 12.6% for deletions and duplications, respectively. Genome STRiP [7], which was the best competing method, achieved a 8.2% (50% reciprocal overlap) FDR for this sample when calculated in exactly the same way, although a lower FDR of 3.7% has been reported [4] based on experimental validation, demonstrating the conservative nature of our validation approach (Table S3 in Additional file 1). By adjusting the posterior probability threshold for calling CNVs we show that our method maintains a low FDR even when making twice as many calls as other methods [7,10,11] (Figure 3b).
Figure 3.
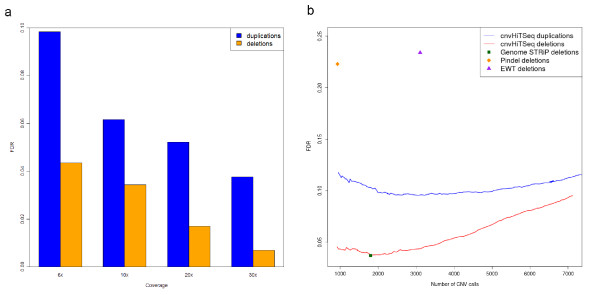
False discovery rate analysis of cnvHiTSeq. (a) FDR of our method for different depths of coverage. FDR determined using segregation analysis on the HapMap CEU trio. (b) FDR of cnvHiTSeq versus the number of detected CNVs. Our method maintains a low FDR even when calling twice as many deletions as competing methods.
Since estimation of FDR using Mendelian inconsistency may be prone to sequencing biases potentially affecting all three samples in the trio, we also applied a strict criterion of requiring true positive predictions to have been observed in only one of the parents. Thus, after excluding common CNVs using the Database of Genomic Variants (DGV) [12], we designated CNVs present in both or neither parent as false positive to obtain an FDR upper bound of 15.2%, which remained better than the equivalent for Genome STRiP (Table S4 in Additional file 1).
We also obtained an independent estimate of FDR using array-CGH data from the High Resolution CNV Discovery project [2]. We identified 166 deletions predicted by cnvHiTSeq for NA12878, which encompassed at least 4 CGH probes and were thus considered for validation. CGH analysis validated 155 of these 166 regions (FDR = 6.8%; Table S1 in Additional file 1), while 101 of the 166 deletions were also identified by Genome STRiP, confirming the higher sensitivity of our approach. The validated deletions range from 541 bp to 143,379 bp in length, with a median of 4,170 bp and cnvHiTSeq maintains a low FDR across CNV lengths, while being slightly more accurate for longer variants (Figure S3 in Additional file 1).
Sensitivity analysis
In order to estimate the sensitivity of our method, we applied cnvHiTSeq to the low coverage NA12156 sample and then compared the results to the gold standard dataset used by the 1000 Genome Structural Variant discovery study [4]. This gold standard comprises three heterogeneous CNV call sets for sample NA12156 that were obtained using different technologies and are therefore more sensitive to CNV events of different sizes, covering a wide range of the CNV spectrum. Smaller CNVs are examined using capillary read data (median = 0.2 kb) [13], medium-sized CNVs are examined using array-comparative genomic hybridization (array-CGH; median = 2 kb) [14] and larger variants using fosmid sequencing (median = 6 kb) [15]. Both our reported sensitivity results and those of competing methods are based on a 1-bp overlap criterion. Using low-coverage WGS data, we report an overall sensitivity of 80.1%, with cnvHiTSeq performing consistently better than competing methods on the same data [4] (Figure 4a; Table S5 in Additional file 1). Specifically, on the fosmid dataset cnvHiTSeq achieves a sensitivity of 88%, compared to 63% for Genome STRiP. For array-CGH we report a sensitivity of 86%, while the next best result of 70% is achieved by event-wise testing (EWT) [10]. cnvHiTSeq also outperforms other methods on the capillary read data, for which we report a sensitivity of 48% compared to Genome STRiP's 21%.
Figure 4.
