

ABSTRACT
Staphylococcal enterotoxin B (SEB) is a potent toxin that is produced by Staphylococcus aureus strains and is classified as a category B select agent. We have previously shown that monoclonal antibody (MAb) 20B1, a murine anti-SEB IgG1, successfully treats SEB-induced lethal shock (SEBILS) and bacteremia that is caused by SEB-producing S. aureus. In this study, we have generated two isotype switch variants of the original IgG1 MAb 20B1, an IgG2a and IgG2b, both bearing the same variable region sequence, and compared their neutralizing and protective activity in in vitro and in vivo assays, respectively. All 3 isotypes demonstrated comparable affinity to SEB and comparable 50% inhibitory concentrations (IC50s) in T cell proliferation assays. In vivo, however, the IgG2a isotype variant of 20B1 exhibited significantly greater protection than IgG1 or IgG2b in murine SEB intoxication and S. aureus sepsis models. Protection was associated with downmodulation of inflammatory host response. Our data demonstrate that changing the isotype of already protective MAbs, without affecting their antigen specificity or sensitivity, can result in an enhancement of their protective ability. Isotype selection, therefore, should be carefully considered in the development of toxin-neutralizing MAbs and the design of antibody therapeutics.
IMPORTANCE
The purpose of this study was to enhance the protective efficacy of an existing, protective monoclonal antibody against staphylococcal enterotoxin B. Using two in vivo mouse models, our study demonstrates that the protective efficacy of a monoclonal antibody may be improved by inducing an isotype switch at the Fc region of an antibody, without altering the antigen specificity or sensitivity of the antibody. The development of therapeutic MAbs with higher efficacy may allow for the achievement of equal therapeutic benefit with a lower dosage. In turn, the use of lower doses may reduce the cost of these therapies, while reducing the potential for adverse side effects.
INTRODUCTION
Staphylococcal enterotoxin B (SEB) is a clinically relevant staphylococcal superantigen, as well as a class B biological warfare agent. Passive immunotherapy with anti-SEB monoclonal antibodies (MAbs) has successfully neutralized the SEB toxin in vitro, as well as protected mice from SEB-induced lethal shock (SEBILS) in in vivo studies (1–6). In addition, recent work in murine infection models has demonstrated that SEB-specific MAb 20B1 successfully treats sepsis and deep-seated tissue infection that is caused by SEB-producing Staphylococcus aureus strains (7).
In recent decades, the use of MAbs has increased dramatically and represents one of the most lucrative and fast-growing classes of drugs (8). The majority of MAbs that are FDA licensed to date are used to treat either oncological or autoimmune diseases. Anti-infective antibodies, however, remain scarce, as only two such MAbs are licensed to treat infectious disease (9, 10). Although great progress has been made in antibody (Ab) technology, especially with respect to generating human and humanized antibodies, it remains unclear to what extent the constant region affects Ab efficacy. To date, 36 MAbs are FDA licensed, of which 16 are humanized, 11 are human, 6 are chimeric, and only 3 are murine.
The ability of Abs to affect host-pathogen interactions is not reliant solely on the function of their variable region, which binds to the target antigen. Their constant domains also mediate biological properties through Fc receptor (FcR) binding, complement activation, and effects on avidity and serum half-life (11). These biological properties can differ significantly between the different isotypes. For instance, the Fc portion of mouse immunoglobulin G2a (IgG2a) Abs interacts with complement components (12) and high-affinity activatory FcRs (13), whereas the Fc portion of mouse IgG1 antibodies mediates a relatively lower affinity interaction with activatory FcRs and does not stimulate FcR-mediated immune responses as effectively (14, 15). These properties have not been fully exploited for therapeutic antibody purposes, as the majority (73%) of currently licensed MAbs are of the human IgG1 isotype, which resembles mouse IgG2a in its similar effector functions. For the efficacy of anti-infective Abs, isotype selection may be of particular importance, as pathogens not only harm the host directly but also indirectly provoke immune responses by eliciting uncontrolled damage (16). In this regard, certain isotypes of anti-infective Abs could either enhance or decrease the host response through their interaction with specific Fc receptors (17).
To investigate the effect of isotype on protective efficacy, we generated isotype switch variants of a murine IgG1, MAb 20B1 (18). These isotype switch variants encode a variable region identical to their parent IgG1, and they differ only in their Fc binding region. In previous studies, we have shown that passive immunotherapy with MAb 20B1 successfully treats SEBILS (2) and S. aureus-mediated sepsis (7). Our data demonstrate that isotype switch variants of the IgG2a isotype are superior in efficacy compared to the IgG1 and IgG2b isotypes. Specifically, lower doses of the IgG2a isotype switch variant are required to enhance the survival of mice in both the SEBILS and S. aureus sepsis models. Cytokine analysis documented that enhanced efficacy was associated with early inhibition of proinflammatory cytokines. In summary, these results provide valuable data that encourage systematic evaluation of specific isotypes for the development of anti-infective MAbs.
RESULTS
Generation of IgG2a and IgG2b switch variants.
Isotype switch variants of SEB-specific IgG1 MAb 20B1 were generated. We used the enzyme-linked immunosorbent spot assay (ELISPOT) to detect spontaneously arising variant cells producing new downstream isotypes of IgG1. IgG2a- and IgG2b-producing cells spawned at rates of 6 × 10−5 and 6 × 10−4, respectively. Next, we attempted to enrich each of the resulting two isotype variant populations by Sib selection, which allowed us to successfully obtain IgG2b-producing variants. However, despite successive rounds of enrichment, the IgG2a variants remained too rare to recover through this approach. Consequently, we used fluorescence-activated cell sorting (FACS) with immunofluorescence staining for surface-associated Ab to identify a cell fraction significantly enriched for the IgG2a isotype. Small fractions of viable cells stained for surface-associated IgG2a were isolated by cell sorting on the basis of surface fluorescence intensity. This step enriched the fraction of hybridomas producing IgG2a variant cells and sufficiently reduced the dominant fraction of IgG1 producers. Two rounds of soft agar cloning were performed to stabilize each variant cell type. Analysis of supernatants from the IgG2a- and IgG2b-secreting clones revealed no trace of IgG1 by enzyme-linked immunosorbent assay (ELISA). Comparative alignment of nucleotide sequences derived from each isotype variant confirmed identical heavy- and light-chain variable sequences to the parent IgG1 from which they were derived (data not shown).
