

ABSTRACT
Pseudomonas aeruginosa is a Gram-negative pathogen commonly associated with nosocomial infections such as hospital-acquired pneumonia. It uses a type III secretion system to deliver effector proteins directly into the cytosol of host cells. Type III secretion in P. aeruginosa has been linked to severe disease and worse clinical outcomes in animal and human studies. The majority of P. aeruginosa strains secrete ExoS, a bifunctional toxin with GTPase-activating protein and ADP-ribosyltransferase activities. Numerous in vitro studies have investigated the targets and cellular effects of ExoS, linking both its enzymatic activities with inhibition of bacterial internalization. However, little is known about how this toxin facilitates the progression of infection in vivo. In this study, we used a mouse model to investigate the role of ExoS in inhibiting phagocytosis during pneumonia. We first confirmed previous findings that the ADP-ribosyltransferase activity of ExoS, but not the GTPase-activating protein activity, was responsible for bacterial persistence and decreased host survival in this model. We then used two distinct assays to demonstrate that ExoS inhibited phagocytosis during pneumonia. In contrast to the findings of several in vitro studies, this in vivo inhibition was also dependent on the ADP-ribosyltransferase activity, but not the GTPase-activating protein activity, of ExoS. These results demonstrate for the first time the antiphagocytic function of ExoS in the context of an actual infection and indicate that blocking the ADP-ribosyltransferase activity of ExoS may have potential therapeutic benefit.
IMPORTANCE
Pseudomonas aeruginosa is a major cause of hospital-acquired infections. To cause severe disease, this bacterium uses a type III secretion system that delivers four effector proteins, ExoS, ExoT, ExoU, and ExoY, into host cells. The majority of P. aeruginosa strains secrete ExoS, a bifunctional toxin with GTPase-activating protein and ADP-ribosyltransferase activities. In cell culture models, both enzymatic activities have been associated with decreased bacterial internalization. However, our study is the first to examine a role for ExoS in blocking phagocytosis in an animal model. We report that ExoS does inhibit phagocytosis during pneumonia. The ADP-ribosyltransferase activity, but not the GTPase-activating protein activity, of ExoS is necessary for this effect. Our findings highlight the ability of P. aeruginosa to manipulate the inflammatory response during pneumonia to facilitate bacterial survival.
INTRODUCTION
Pseudomonas aeruginosa is an important nosocomial pathogen, causing approximately 51,000 infections per year in the United States (1). Hospital-acquired pneumonia makes up a significant portion of these infections, with P. aeruginosa reported to cause 15 to 20% of these infections (2–4). This pathogen produces a spectrum of virulence factors, including a type III secretion system that makes a major contribution to its ability to cause disease. This system forms a needle-like apparatus that delivers toxic effector proteins directly from the bacterium into the cytosol of the host cell (5). An important role for the type III secretion system in P. aeruginosa infections is supported by results from studies of animal models (6–8) and by epidemiological studies of human patients (9, 10).
The type III secretion system of P. aeruginosa secretes four known effectors: ExoS, ExoT, ExoU, and ExoY. Approximately two-thirds of clinical strains contain the gene encoding ExoS (11). This effector protein is a bifunctional toxin, with distinct domains for GTPase-activating protein (GAP) and ADP-ribosyltransferase (ADPRT) activities. The GAP domain targets Rho GTPases (12), whereas the ADPRT domain modifies a variety of host cell substrates, including Ras, ezrin/radixin/moesin (ERM), Rab5, and cyclophilin A (13–16). These activities result in disruption of the actin cytoskeleton and inhibition of bacterial internalization by eukaryotic cells in cell culture (17–20), induction of lymphocyte proliferation (21), and apoptotic death of epithelial cells, fibroblasts, and neutrophils (22–24). Due to the difficulties in examining bacterial toxin activities in vivo, studies of the mechanism of action of ExoS have primarily relied on in vitro assays. As a result, the enzymatic and cellular activities of ExoS have been well defined, but it is not known whether these same effects occur in animal models of infection (7). In other words, the pathophysiological role of ExoS during an actual infection remains unclear.
In the present study, we examined the pathogenic role of ExoS during pneumonia in a mouse model. We characterized bacterial persistence and internalization of P. aeruginosa strains secreting wild-type ExoS or enzymatic variants of ExoS lacking GAP or ADPRT activity. Our findings indicate that the ADPRT domain of ExoS plays a major role in bacterial persistence and virulence during infection by preventing bacterial internalization within lung phagocytes. In contrast, disruption of GAP activity had at most a limited effect on bacterial internalization and virulence. These observations support a model whereby the ADP-ribosylation of host proteins by ExoS blocks phagocytosis of P. aeruginosa during acute pneumonia, which in turn leads to bacterial persistence and more-severe infection. Interventions that inhibit the ADPRT activity of ExoS or enhance the phagocytic activity of neutrophils and macrophages would therefore be expected to have potential therapeutic benefit in the treatment of P. aeruginosa pneumonia.