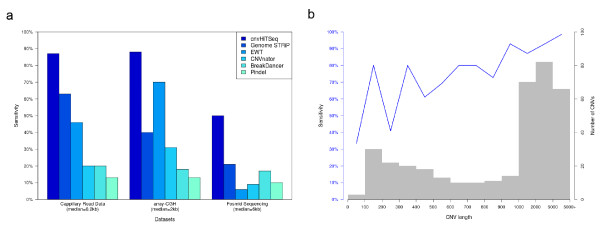
Sensitivity analysis of cnvHiTSeq. (a) Sensitivity of cnvHiTSeq for sample NA12156 compared to competing methods for three gold standard datasets. (b) Sensitivity of cnvHiTSeq for the CEU trio (downsampled to 6× coverage), based on data from the High-Resolution CNV Discovery project. The histogram represents the number of CNVs in the call set, and the blue line denotes the sensitivity.
To provide context for our FDR results we also estimated our method's sensitivity on the downsampled CEU trio. The previously used gold standard datasets contain CNV calls for the child of the trio (NA12878). For this sample we report an overall sensitivity of 78.4% and individual results consistent with those for sample NA12156 (Table S5 in Additional file 1). Furthermore, we created a CNV call set for the entire trio by analyzing the raw array-CGH intensity data from the High Resolution CNV Discovery project [2] using cnvHap. On this set, which contains both duplication and deletion events as small as 100 bp, cnvHiTSeq achieves an overall sensitivity of 87.8% (Figure 4b).
Genotyping accuracy
In order to explore the extra benefits available from population level modeling of NGS features, we applied cnvHiTSeq to 94 low-coverage CEU samples at the sites of 18 common deletions characterized by PCR, which have been previously used to benchmark CNV genotyping accuracy [9,16]. We used cnvHiTSeq in two different configurations: single-sample, in which each sample is analyzed separately, and population-aware, in which the model parameters are updated via ten iterations of expectation maximization. The population-aware mode achieved perfect genotyping concordance with the reference in 14 of 18 deletions (98.2% genotyping accuracy, 0.5% missing rate) and outperforms the single-sample mode at all deletions. Furthermore, the population-aware mode is shown to be as good as, or superior to, the results obtained from Illumina 1M genotyping arrays in 16 of 18 deletions (Table 1).
Table 1.
Genotyping accuracy on a subset of the HapMap CEU population
| cnvHiTSeq | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cnvHiTSeq* | cnvHiTSeq† | |||||||||
| Location | Predicted Location | Predicted length (bp) | cnvHap r2 | SCIMM r2 | r2 | Accuracy | Missing rate | r2 | Accuracy | Missing rate |
| chr1:35098051-35115368 | chr1:35100671-35112111 | 11,440 | 1.00 | 1.00 | 0.77 | 0.88 | 0.23 | 1.00 | 1.00 | 0.00 |
| chr1:152759872-152770356 | chr1:152760173-152770753 | 10,580 | 0.90 | 0.94 | 0.85 | 0.82 | 0.23 | 1.00 | 1.00 | 0.00 |
| chr3:151625213-151657165 | N/A | N/A | 0.00 | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| chr7:97395305-97402641 | chr7:97395365-97402646 | 7,281 | 1.00 | 1.00 | 0.66 | 0.81 | 0.05 | 1.00 | 1.00 | 0.00 |
| chr7:115930472-115941073 | chr7:115931453-115941632 | 10,179 | 1.00 | 1.00 | N/A | 0.95 | 0.00 | 1.00 | 1.00 | 0.00 |
| chr8:51030941-51038331 | chr8:51031082-51038282 | 7,200 | 0.92 | 0.93 | 0.63 | 0.94 | 0.11 | 1.00 | 1.00 | 0.00 |
| chr8:144700485-144714694 | chr8:144700505-144714606 | 14,101 | 1.00 | 0.97 | 0.54 | 0.67 | 0.00 | 1.00 | 1.00 | 0.00 |
| chr10:71280989-71291079 | chr10:71280949-71291070 | 10,121 | 0.90 | 0.82 | 0.58 | 0.86 | 0.05 | 0.89 | 0.95 | 0.00 |
| chr11:5783630-5809284 | chr11:5784450-5809211 | 24,761 | 1.00 | 1.00 | 0.61 | 0.83 | 0.00 | 0.94 | 0.94 | 0.00 |
| chr11:107238222-107244154 | chr11:107238422-107244103 | 5,681 | 0.94 | 0.97 | 0.83 | 0.90 | 0.00 | 1.00 | 1.00 | 0.00 |
| chr15:34694542-34817215 | chr15:34701483-34817043 | 115,560 | 0.80 | 1.00 | 0.69 | 1.00 | 0.05 | 1.00 | 1.00 | 0.00 |
| chr15:76884597-76907042 | chr15:76884597-76896918 | 12,321 | 0.56 | N/A | 0.96 | 0.95 | 0.00 | 1.00 | 1.00 | 0.00 |