Binding affinity of isotype switch variants to SEB.
Initial assessment of binding affinities by ELISA demonstrated comparable binding to SEB by both the isotype variants and their 20B1 parent (Fig. 1A). Standard measurement of dissociation constants (Kd) using Biacore could not be performed with wild-type SEB due to federal regulations that restrict the use of select agents. We instead performed BLItz analysis to obtain Kd values and thus investigate whether changes to the constant regions affected the fine specificity of these MAbs toward SEB. This method determined the Kd of 20B1 IgG1, IgG2a, and IgG2b toward SEB to be 16.25 + 1.8 nM, 66.27 + 2.7 nM, and 47.15 + 13.9 nM, respectively (Fig. 1B to D). These Kd values are considered comparable, as they are within the experimental error range of the assay.
FIG 1 .

(A) Comparable binding of three IgG subclasses of SEB-specific MAb 20B1 isotype switch variants 20B1 IgG1, 20B1 IgG2a, and 20B1 IgG2b. Experiment was performed in triplicates. Each point represents the mean value of triplicates, and bars represent the standard error derived from measurements in the same experiment. (B, C, D) Dose response curves for binding of murine MAb 20B1 isotypes to biotinylated SEB were captured by streptavidin biosensors using BLItz. Analyses were performed using Forte Bio’s software to determine R equilibrium. Measured kinetic constants are reported.
Inhibition of SEB-induced T-cell proliferation by MAb 20B1 is independent of IgG subclass.
In vitro, we compared the ability of all three 20B1 isotypes to inhibit SEB-induced proliferation of murine splenocytes. All three isotypes of 20B1 demonstrated comparable levels of inhibition after 96 h of SEB stimulus and consistently showed 90 to 100% inhibition with subnanomolar 50% inhibitory concentrations (IC50s) (IgG1, 0.27 nM; IgG2a, 0.68 nM; and IgG2b, 1.04 nM) (Fig. 2A to C).
FIG 2 .
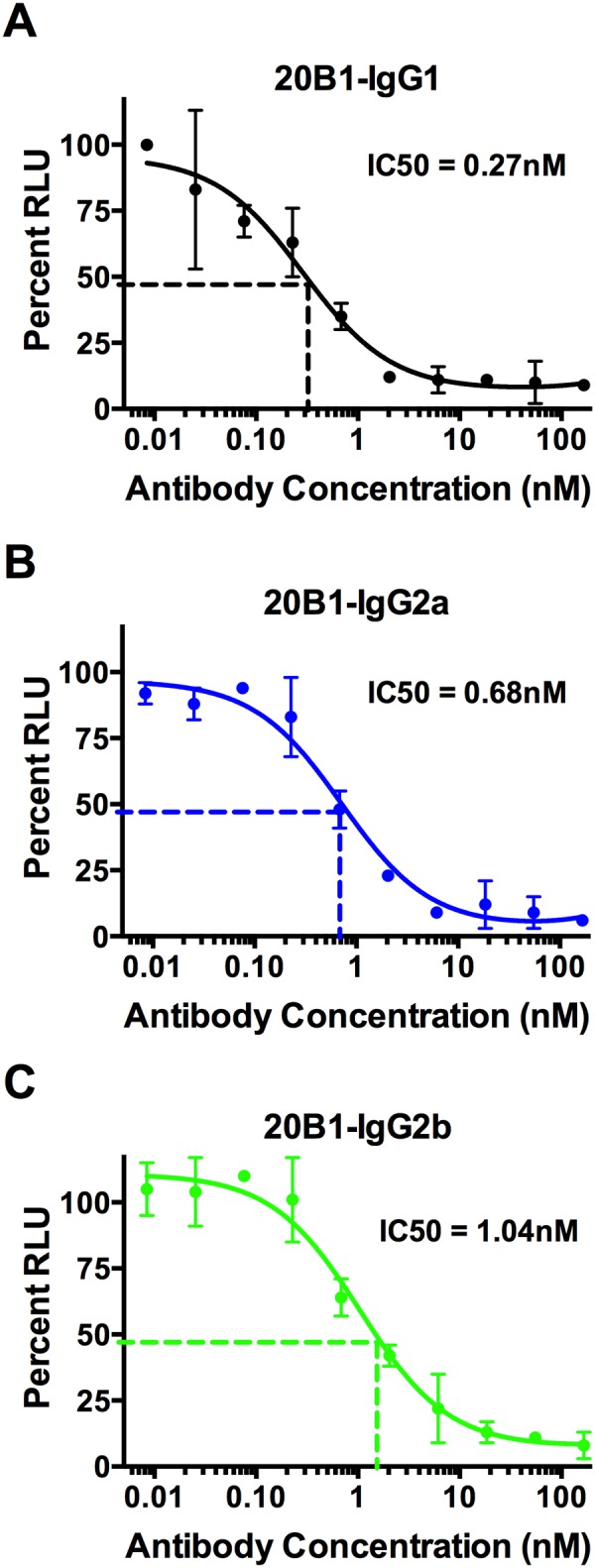
IgG subclasses of MAb 20B1 showed comparable inhibition of SEB-induced proliferation in murine splenocytes. Calculated IC50 of MAb 20B1 IgG1 was 0.28 nM (A), 20B1 IgG2a was 0.68 nM (B), and 20B1 IgG2b was 1.04 nM (C). Experiments performed in triplicates. The circles represent an average from triplicates of the relative luminescence units (RLU). Bars represent the standard deviations derived from measurements in triplicate wells in the same experiment. IC50s were determined from concentration-response curve analysis (GraphPad Prism 6).
Isotype variants provide enhanced protection from SEB-induced lethal shock.