RESULTS
An ExoS-secreting strain of P. aeruginosa persists at higher numbers than a nonsecreting strain in the lungs of a mouse model of pneumonia.
We previously showed that ExoS contributes to the overall virulence of P. aeruginosa in a mouse model of pneumonia and that the ADPRT domain is responsible for most of this effect (7). However, the kinetics of bacterial persistence within the lungs during the early hours of pneumonia have not been characterized. We infected mice with P. aeruginosa strain PA99S, which secretes ExoS, or strain PA99null, which is identical to PA99S except that it harbors a deletion in the exoS gene. At 3, 12, 18, and 24 h postinfection, the lungs were extracted, and lung homogenates were plated for enumeration of bacteria. Strain PA99S persisted in the lungs at higher numbers than strain PA99null did throughout the first 24 h of infection (Fig. 1). Bacterial numbers were the same for both strains at 3 h postinfection, indicating that initial colonization of the lungs is not dependent on ExoS. Thus, ExoS prevents P. aeruginosa from being cleared from the lungs during early pneumonia.
FIG 1 .
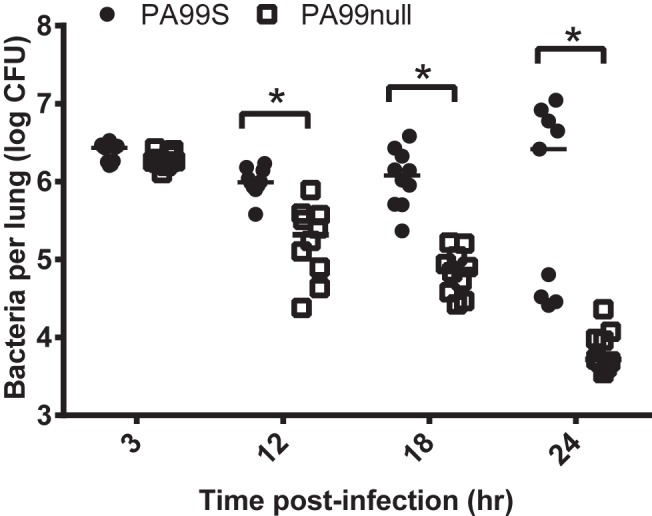
ExoS enhances persistence of P. aeruginosa in the lungs of a mouse model of pneumonia. Mice were inoculated with either P. aeruginosa PA99S or PA99null, and bacterial numbers in the whole lungs of individual mice were measured at various times. The bacterial loads in mice infected with strain PA99S were significantly higher than those in mice infected with strain PA99null at 12, 18, and 24 h postinfection (the values that were significantly different [P < 0.001] are indicated by a bracket and asterisk). Each symbol represents the bacterial load in the whole lungs from an individual mouse (at least nine mice were tested for each strain). Bars represent medians for the groups of mice. Each experiment was performed twice with cumulative results shown.
To examine the contributions of the GAP and ADPRT activities of ExoS to bacterial persistence, PA99null strains complemented with exoS alleles that expressed wild-type ExoS, ExoS deficient in GAP activity (GAP− ExoS), or ExoS deficient in ADPRT activity (ADPRT− ExoS) were used. An allele expressing wild-type ExoS was used to generate strain PA99null+S. Likewise, alleles that expressed an ExoS variant with an R146A substitution, which disrupts GAP activity (12), and an ExoS variant with both E379A and E381A substitutions, which disrupt ADPRT activity (25), were used to generate PA99null+S(R146A) and PA99null+S(E379A/E381A), respectively. In each of these strains, the exoS allele was inserted at a neutral chromosomal site and was expressed from its endogenous promoter. Secretion of ExoS by each complemented strain was confirmed by immunoblot analysis of culture supernatants (see Fig. S1 in the supplemental material). These complemented strains were used to assess the relative contributions of the GAP and ADPRT activities of ExoS to virulence.
An intranasal aspiration mouse model was used to examine the persistence of each of the ExoS-secreting strains during acute pneumonia. Since the majority of P. aeruginosa bacteria are found in the airways and alveoli during pneumonia (26), we collected bronchoalveolar lavage fluid (BALF) from infected mice at 18 and 24 h postinfection and plated it for enumeration of viable bacteria. Strain PA99null+S(E379A/E381A), which secretes ExoS deficient in ADPRT activity, was cleared from the lungs, similar to strain PA99null at both 18 and 24 h postinfection (see Fig. S2A and S2B in the supplemental material). In contrast, strain PA99null+S(R146A), which secretes ExoS deficient in GAP activity, persisted, similar to strain PA99null+S. Therefore, the GAP domain of ExoS did not contribute significantly to the persistence of P. aeruginosa in the lungs; nearly all of the increased bacterial persistence associated with ExoS was attributable to the ADPRT domain.