| chr19:35851153-35861684 | chr19:35851134-35863213 | 12,079 | 1.00 | 1.00 | 0.81 | 0.90 | 0.05 | 1.00 | 1.00 | 0.00 |
| chr19:52132525-52148984 | chr19:52132606-52149186 | 16,580 | 0.96 | 0.96 | 0.88 | 0.86 | 0.00 | 1.00 | 1.00 | 0.00 |
| chr20:1558407-1585809 | chr20:1561187-1585928 | 24,741 | 0.90 | N/A | 0.95 | 0.94 | 0.00 | 1.00 | 1.00 | 0.00 |
| chr22:23154417-23243496 | chr22:23186037-23241798 | 55,761 | 0.22 | N/A | 0.61 | 0.73 | 0.00 | 0.77 | 0.80 | 0.09 |
| chr22:24323894-24418396 | chr22:24343395-24397295 | 53,900 | 0.00 | N/A | 0.91 | 0.86 | 0.00 | 1.00 | 1.00 | 0.00 |
| chr22:39366812-39386139 | chr22:39358773-39383652 | 24,879 | 1.00 | 0.96 | 0.46 | 0.85 | 0.00 | 1.00 | 1.00 | 0.00 |
Genotyping accuracy as measured by the concordance between copy number estimates on 22 HapMap CEU samples from the low-coverage pilot of the 1000 Genomes Project and reference copy number estimates obtained using PCR. Concordance is quantified using two different metrics: the correlation coefficient r2 between the reference and the predicted genotypes as well as the fraction of calls with the correct genotype for both alleles. r2 measurements for SCIMM (SNP-Conditional Mixture Modeling) were obtained from the supplementary material of [16]. r2 measurements for cnvHap were obtained from the supplementary material of [9]. Two different versions of cnvHiTSeq were used: cnvHiTSeq*, which is a single-sample version of the algorithm that does not take advantage of the population modeling capabilities, and cnvHiTSeq†, which trains the parameters of the model using the entire low-coverage HapMap CEU population from the 1000 Genomes Project (currently consisting of 94 samples). The genotyping accuracy was calculated using 22 of the 94 samples, since these were the only samples for which PCR copy number estimates were available. When all the samples are predicted to be copy neutral for a given location, the accuracy and r2 are undefined and denoted by N/A. cnvHiTSeq calls with posterior probabilities lower than 80% were excluded and declared as missing.
We also tested the genotyping accuracy of population-aware cnvHiTSeq on a larger genotyping dataset obtained from the High Resolution CNV Discovery study [4]. This dataset consists of approximately 5,000 CNV regions genotyped for 450 HapMap samples and was created using a custom array-CGH. We applied cnvHiTSeq on a randomly chosen subset of 150 CNVs for 91 CEU samples that were common with the low-coverage phase of the 1000 Genomes Project. These CNVs range from 462 bp to 48,748 bp in length, with a median of 2,550 bp. We report a genotyping concordance of 96.0% (2.7% missing rate), which is consistent with our previous results considering the limitations of array-CGH platforms in identifying complex and nested copy number events (Figure S4 in Additional file 1).
Discussion
In our study, we have demonstrated that our novel CNV detection framework maintains a low FDR and high sensitivity while identifying considerably more variants than other methods in low-coverage WGS data. By adopting a unifying approach we are able to detect both deletions and duplications/insertions across a wide range of sizes, without deviating from previously reported length distributions [4] (Figure S2 in Additional file 1).
Our results indicate that almost all CNVs that are detectable by microarray technologies can also be identified using low-coverage sequencing with similar, if not greater, genotyping accuracy. And since the proportion of the genome that can be interrogated by sequencing is much higher than that by microarrays, low-coverage NGS constitutes a natural choice of platform for CNV association studies.