An in vivo murine SEBILS model was used to test and compare the protective efficacy of the MAb 20B1 IgG1 parent and its isotype variants at equivalent doses. We have reported earlier that MAb 20B1 IgG1 was an excellent lead candidate to treat SEBILS (2) and SEB-secreting S. aureus sepsis (7). In this study, various doses of MAbs ranging between 50 µg and 500 µg were compared for their efficacy. Results from these experiments showed that switching the isotype of the parent IgG1 enhanced protection, with relative efficacies of IgG2a > IgG2b = IgG1 (Fig. 3A to C), in a dose-dependent manner.
FIG 3 .

BALB/c mice (n = 10 per group) were treated with various IgG subclasses of MAb 20B1 at different doses or PBS intraperitoneally (i.p.) and challenged with 20 µg of SEB and sensitized with 25 mg of galactosamine (i.p.). Mice treated with 250-µg doses of 20B1 isotypes showed 100% survival (A), variable protection with the 100-µg dose (B), and IgG2a was 100% effective at the 50-µg dose, but IgG2b and IgG1 failed to protect (C). (D) Enhanced protection was observed when 20B1 isotypes (50 µg) were combined with 14G8 IgG1 (50 µg) compared to PBS treatment (20B1 IgG1 and 14G8, P = 0.003; 20B1 IgG2b and 14G8, P = 0.001; 20B1 IgG2a and 14G8, P = 0.0001). Analysis of survival data was performed using log rank (Mantel-Cox test).
We further explored the protective efficacy of these isotype variants in combination with 14G8 IgG1, an anti-SEB MAb that we previously reported does not provide protection and does not compete or share epitopes with MAb 20B1 (2). Improved protective efficacy was observed when MAb 20B1 IgG1 or the isotype variants at doses as low as 50 µg were administered in combination with MAb 14G8 IgG1 in comparison to 20B1 alone (Fig. 3D). Survival efficacy increased from 0% to 60% when 20B1 IgG1 was combined with 14G8 IgG1, and from 10% to 40% after combined treatment with 20B1 IgG2b and 14G8 IgG1. Increased survival was also observed when a low dose of 25 µg of 20B1 IgG2a was administered in combination with MAb 14G8, a dose that was partially protective (50%) when 20B1 IgG2a was used alone (data not shown).
Enhanced protection correlated with more potent inhibition of proinflammatory cytokines. Multiplex cytokine analysis of sera at 2 h postchallenge showed significantly lower levels of all seven proinflammatory cytokines examined (interleukin 6 [IL-6], gamma interferon [IFN-γ], tumor necrosis factor alpha [TNF-α], IL-10, IL-1β, keratinocyte-derived chemokine/growth-related oncogene [KC-GRO], and IL-12 p70) in mice treated with 20B1 IgG2a than in those treated with phosphate-buffered saline (PBS) or the IgG2b isotype of MAb 20B1 (Fig. 4A to F). Levels of cytokines IL-6, IFN-γ, and KC-GRO were also significantly lower after treatment with 20B1 IgG2a than after treatment with the parental IgG1 isotype.
FIG 4 .

Multiplex cytokine analysis of serum (after 2 h) from mice (n = 5) treated with MAb 20B1 isotypes (50 µg) or PBS and challenged with SEB in administration with galactosamine. Levels of proinflammatory cytokines (IL-6, IFN-γ, TNF-α, IL-10, IL-1β, and KC-GRO) were significantly lower in mice treated with 20B1 IgG2a than in mice treated with PBS or the IgG2b isotype of MAb 20B1 (P < 0.05). Cytokines IL-6, IFN-γ, and KC-GRO were significantly lower (P < 0.05) in 20B1 IgG2a-treated mice than in those treated with the parental 20B1 IgG1. All controls and samples were done in duplicates. Bars are represented as mean values + standard deviations. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc multiple-comparison test to compare the mean of each data set with the mean of every other datasets using GraphPad Prism 6 software, and data were considered significant as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. NS, not significant.
Isotype switch variants have comparable effects on SEB clearance in vivo.
We further explored whether isotypes differentially affect the clearance of SEB from serum. Accordingly, SEB levels were quantified at 2 h (Fig. 5A) and 6 h (Fig. 5B) postchallenge in serum and organs (liver, spleen, and kidney) of mice treated with individual subclasses of MAb 20B1 IgG followed by challenge with SEB. Treatment with any of the three isotypes resulted in comparable clearance of SEB. As previously published, immunocomplex formation with Abs slows the clearance of the toxin through the kidney (19), and therefore significantly lower clearance is observed than SEB clearance in mice treated with PBS (20).
FIG 5 .
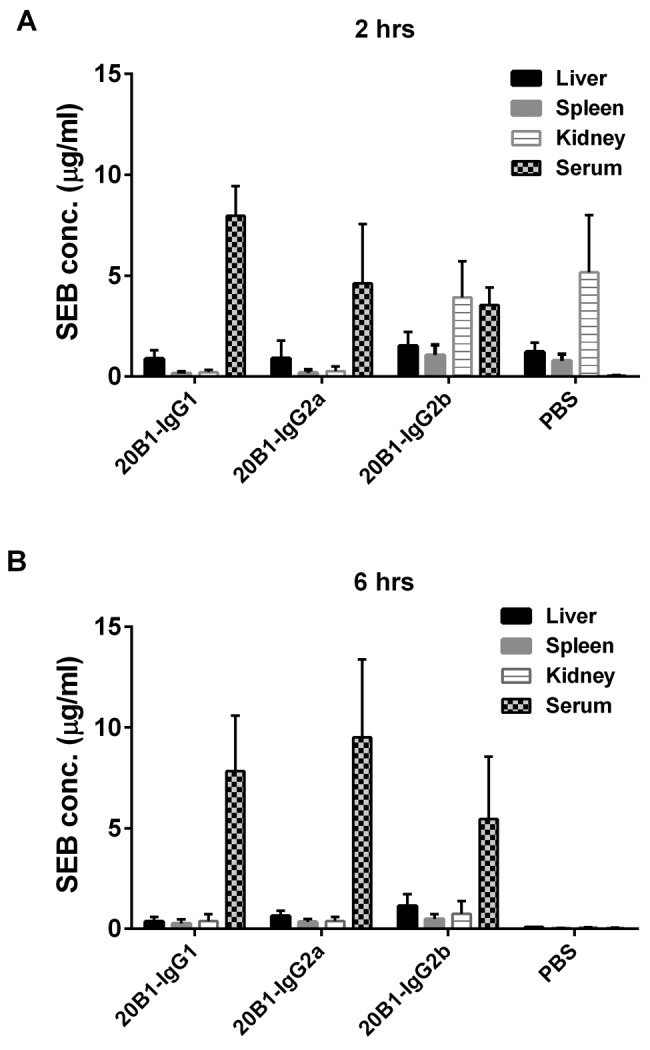
Eight different sets of mice (n = 5) were treated either with isotypes of MAb 20B1 (500 µg) or PBS and challenged with SEB. ELISAs were performed to measure the SEB levels from sera, kidney, liver, and spleen at 2 h and 6 h after SEB challenge. SEB level was higher in blood at the initial time point at 2 h, as well as at 6 h in mice treated with all isotypes of MAb 20B1. As expected, higher SEB clearance was observed from sera of mice treated with PBS only. Bars represent the standard error derived from the SEB measurement of a group of 5 mice in each data set.