We next sought to confirm previous reports that the ADPRT activity of ExoS is primarily responsible for the decreased host survival associated with ExoS-secreting strains of P. aeruginosa. We first verified that the complemented strain PA99null+S behaved similarly to strain PA99S by showing that both strains caused similar survival curves in the mouse model of pneumonia (see Fig. S3A in the supplemental material). We next examined the effect of the GAP and ADPRT activities on survival in the mouse model of pneumonia. As demonstrated previously (7), disruption of the GAP activity of ExoS did not appreciably enhance survival of mice (Fig. S3B). In contrast, disruption of the ADPRT activity did result in a significant improvement in survival. These observations confirm previous reports that the ADPRT activity is responsible for most or all of the virulence associated with ExoS.
Secretion of ExoS causes a heightened inflammatory response in the lungs.
One possible explanation for how ExoS contributes to bacterial persistence and virulence is that it prevents timely recruitment of phagocytes to the lungs. To test this hypothesis, we characterized the cellular inflammatory response within the lungs of mice infected with strain PA99S or PA99null at 3, 12, 18, and 24 h postinfection. The lungs were extracted and minced, and leukocytes were quantified by flow cytometry. ExoS was associated with a heightened cellular inflammatory response within the lungs. Increased numbers of leukocytes were observed in the lungs at 12, 18, and 24 h postinfection (Fig. 2A). The majority of these leukocytes were neutrophils and monocytes (Fig. 2B and C). The numbers of resident macrophages remained the same for the two infections throughout the entire time course. Histological examination also demonstrated a greater inflammatory infiltrate within the lungs of PA99S-infected mice compared to PA99null-infected mice (see Fig. S4 in the supplemental material). These results show that secretion of ExoS is not associated with a paucity of inflammatory cells but rather that bacteria persist despite the robust recruitment of phagocytic cells into the lungs.
FIG 2 .
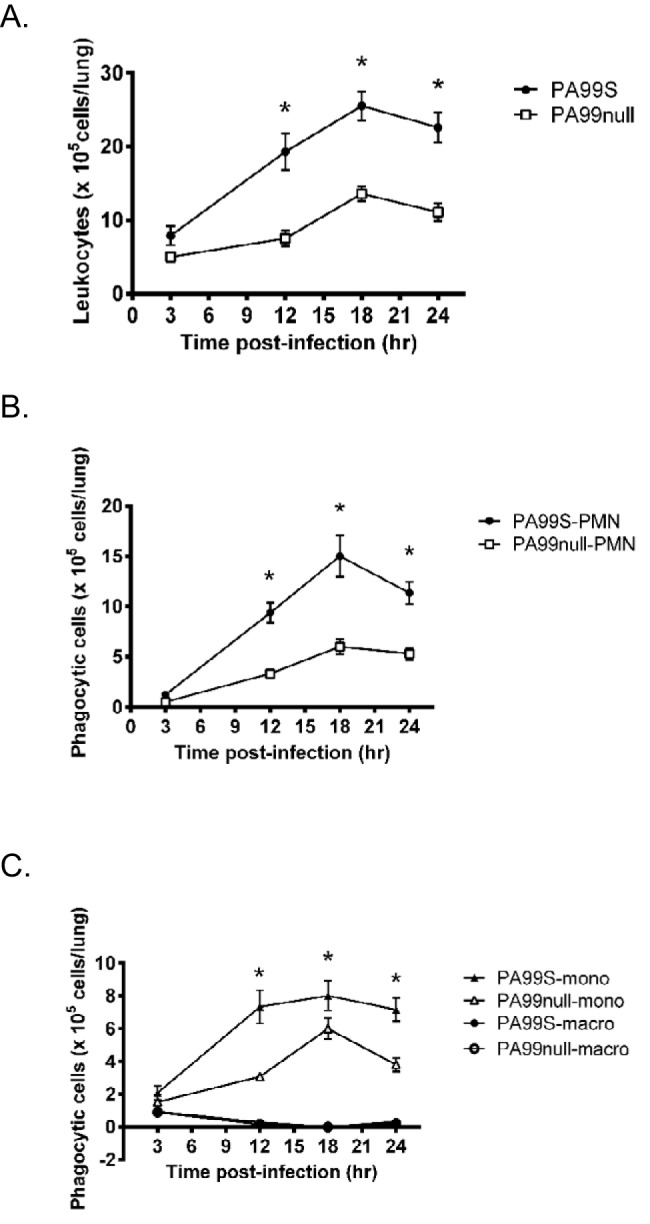
Secretion of ExoS is associated with a robust inflammatory response in the lungs. (A to C) The average number of CD45+ leukocytes (A) or leukocyte subsets (B and C) from the lungs of infected mice were quantified by flow cytometry. A minimum of 13 mice pooled from three separate experiments were used for each condition. At 12, 18, and 24 h postinfection, neutrophil (polymorphonuclear leukocyte [PMN]) and monocyte (mono) numbers, but not macrophage (macro) numbers, were significantly higher with P. aeruginosa PA99S infections than with P. aeruginosa PA99null infections (*, P < 0.01). Error bars represent standard errors of the means.