Our method's modular framework makes it readily extendible to additional data sources as they become available. Furthermore, as cnvHiTSeq constitutes a natural extension of cnvHap, it can take full advantage of cnvHap's microarray-based CNV detection framework. Synthesizing the two technologies will achieve the most comprehensive results and allow us to impute sequencing-derived CNVs onto existing genotyped datasets.
High-throughput sequencing technologies are still in active development. Over the course of the past few years, there has been tremendous progress that allowed for faster, more affordable sequencing on a genome-wide scale. As a result, we are now in an era in which hundreds of animal and plant species have been, or are being sequenced, while thousands are being planned. NGS is the driving force behind population re-sequencing projects, which rely on existing reference genomes to identify and catalogue genetic variation, construct the pan-genome [17], and make inference on population structure and demographic history. Population re-sequencing projects are underway in multiple human populations, in Arabidposis thaliana [18], as well as rice [19] and soybean [20]. Such projects typically attempt to sequence more individuals by lowering the coverage per individual to between 4× and 8×. As these low-coverage re-sequencing cohorts become available, cnvHiTSeq will provide the means for interrogating the role of both deletions and duplications on the phenotypic diversity of multiple species. Considering the added ability to accurately distinguish between homozygous and heterozygous events, cnvHiTSeq offers a complete solution to sequencing-based CNV detection and genotyping, aiming to further our understanding of CNV impact on disease and evolution.
Conclusions
We have presented a novel approach to detect and genotype CNVs using high-throughput sequencing data. By combining evidence from various sequencing features, our method offers a substantial improvement in CNV detection sensitivity over existing methods, while maintaining a low FDR. The population modeling aspects of cnvHiTSeq also allow it to achieve a high genotyping accuracy even from low-coverage data. Therefore, our method is especially well-suited for low-coverage re-sequencing cohorts and can provide valuable insights into CNV prevalence and importance.
Materials and methods
Samples and datasets
Development and benchmarking of cnvHiTSeq were accomplished using the publicly available data from the recently completed low-coverage and trio phases of the 1000 Genomes Project [1]. These datasets were sequenced using the Illumina Genome Analyzer platform and aligned to the GRCh37 reference genome using the Burrows-Wheeler algorithm (BWA) [21]. The resulting alignments, encoded in the BAM format [22], comprise the input of our algorithm.
To evaluate our method's FDR we used raw array-CGH intensity data from the High Resolution CNV Discovery project [2] (available online at [23]). For our sensitivity analysis we obtained three gold standard datasets from 'Additional file 1, Material' of the 1000 Genome Structural Variant discovery study [4]. Our genotyping accuracy was assessed using the reference CNV genotypes that were generated by the Genome Structural Variation Consortium [2] (available online at [24]).
Data pre-processing and normalization
The first step of our pipeline is to calculate normalized summary statistics for each of the three data sources across the genome (Figure S1 in Additional file 1), which form the input to the cnvHiTSeq HMM. We carry out separate data processing steps to get RD, RP and SR summary statistics. Each of these summary statistics is sampled at a user-defined frequency (with a default of 20 bp) across the chromosome. Different data sources are offset by 1 bp to avoid obtaining measurements from different sources at the same position. The computational requirements of the pre-processing steps are outlined in Table S2 in Additional file 1.
Read depth pre-processing
The RD pre-processing proceeds by first filtering the BAM alignment files to discard unmapped and duplicate reads. The results of the filtering process are reformatted as BAM files. The filtered BAM files are then sorted and converted to the pileup format using the SAMtools software package [22]. The pileup format comprises a summary of the alignment by chromosomal position, thus allowing us to extract the desired RD information. However, even in the summarized pileup format the alignments are of prohibitive size (>10 GB, for low-coverage samples) both for storage and analysis. Therefore, we split the files up by chromosome and developed a compression scheme that combines run-length encoding and ASCII-to-binary conversion that achieves a 60-fold decrease in pileup file size.