IgG2a isotype enhances protection from S. aureus sepsis.
We further explored the efficacy of each isotype switch variant in a murine model of S. aureus sepsis. Previous work using this model has shown that treatment with the 20B1 IgG1 isotype at 500 µg resulted in 80 to 100% survival, whereas all untreated mice died (7). Here, we tested a lower dose of 300 µg of each IgG subclass and compared treatment efficacies. At this lower dose, 50% of mice survived after treatment with the IgG2b isotype, 40% survived after treatment with the IgG1 isotype, and 100% survived after treatment with the IgG2a isotype (Fig. 6).
FIG 6 .
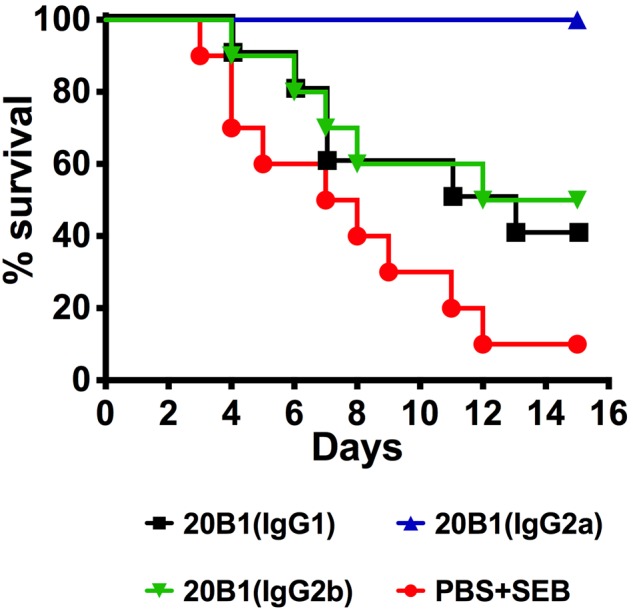
Different IgG subclasses of MAb 20B1 (i.v.) (300 µg) were injected into mice and challenged with S. aureus (MRSA strain 38, i.v. 5 × 107 CFU) in a sepsis model. Treatment with the IgG2a isotype resulted in significantly longer survival than that in mice treated with 20B1 IgG2b (P = 0.01), 20B1 IgG1 (P = 0.004), or PBS (P = 0.0001). Analyses of survival data were performed using log rank (Mantel-Cox test).
DISCUSSION
We conclude from our results that the constant region of anti-SEB MAbs can significantly affect toxin neutralization efficacy through modulation of the host immune response. Most importantly, the IgG2a isotype exhibits enhanced efficacy in comparison to that of other isotypes in both an SEB intoxication model and an SEB-secreting methicillin-resistant S. aureus (MRSA) sepsis model. Our findings underscore the importance of efforts to investigate Fc domain engineering as a strategy for optimizing therapeutic Abs. Enhancement of Ab efficacy could ultimately permit lower dosing of Abs, leading to a more reasonable pricing of this treatment modality and, therefore, substantially increase the development and use of anti-infective Abs.
Many human pathogens, including S. aureus, exhibit a diverse set of virulence factors, including toxins that facilitate evasion of the host’s sophisticated immune response (21–23). Many of these toxins directly damage host cells (24); however, staphylococcal enterotoxins like SEB have more of an indirect toxic effect. Specifically, they overstimulate immune cells, resulting in a cytokine storm that can ultimately kill the host (25). Historically, the view has been that Abs mediate SEB neutralization by interfering with binding of SEB to its targets, the major histocompatibility complex class II (MHC-II) molecule and the V-β chain of the T-cell receptor (TCR) (26). Interruption of this trimer formation is dependent on Ab specificity. Only high-affinity Abs or soluble V-β mimics have been successful in neutralizing SEB, largely due to the rapid and high-affinity binding between SEB and its in vivo targets, MHC-II and TCR V-β (27–29). To investigate the role of the Ab constant region in toxin neutralization, we generated IgG2a and IgG2b variants of SEB-specific murine MAb 20B1, which is of the IgG1 isotype. Isotype switching in mice progresses through an IgM → IgG3 → IgG1 → IgG2b → IgG2a spontaneous recombination process that generates Abs of different isotypes but that continue to share the identical variable region to the original Ab. Earlier studies on the generation of isotype switch variants from murine B cell lymphomas (30) have reported challenges in generating IgG2a isotypes. In this study, cytokine treatment in addition to FACS and immunofluorescence staining was required to identify and enrich populations of IgG2a-producing hybridoma cells.