In order to avoid biases arising from highly repetitive portions of the reference sequence (including telomeres and centromeres), we filter genomic regions based on 'sequence uniqueness'. The standard metric used by the ENCODE pilot project [25] to quantify sequence uniqueness is the alignability score. This score is calculated by mapping sliding windows of k-mers to the genome allowing up to two mismatches and assigning a score to each window equal to the inverse of the number of matches across the genome. Thus, alignability ranges from 0 for redundant sequences to 1 for perfectly unique sequences. Regions with alignability scores lower than 0.05 (corresponding to at least 20 genomic matches) were masked out. We also used the ENCODE Data Analysis Consortium (DAC) blacklist to exclude 9.8 Mb from further analysis. This list was designed to complement the alignability metric with pathological elements such as pericentromeric and subtelomeric repeats that have proven troublesome for short read alignment. In total we excluded approximately 13.6% of the reference sequence, which is in almost perfect accordance with the 'accessible genome' for low-coverage analysis as defined by the 1000 Genomes Pilot study [1].
Next, the RD is normalized to correct for the documented NGS biases in GC-rich and GC-poor regions [26]. Even after correction, RD data exhibit spatial autocorrelation patterns, known as 'wave artifacts', that cannot be fully explained by the GC content bias [27]. To minimize the effects of such artifacts we fit a LOESS curve to the RD data using second degree local polynomials. The LOESS smoothed data are then normalized by the average per-sample chromosome depth to account for differences in coverage among samples. Finally, we calculate the average of these values in non-overlapping 20-bp windows as the RD summary statistic.
Read pair pre-processing
RP data are extracted from BAM alignment files after a rigorous filtering process. In addition to duplicate and unaligned reads, we also filter out reads with non-unique alignments as they would introduce ambiguity. Both reads of a pair are required to have a high mapping quality (Phred Quality Score >20) and originate from a sequencing library of known insert size. Since RP data are extracted from different libraries, we account for the different insert size distributions by quantile-normalizing them to an arbitrary Gaussian reference (N(200,15) in our case). To that end, we use a kernel quantile estimator, with a standard Gaussian kernel function to avoid biases in libraries containing only a small number of RPs. Finally, we record two RP summary statistics at every 20 bp across the genome: the average normalized distance of reads that span the given position (RPS) and the total count of spanning read pairs (RPC), which may be different from the overall RD in the presence of single-end libraries.
Split read pre-processing
SR discovery involves a non-trivial computational problem that is related to sequence alignment. To that end, we look for RPs with one read properly mapped and the other not. The mapped read is going to provide the 'anchor' and limit the search space. The unmapped read of the pair is exhaustively split in all plausible combinations and the results are stored in paired FASTQ files. Since some of the resulting split fragments may be quite short, we eliminate fragments of low-complexity (for example, highly repetitive fragments) using the DUST algorithm [28]. The split reads are then aligned to the reference using BWA, allowing up to two mismatches but no gaps. All the alternative mappings of the paired split fragments are retained in order to find the best combination. The results are filtered to discard fragments aligning more than 1 Mb away from the anchor read. This creates the final SR discovery set. Note that this discovery set may contain alternative splits of the same read, which are used to fine-map the breakpoints of a deletion.
The SR mappings are used in two separate configurations. First, we exploit the fact that the anchor read was originally orphaned but is now properly paired. These newly rescued pairs are especially informative in repetitive regions, where there is a higher chance of splits occurring, and are thus incorporated into the previously described RP framework (Figure 1f). Second, we keep a count of the actual splits that span any given genomic position (Figure 1c), which is sampled every 20 bp. This sampled count constitutes the split read count (SRC) summary statistic.
cnvHiTSeq hidden Markov model
First, cnvHiTSeq builds a haploid HMM (Figure 2a). This HMM comprises one hidden state smc per haploid copy number c, up to a user-defined maximum c∈{0, ..., Z-1}, at each measured position m∈{1, ..., M}. Two copies of this HMM are paired to create a diploid HMM (or n copies for a n-ploid sample) (Figure 2b). The computational cost of modeling increased copy number and polyploidy has been discussed previously for cnvHap [9]. The diploid HMM states have emission distributions specific to the data source measured at a given position (Figure 2c), thus interweaving different sources in the same model. In more detail, we represent the emission data for sample j by Ijm, so that Ijm = {RDjm} for read depth, Ijm = {RPSjm ,RPCjm} for read pair, and Ijm = {SRCjm} for split read. As noted above, each of these data sources is measured at pre-defined resolution (default 20 bp) across the genome, with an offset of 1 bp between sources. The emission probability of a list of unordered states sm={l1,..., lN} depends only on the total copy number:
| (1) |
where c = c(sm) represents the total copy number for an unordered list of haploid states, θm represents the parameters of the emission distributions at position m, NB represents the negative binomial distribution, Ν the normal distribution and SN the skew normal distribution. For the single-sample mode, there is only one global set of emission parameters, so, for example .
RD is modeled using the negative binomial distribution in order to account for the observed overdispersion of RD due to the non-uniform distribution of mapped reads across the genome [26]. The initial parameters of NB reflect the mode of the expected distribution (for example, a single copy deletion has an expected normalized mode of one read per fold coverage). We followed the parameterization adopted in [29] according to which, if Y is a negative binomial random variable with mean parameter η and dispersion parameter κ, it has a probability mass function given by:
| (2) |
We model the average read pair span RPS as a normal distribution. The initial parameters for the normal 2 copy number state are set to reflect the size and random variation of the paired-end insert library (for example, 200 ± 15 bp insert). The initial parameters for a homozygous (heterozygous) deletion/insertion are set so that the mean is 3 (2) standard deviations above/below the library insert size, respectively. The spanning RP count (RPC) is again modeled with NB as in Equation 2 to account for overdispersion. We assume that RPS and RPC are independent, conditional on the hidden copy number, and hence the joint conditional probability is the product of their individual probabilities. In this way, cnvHiTSeq avoids using a hard threshold on the number of supporting RPs, as is the case for competing algorithms, and instead assigns more confidence to distances that arise from RP counts closer to the average coverage. This is especially important for detecting deletion events in low-coverage data, as the scarcity of RPs should not be interpreted as absence of CNV. On the other hand, our approach also downweights regions that appear to have extremely high RP counts, since they most likely correspond to sequencing artifacts or highly redundant reference sequences.
Since SR events are less common than the evidence provided by the other sequencing data sources, the presence of a few overlapping SRs is usually sufficient to make confident CNV calls. Therefore, we model the SR count using a positively skewed normal distribution with initial parameters depending on the depth of coverage. As with RPC, cnvHiTSeq eliminates the need for a minimum number of supporting SRs by directly modeling the SRC. By avoiding hard thresholds and allowing for alignment mismatches, our method achieves higher flexibility than existing methods, especially for low-coverage samples where SRs are infrequent.
The transition probability between unordered pairs of states is given by:
| (3) |
where T(N) represents all possible permutations of a list of length N. The haploid transition is calculated as:
| (4) |
where Q is a global reversible transition rate matrix between copy number, rm is a scalar representing the local rate of transitions at position m, and d(m) represents the base-pair coordinate of position m. The user can either specify the steady-state distribution of this rate matrix (for example, reflecting an expectation of the relative number of different copy number states in the genome) or specify this rate matrix directly.
Model training
Model training is accomplished via a generalized expectation maximization algorithm. In the expectation step, cnvHiTSeq applies the forward-backward algorithm to each sample separately to calculate the expected counts of transitions between states k,l of the haploid HMM Etmkl, as well as the posterior probability of each copy number at each position p(cjm|Ij). The parameters of the transition model are updated based on the Etmkl, as described in more detail in [9]. The posterior probability of each copy number state at each position is also recorded as wjm(CN) = p(cjm = CN|Ij), and can be thought of as the assignment weight of the data at position m to state CN, using the current parameterization of the model.
We optimize the parameters of the emission distribution for copy number c based on maximization of the log-likelihood:
| (5) |
where Equation 1 is used to calculate the emission probabilities given new parameters. The maximization is carried out using a gradient descent algorithm. In single-sample mode, there is only one global set of emission distributions, and the sum is performed across all sites m. In order to avoid overfitting, we introduce pre-defined counts of pseudo-observations I'mj , sampled from the initial emission distributions (Equation 1).
Figure 2d,e presents examples of the trained haploid HMM for the characteristic CNVs introduced in Figure 1. After ten training iterations, the Viterbi algorithm is used to calculate the most likely CNV segmentation (conditional on the trained parameters) for each sample.