The variable regions of all isotype variants were sequenced to ensure that no somatic mutation had occurred. All three isotype switch variants expressed identical variable regions, and BLItz analysis revealed comparable Kd values in the nanomolar range for all isotypes. Of note is that in vitro T-cell assays did not demonstrate differences in neutralization efficacy, as the effective IC50s were comparable between all isotypes. This finding highlights the limitations of commonly used screening assays and is in accordance with previously published observations that SEB-specific MAbs, which neutralize the toxin in vitro, may not have the same effect in vivo (2). In our SEBILS model, significant differences in efficacy of Ab-mediated SEB neutralization were observed between the isotype switch variants. Efficacy was most potent for IgG2a, followed by IgG2b and IgG1. The IgG2a MAb also prevents SEBILS at lower doses than the IgG1 and IgG2b MAbs. As previously shown with modified capture ELISAs, MAb 20B1 binds to a single epitope on the toxin (2). Hence, only one MAb at a time can bind an SEB molecule. Furthermore, it has been shown that binding of SEB-specific MAbs to the toxin, in vivo, results in slower clearance of SEB toxin from the blood (2) because the immunocomplex is cleared not as efficiently through the glomerular filter of the kidney. Theoretically, treatment with different isotype variants, which commonly exhibit significant differences in their pharmacokinetics (31), could result in different degrees of SEB clearance. However, at least in the SEBILS model, toxin clearance is rapid (20), and therefore differences in half-life time of the various isotypes would likely not be relevant. Consequently, we found no difference in SEB levels in the sera of mice treated with different Abs. SEBILS was even more efficiently prevented when MAb 20B1 was combined with MAb 14G8, which is a SEB-specific MAb that is not protective on its own but possibly enhances the efficacy of other SEB-specific MAbs by facilitating uptake and clearance through cross-linking of Fcγ receptors (FcγR) (2). Most importantly, our data demonstrate that low doses of the IgG2a isotype prevent death from sepsis after infection with SEB-secreting MRSA. In summary, our findings support the concept that the IgG2a isotype of MAb 20B1 further enhances the efficacy of this protective Ab. Earlier studies have found that MAbs of the IgG2a isotype are more potent than other IgG isotypes in their ability to suppress tumor growth in a number of animal models (32–34). It has also been described that the IgG2a isotype has superior iodine radiolabeling characteristics compared to those of the parent IgG1 Ab (35). Furthermore, in viral infections, MAbs of the IgG2a subclass are more efficacious in providing Ab-mediated protection (36, 37). Our results are also consistent with similar studies involving isotype switch variants to the cryptococcal polysaccharide capsule (38). Those studies demonstrated that a nonprotective IgG3 MAb could be converted to a protective MAb by switching the constant region. Furthermore, the protective efficacy of an IgG1 MAb was increased by isotype switching to IgG2a or IgG2b (38). Differences in efficacy have also been observed in Abs that treat HIV infections, where switch variants of human anti-CD4-binding site MAb F105 differed in their ability to neutralize infection (39, 40).
In a study that generated IgG2a and IgG2b variants of the Bacillus anthracis protective antigen-binding IgG1 MAb 19D9 (41), variants expressed identical variable regions and affinities to the target. Their readout of efficacy similarly determined that neutralization activity was highest by IgG2a, followed by IgG2b and IgG1. In that study, it was found that Ab protection required engagement of FcγR. Engagement of FcγR is not essential for neutralization of SEB, as Fab fragments of MAb 20B1 also prevent death of mice from SEBILS (unpublished data), and a humanized MAb variant of MAb 20B1 containing a mutated constant region that prevents FcγR interaction is also effective (42). Enhanced protection in our SEBILS model correlated with inhibition of proinflammatory cytokines. We hypothesize that these effects are indeed mediated through engagement of the different FcγRs or even different degrees of complement activation (12, 13). We, however, did not pursue experiments in FcγR-deficient mice, as they are not sensitive to SEBILS. It is noteworthy that isotype switching can also result in loss of protective activity due to a change in binding of MAb to its target, as recently described with anticapsular Abs to anthrax (43). These and other findings emphasize that changes distant from the Ag binding site can have an effect on Ab efficacy due to cross-domain interrelationships, whereby constant region changes may result in significant overall structural differences (43, 44).
Most FDA-licensed MAbs are human, humanized, or chimeric IgG1. The first MAb to be licensed by the FDA is a murine anti-CD3 IgG2a that is still being used. Currently, only three murine MAbs (two IgG2a and one IgG1) are licensed. One reason why isotype subclasses have not been consistently explored in preclinical trials may be that most novel therapeutic Abs are either humanized or chimeric. Given the differences between the FcγR of mice and humans, it has been difficult to study the interaction of human Ab Fc regions with FcγRs of mice. Newer studies with mice that express human FcγRs promise to elucidate the contribution of FcγR-mediated pathways to the neutralizing activity of MAbs (45). Our findings highlight unique properties of specific isotypes and underscore the importance of isotype selection in the design of antibodies for therapeutic applications.
MATERIALS AND METHODS
S. aureus toxins.
SEB toxin was purchased from Toxin Technology (Sarasota, FL). SEB toxin was handled in the laboratory in accordance with the Centers for Disease Control and Prevention biosafety regulations. A single colony of clinical MRSA strain 38 (46) streaked on a brain heart infusion (BHI) agar plate was transferred to 50 ml liquid BHI medium grown overnight at 37°C in a shaking incubator. Bacteria were centrifuged at 2.9 × g at 4°C and washed in sterile PBS. Inocula of MRSA were prepared in sterile PBS, and dilutions were plated to verify CFU.
Isolation of isotype-switched variant hybridomas.
The development of murine MAb 20B1 IgG1 and MAb 14G8 IgG1 that bind to SEB has been described earlier (2). MAbs 20B1 IgG2a and IgG2b isotype switch variants were identified using the ELISPOT and isolated by the Sib selection technique (47, 48). Briefly, 5 × 105 cells were treated with lipopolysaccharide (LPS) and/or interleukin 4 (IL-4) and further examined for IgG2a or IgG2b switch variants using ELISPOT after 3 days. A dissecting microscope was used to count spots, and then the median frequencies of switching were calculated. The corresponding well with the highest number of spots was subsequently plated out in a new 96-well plate at progressively lower cell densities. Those plates were then screened again for the frequency of switch variants. Later, cell sorting for specific isotypes was performed by FACS analysis using a FACSAria (BD) and coherent sapphire with a 100-mm quartz nozzle as described elsewhere (41). Soft agar subcloning was performed and random clones were picked and characterized further for specific isotypes (49).