Mendelian inconsistency analysis
To obtain an estimate of cnvHiTSeq's FDR we applied a Mendelian inconsistency approach on the CEU trio using standard and strict criteria. For the standard criterion, we required the CNVs detected in the child (NA12878) to overlap CNVs of the same class in at least one parent. For the strict criterion, we consider CNVs that have been detected in all three members of the trio as candidate false positive calls corresponding to systematic biases. The child CNV is required to have at least 50% overlap with both parents to be included in this category. This candidate false positive list is filtered to exclude common CNVs that were present in the DGV (ignoring those predicted for NA12878 as well as those from early bacterial artificial clone (BAC) studies) as these are likely to recur in the population. We also required that a DGV variant covered at least 90% of our corresponding CNV call. After filtering, the remaining false positives were included in our Mendelian inconsistency calculation to obtain a conservative upper bound of the FDR. To facilitate comparison we applied the exact same procedure to Genome STRiP calls on the CEU trio.
Abbreviations
bp, base pair; BWA, Burrows-Wheeler aligner; CGH, comparative genomic hybridization; CNV, copy number variation/variant; DGV, Database of Genomic Variants; EWT, event-wise testing; FDR, false discovery rate; HMM, hidden Markov model; NGS, next-generation sequencing; PCR, polymerase chain reaction; RD, read depth; RP, read pair; RPC, read pair count; RPS, read pair span; SR, split read; SRC, split read count; WGS, whole-genome sequencing.
Authors' contributions
EB, MRJ and LJMC conceived the study. EB and LJMC designed the algorithm. EB implemented and benchmarked the software. EB and LJMC drafted the manuscript. MRJ critically reviewed the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Supplementary material. This file contains Figures S1, S2, S3 and S4, and Tables S1, S2, S3, S4 and S5. Figure S1 presents a schematic of our pre-processing pipeline. Figure S2 presents the length distribution of our CNV calls. Figure S3 presents the cumulative length distribution of cnvHiTSeq calls that were validated with array-CGH data. Figure S4 presents a heatmap of the genotyping concordance between cnvHiTSeq and a benchmark dataset. Table S1 presents the array-CGH validation results. Table S2 describes the computational requirements of our pipeline. Table S3 presents a comparison of our deletion calls with those of Genome STRiP for sample NA12878. Table S4 presents a comparison of our Mendelian inconsistency results for different criteria. Table S5 presents a comparison of the sensitivity of various methods on low-coverage samples.
This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Contributor Information
Evangelos Bellos, Email: evangelos.bellos09@imperial.ac.uk.
Michael R Johnson, Email: m.johnson@imperial.ac.uk.
Lachlan J M Coin, Email: l.coin@imperial.ac.uk.
Acknowledgements
This study makes use of publicly available data generated by the 1000 Genomes Project. We also acknowledge the Genome Structural Variation Consortium (PIs Nigel Carter, Matthew Hurles, Charles Lee and Stephen Scherer) for pre-publication access to their CNV discovery and genotyping data. This work was supported by grant BB/H024808/1 from the UK Biotechnology and Biological Sciences Research Council (BBSRC). The authors would also like to thank Simon Burbidge of the Imperial College High Performance Computing Service for his assistance.
References
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;13:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, MacArthur DG, MacDonald JR, Onyiah I, Pang AWC, Robson S, Stirrups K, Valsesia A, Walter K, Wei J. The Wellcome Trust Case Control Consortium. Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME. Origins and functional impact of copy number variation in the human genome. Nature. 2010;13:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, Falchi M, Chen F, Andrieux J, Lobbens S, Delobel B, Stutzman F, El-Sayed Mousafa JS, Chevre JC, Lecoeur C, Vatin V, Bouquillon S, Buxton JL, Boute O, Holder-Espinasse M, Cuisset JM, Lemaitre MP, Ambresin AE, Brioschi A, Gaillard M, Guisti V, Fellman F, Ferrarini A, Hadjikhani N, Campion D. et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;13:671–675. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla RK, Chinwalla A, Conrad DF, Fu Y, grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Krurana E, Kiral D, Lam HY, leng J, Li R, Li Y, Lin CY, Luo R. et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;13:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, Sahinalp SC, Gibbs RA, Eichler EE. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;13:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009;13:S13–20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- Handsaker RE, Korn JM, Nemesh J, McCarroll SA. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet. 2011;13:269–276. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;13:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin LJ, Asher JE, Walters RG, Moustafa JS, de Smith AJ, Sladek R, Balding DJ, Froguel P, Blakemore AI. cnvHap: an integrative population and haplotype-based multiplatform model of SNPs and CNVs. Nat Methods. 2010;13:541–546. doi: 10.1038/nmeth.1466. [DOI] [PubMed] [Google Scholar]
- Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;13:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;13:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;13:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;13:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, Elliott AL, Parkin M, Hubbell E, Webster T, Mei R, Veitch J, Collins PJ, Handsaker R, Lincoln S, Nizzari M, Blume J, Jones KW, Rava R, Daly MJ, Gabriel SB, Altshuler D. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;13:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, Haugen E, Zerr T, Yamada NA, Tsang P, Newman TL, Tuzun E, Cheng Z, Ebling HM, Tusneem N, David R, Gillett W, Phelps KA, Weaver M, Saranga D, Brand A, Tao W, Gustafson E, McKerman K, Chen L, Malig M. et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;13:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Zerr T, Kidd JM, Eichler EE, Nickerson DA. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat Genet. 2008;13:1199–1203. doi: 10.1038/ng.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Zheng H, Luo R, Zhu H, Li Q, Qian W, Ren Y, Tian G, Li J, Zhou G, Zhu X, Wu H, Qin J, Jin X, Li D, Cao H, Hu X, Blanche H, Cann H, Zhang X, Li S, Bolund L, Kristiansen K, Yang H, Wang J, Wang J. Building the sequence map of the human pan-genome. Nat Biotechnol. 2010;13:57–63. doi: 10.1038/nbt.1596. [DOI] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, Gunther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, Wang X, Ott F, Muller J, Alonso-Blanco C, Borgwardt K, Schmid KJ, Weigel D. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;13:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- Xu X, Liu X, Ge S, Jensen JD, Hu F, Li X, Dong Y, Gutenkunst RN, Fang L, Huang L, Li J, He W, Zhang X, Wang J, Wright M, McCouch S, Nielsen R, Wang J, Wang W. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol. 2011;13:105–111. doi: 10.1038/nbt.2050. [DOI] [PubMed] [Google Scholar]
- Lam HM, Xu X, Liu X, Chen W, Yang G, Wong FL, Li MW, He W, Qin N, Wang B, Li J, Jian M, Wang J, Shao G, Wang J, Sun SS, Zhang G. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet. 2010;13:1053–1059. doi: 10.1038/ng.715. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;13:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Proc GPD. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;13:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EBI ArrayExpress Archive. E-MTAB-142. http://www.ebi.ac.uk/arrayexpress/files/E-MTAB-142
- Wellcome Trust Sanger Institute. High resolution CNV discovery (Conrad et al, 2010) http://www.sanger.ac.uk/research/areas/humangenetics/cnv/highres_discovery.html
- ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;13:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetam R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;13:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni JC, Thorne NP, Valsesia A, Fitzgerald T, Redon R, Fiegler H, Andrews TD, Stranger BE, Lynch AG, Dermitzakis ET, Carter NP, Tavare S, Hurles ME. Breaking the waves: improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol. 2007;13:R228. doi: 10.1186/gb-2007-8-10-r228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgulis A, Gertz EM, Schaffer AA, Agarwala R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J Comput Biol. 2006;13:1028–1040. doi: 10.1089/cmb.2006.13.1028. [DOI] [PubMed] [Google Scholar]
- Saha K, Paul S. Bias-corrected maximum likelihood estimator of the negative binomial dispersion parameter. Biometrics. 2005;13:179–185. doi: 10.1111/j.0006-341X.2005.030833.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material. This file contains Figures S1, S2, S3 and S4, and Tables S1, S2, S3, S4 and S5. Figure S1 presents a schematic of our pre-processing pipeline. Figure S2 presents the length distribution of our CNV calls. Figure S3 presents the cumulative length distribution of cnvHiTSeq calls that were validated with array-CGH data. Figure S4 presents a heatmap of the genotyping concordance between cnvHiTSeq and a benchmark dataset. Table S1 presents the array-CGH validation results. Table S2 describes the computational requirements of our pipeline. Table S3 presents a comparison of our deletion calls with those of Genome STRiP for sample NA12878. Table S4 presents a comparison of our Mendelian inconsistency results for different criteria. Table S5 presents a comparison of the sensitivity of various methods on low-coverage samples.