High-scale MAb production and purification.
Large-scale production of 20B1 MAbs (IgG1, IgG2b, and IgG2a) was performed in the CELLine bioreactor CL 1000 (Argos Technologies) according to the manufacturer’s instructions. Briefly, 2.5 × 107 viable hybridoma cells were inoculated into the lower cell compartment followed by ~800 to 900 ml of 5% Dulbecco’s modified Eagle’s medium (DMEM) into the upper chamber. Hybridoma cells were monitored and their supernatants were harvested every week. The concentration of MAb was quantified by ELISA, purified by protein G chromatography (Thermo, Fisher Scientific), and sterilized by filtering through a 0.2-mm-pore-size syringe filter (Thermo Scientific).
Relative affinity and kinetic measurements.
SEB binding analysis to MAbs was established by decreasing titers of MAb in an ELISA. Briefly, ELISAs were performed by coating a 96-well plate with SEB (0.5 µg/ml), followed by the serial dilution of MAb 20B1 (IgG1 or IgG2a or IgG2b) and further detected by alkaline phosphatase-conjugated anti-mouse isotype MAb. These experiments were done in triplicates. Kinetic rate constants to compare binding of different isotypes of MAbs 20B1 to SEB were measured using a Forte Bio BLItz instrument (Forte Bio, Menlo Park, CA). Biotinylated SEB was immobilized on a streptavidin sensor, and MAbs were allowed to bind SEB at different dilutions (0 nM to 100 nM). The final MAb-SEB binding was analyzed using Forte Bio’s analysis software to determine R equilibrium as described by the manufacturer.
T-cell proliferation assay.
Splenocytes (1 × 105 per well) from 6- to 8-week-old female BALB/c mice were seeded in a 96-well plate, with a final volume of 100 µl. Serial dilutions of isotype variants of MAb 20B1 were added starting from 100 µg/ml followed by SEB (25 ng/ml) and incubated for 96 h. Experiments were performed in triplicates. T-cell proliferation was measured using the Via-Light HS cell proliferation kit (Lonza, USA) according to the manufacturer’s instructions. IC50s were determined from concentration-response curve analysis using GraphPad Prism version 6.
Animal experiments.
All animal experiments were carried out with the approval of the Animal Institute Committee (AIC), in accordance with the rules and regulations set forth by the Albert Einstein College of Medicine. Six- to eight-week-old female BALB/c mice were purchased from National Cancer Institute (Bethesda, MD). Protective efficacy of isotype switch variants of MAb 20B1 was tested in vivo in the SEB-induced lethal shock (SEBILS) model as previously described (2). Mice (n = 5) were treated intraperitoneally (i.p.) at indicated doses of isotype variants of MAb 20B1 (50 µg, 100 µg, 250 µg, or 500 µg) 10 min prior to challenge with SEB (20 µg) and d-galactosamine (25 mg). Survival of mice was monitored for 5 days. In another subset of experiments, blood was drawn after 2 h from mice (n = 5) treated with a low dose of MAb 20B1 isotypes (50 µg) followed by challenge with SEB. Sera were analyzed for mouse cytokines using a proinflammatory 7 ultrasensitive kit (Meso Scale Discovery, Gaithersburg, MD) as per the manufacturer’s instructions. Data were further analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc multiple comparison tests using GraphPad Prism 6. All samples were measured in duplicates. SEB clearance from blood, kidney, spleen, and liver was measured at 2 h and 6 h after mice were challenged with SEB and treated with 500 µg of MAb 20B1 isotypes.
Protective efficacy of MAb 20B1 isotypes was further investigated in the S. aureus sepsis model induced by intravenous injection (i.v.) of 5 × 107 CFUs of MRSA strain 38 as previously described (7). Mice were given prophylactic treatment of MAb 20B1 isotypes (300 µg) 2 h prior to the SEB-secreting MRSA infection. Control mice were treated with PBS only.
ACKNOWLEDGMENTS
We thank the hybridoma facility, especially Manxia Fan at AECOM for technical support and Kaushik Dutta for helping in the analysis of BLItz experiments.
This work was supported by the NIH-funded Northeast Biodefense Center, U54-AI057158-Lipkin.
There are no potential conflicts of interest to report.
Footnotes
Citation Varshney AK, Wang X, Aguilar JL, Scharff MD, Fries BC. 2014. Isotype switching increases efficacy of antibody protection against staphylococcal enterotoxin B-induced lethal shock and Staphylococcus aureus sepsis in mice. mBio 5(3):e01007-14. doi:10.1128/mBio.01007-14.
REFERENCES
- 1. Hamad AR, Herman A, Marrack P, Kappler JW. 1994. Monoclonal antibodies defining functional sites on the toxin superantigen staphylococcal enterotoxin B. J. Exp. Med. 180:615–621. 10.1084/jem.180.2.615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Varshney AK, Wang X, Cook E, Dutta K, Scharff MD, Goger MJ, Fries BC. 2011. Generation, characterization, and epitope mapping of neutralizing and protective monoclonal antibodies against staphylococcal enterotoxin B-induced lethal shock. J. Biol. Chem. 286:9737–9747. 10.1074/jbc.M110.212407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. LeClaire RD, Hunt RE, Bavari S. 2002. Protection against bacterial superantigen staphylococcal enterotoxin B by passive vaccination. Infect. Immun. 70:2278–2281. 10.1128/IAI.70.5.2278-2281.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tilahun ME, Rajagopalan G, Shah-Mahoney N, Lawlor RG, Tilahun AY, Xie C, Natarajan K, Margulies DH, Ratner DI, Osborne BA, Goldsby RA. 2010. Potent neutralization of staphylococcal enterotoxin B by synergistic action of chimeric antibodies. Infect. Immun. 78:2801–2811. 10.1128/IAI.01121-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Larkin EA, Stiles BG, Ulrich RG. 2010. Inhibition of toxic shock by human monoclonal antibodies against staphylococcal enterotoxin B. PLoS One 5:e13253. 10.1371/journal.pone.0013253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drozdowski B, Zhou Y, Kline B, Spidel J, Chan YY, Albone E, Turchin H, Chao Q, Henry M, Balogach J, Routhier E, Bavari S, Nicolaides NC, Sass PM, Grasso L. 2010. Generation and characterization of high affinity human monoclonal antibodies that neutralize staphylococcal enterotoxin B. J. Immune Based Ther. Vaccines 8:9. 10.1186/1476-8518-8-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Varshney AK, Wang X, Scharff MD, Macintyre J, Zollner RS, Kovalenko OV, Martinez LR, Byrne FR, Fries BC. 2013. Staphylococcal enterotoxin B-specific monoclonal antibody 20B1 successfully treats diverse Staphylococcus aureus infections. J. Infect. Dis. 208:2058–2066. 10.1093/infdis/jit421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leavy O. 2010. Therapeutic antibodies: past, present and future. Nat. Rev. Immunol. 10:297. 10.1038/nri2763 [DOI] [PubMed] [Google Scholar]
- 9. The IMpact-RSV Study Group. 1998. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 102:531–537 [PubMed] [Google Scholar]
- 10. Goldenberg MM. 2013. Pharmaceutical approval update. PT 38:86–95 [PMC free article] [PubMed] [Google Scholar]
- 11. Ravetch JV, Kinet JP. 1991. Fc receptors. Annu. Rev. Immunol. 9:457–492. 10.1146/annurev.iy.09.040191.002325 [DOI] [PubMed] [Google Scholar]
- 12. Neuberger MS, Rajewsky K. 1981. Activation of mouse complement by monoclonal mouse antibodies. Eur. J. Immunol. 11:1012–1016. 10.1002/eji.1830111212 [DOI] [PubMed] [Google Scholar]
- 13. Gessner JE, Heiken H, Tamm A, Schmidt RE. 1998. The IgG Fc receptor family. Ann. Hematol. 76:231–248. 10.1007/s002770050396 [DOI] [PubMed] [Google Scholar]
- 14. Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. 2005. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity 23:41–51. 10.1016/j.immuni.2005.05.010 [DOI] [PubMed] [Google Scholar]
- 15. Nimmerjahn F, Ravetch JV. 2005. Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science 310:1510–1512. 10.1126/science.1118948 [DOI] [PubMed] [Google Scholar]
- 16. Pirofski LA, Casadevall A. 2008. The damage-response framework of microbial pathogenesis and infectious diseases. Adv. Exp. Med. Biol. 635:135–146. 10.1007/978-0-387-09550-9_11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nimmerjahn F, Ravetch JV. 2007. Fc-receptors as regulators of immunity. Adv. Immunol. 96:179–204. 10.1016/S0065-2776(07)96005-8 [DOI] [PubMed] [Google Scholar]
- 18. Cook E, Wang X, Robiou N, Fries BC. 2007. Measurement of staphylococcal enterotoxin B in serum and culture supernatant with a capture enzyme-linked immunosorbent assay. Clin. Vaccine Immunol. 14:1094–1101. 10.1128/CVI.00183-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rapoport MI, Hodoval LF, Grogan EW, McGann V, Beisel WR. 1966. The influence of specific antibody on the disappearance of staphylococcal enterotoxin B from blood. J. Clin. Invest. 45:1365–1372. 10.1172/JCI105444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vabulas R, Bittlingmaier R, Heeg K, Wagner H, Miethke T. 1996. Rapid clearance of the bacterial superantigen staphylococcal enterotoxin B in vivo. Infect. Immun. 64:4567–4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Langley R, Patel D, Jackson N, Clow F, Fraser JD. 2010. Staphylococcal superantigen super-domains in immune evasion. Crit. Rev. Immunol. 30:149–165. 10.1615/CritRevImmunol.v30.i2.40 [DOI] [PubMed] [Google Scholar]
- 22. Otto M. 2012. MRSA virulence and spread. Cell. Microbiol. 14:1513–1521. 10.1111/j.1462-5822.2012.01832.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foster TJ. 2005. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3:948–958. 10.1038/nrmicro1289 [DOI] [PubMed] [Google Scholar]
- 24. Goldman DL, Casadevall A. 2008. Anthrax-associated shock. Front. Biosci. 13:4009–4014 [DOI] [PubMed] [Google Scholar]
- 25. Pinchuk IV, Beswick EJ, Reyes VE. 2010. Staphylococcal enterotoxins. Toxins 2:2177–2197. 10.3390/toxins2082177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fraser JD. 2011. Clarifying the mechanism of superantigen toxicity. PLoS Biol. 9:e1001145. 10.1371/journal.pbio.1001145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buonpane RA, Churchill HR, Moza B, Sundberg EJ, Peterson ML, Schlievert PM, Kranz DM. 2007. Neutralization of staphylococcal enterotoxin B by soluble, high-affinity receptor antagonists. Nat. Med. 13:725–729. 10.1038/nm1584 [DOI] [PubMed] [Google Scholar]
- 28. Xia T, Liang S, Wang H, Hu S, Sun Y, Yu X, Han J, Li J, Guo S, Dai J, Lou Z, Guo Y. 2014. Structural basis for the neutralization and specificity of staphylococcal enterotoxin B against its MHC class II binding site. MAbs 6:119–129. 10.4161/mabs.27106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karauzum H, Chen G, Abaandou L, Mahmoudieh M, Boroun AR, Shulenin S, Devi VS, Stavale E, Warfield KL, Zeitlin L, Roy CJ, Sidhu SS, Aman MJ. 2012. Synthetic human monoclonal antibodies toward staphylococcal enterotoxin B (SEB) protective against toxic shock syndrome. J. Biol. Chem. 287:25203–25215. 10.1074/jbc.M112.364075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Whitmore AC, Prowse DM, Haughton G, Arnold LW. 1991. Ig isotype switching in B lymphocytes. The effect of T cell-derived interleukins, cytokines, cholera toxin, and antigen on isotype switch frequency of a cloned B cell lymphoma. Int. Immunol. 3:95–103. 10.1093/intimm/3.1.95 [DOI] [PubMed] [Google Scholar]
- 31. Correia IR. 2010. Stability of IgG isotypes in serum. MAbs 2:221-232. 10.4161/mabs.2.3.11788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bernstein ID, Tam MR, Nowinski RC. 1980. Mouse leukemia: therapy with monoclonal antibodies against a thymus differentiation antigen. Science 207:68–71. 10.1126/science.6965328 [DOI] [PubMed] [Google Scholar]
- 33. Herlyn D, Koprowski H. 1982. IgG2a monoclonal antibodies inhibit human tumor growth through interaction with effector cells. Proc. Natl. Acad. Sci. U. S. A. 79:4761–4765. 10.1073/pnas.79.15.4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seto M, Takahashi T, Nakamura S, Matsudaira Y, Nishizuka Y. 1983. In vivo antitumor effects of monoclonal antibodies with different immunoglobulin classes. Cancer Res. 43:4768–4773 [PubMed] [Google Scholar]
- 35. Wahl RL, Wissing JR, Kaminski MS. 1989. Isotype switch variant anti-idiotype monoclonal antibodies: comparative radiolabeling and in vitro binding. J. Nucl. Med. 30:227–232 [PubMed] [Google Scholar]
- 36. Baldridge JR, Buchmeier MJ. 1992. Mechanisms of antibody-mediated protection against lymphocytic choriomeningitis virus infection: mother-to-baby transfer of humoral protection. J. Virol. 66:4252–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Markine-Goriaynoff D, Coutelier JP. 2002. Increased efficacy of the immunoglobulin G2a subclass in antibody-mediated protection against lactate dehydrogenase-elevating virus-induced polioencephalomyelitis revealed with switch mutants. J. Virol. 76:432–435. 10.1128/JVI.76.1.432-435.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuan RR, Spira G, Oh J, Paizi M, Casadevall A, Scharff MD. 1998. Isotype switching increases efficacy of antibody protection against Cryptococcus neoformans infection in mice. Infect. Immun. 66:1057–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu F, Bergami PL, Duval M, Kuhrt D, Posner M, Cavacini L. 2003. Expression and functional activity of isotype and subclass switched human monoclonal antibody reactive with the base of the V3 loop of HIV-1 gp120. AIDS Res. Hum. Retroviruses 19:597–607. 10.1089/088922203322230969 [DOI] [PubMed] [Google Scholar]
- 40. Cavacini LA, Kuhrt D, Duval M, Mayer K, Posner MR. 2003. Binding and neutralization activity of human IgG1 and IgG3 from serum of HIV-infected individuals. AIDS Res. Hum. Retroviruses 19:785–792. 10.1089/088922203769232584 [DOI] [PubMed] [Google Scholar]
- 41. Abboud N, Chow SK, Saylor C, Janda A, Ravetch JV, Scharff MD, Casadevall A. 2010. A requirement for FcγR in antibody-mediated bacterial toxin neutralization. J. Exp. Med. 207:2395–2405. 10.1084/jem.20100995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Varshney AK, Wang X, Macintyre J, Zollner RS, Kelleher K, Kovalenko OV, Pechuan X, Byrne FR, Fries BC. 5 May 2014. Humanized staphylococcal enterotoxin B (SEB)-specific monoclonal antibodies protect from SEB intoxication and Staphylococcus aureus infections alone or as adjunctive therapy with vancomycin. J. Infect. Dis. 10.1093/infdis/jiu198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hovenden M, Hubbard MA, Aucoin DP, Thorkildson P, Reed DE, Welch WH, Lyons CR, Lovchik JA, Kozel TR. 2013. IgG subclass and heavy chain domains contribute to binding and protection by MAbs to the poly gamma-d-glutamic acid capsular antigen of Bacillus anthracis. PLoS Pathog. 9:e1003306. 10.1371/journal.ppat.1003306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hubbard MA, Thorkildson P, Kozel TR, AuCoin DP. 2013. Constant domains influence binding of mouse-human chimeric antibodies to the capsular polypeptide of Bacillus anthracis. Virulence 4:483-488. 10.4161/viru.25711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bournazos S, Chow SK, Abboud N, Casadevall A, Ravetch JV. 2014. Human IgG Fc domain engineering enhances antitoxin neutralizing antibody activity. J. Clin. Invest. 124:725-729. 10.1172/JCI72676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Varshney AK, Mediavilla JR, Robiou N, Guh A, Wang X, Gialanella P, Levi MH, Kreiswirth BN, Fries BC. 2009. Diverse enterotoxin gene profiles among clonal complexes of Staphylococcus aureus isolates from the Bronx, New York. Appl. Environ. Microbiol. 75:6839–6849. 10.1128/AEM.00272-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Greene G, Hodous J, Dintzis RZ, Dintzis HM. 1990. Modification, optimization and simplification of the spot ELISA technique for the enumeration of cells secreting anti-hapten antibodies. J. Immunol. Methods 129:187–197. 10.1016/0022-1759(90)90438-2 [DOI] [PubMed] [Google Scholar]
- 48. Spira G, Scharff MD. 1992. Identification of rare immunoglobulin switch variants using the ELISA spot assay. J. Immunol. Methods 148:121–129. 10.1016/0022-1759(92)90165-P [DOI] [PubMed] [Google Scholar]
- 49. Zhang W, Bardwell PD, Woo CJ, Poltoratsky V, Scharff MD, Martin A. 2001. Clonal instability of V region hypermutation in the Ramos Burkitt’s lymphoma cell line. Int. Immunol. 13:1175–1184. 10.1093/intimm/13.9.1175 [DOI] [PubMed] [Google Scholar]