ExoS, specifically the ADPRT domain, inhibits neutrophil phagocytic uptake in vitro.
Studies using cell culture-based systems have shown that ExoS prevents bacterial internalization by various mammalian cells (12, 20, 27). However, the effect of ExoS on neutrophils has been relatively unexplored. Since the majority of infiltrating cells during pneumonia were neutrophils, we examined whether ExoS inhibited neutrophil phagocytic function in vitro. We assessed the ability of murine bone marrow neutrophils to take up fluorescently labeled latex beads after infection with ExoS-secreting strains of P. aeruginosa. Following infection with strain PA99null or ADPRT-deficient PA99null+S(E379A/E381A) strain, a higher proportion of neutrophils contained intracellular fluorescent beads than after infection with PA99null+S or GAP-deficient PA99null+S(R146A) (Fig. 3A). This demonstrated that ExoS, specifically the ADPRT domain, inhibits internalization by neutrophils.
FIG 3 .
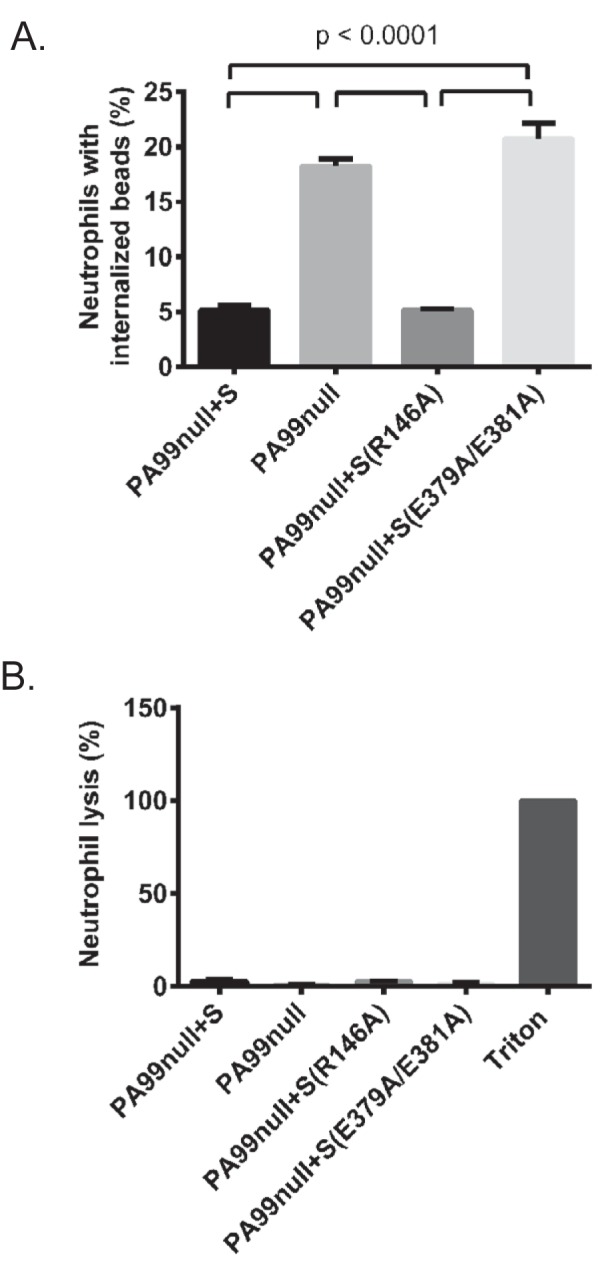
ExoS, specifically the ADPRT domain, inhibits neutrophil phagocytic uptake in vitro. (A) Murine bone marrow neutrophils were infected for 30 min with strain PA99S, PA99null, PA99null+S(R146A), or PA99null+S(E379A/E381A). Following infection, bacteria were removed, and cells were incubated with fluorescently labeled latex beads for 30 min. Phagocytosis of beads was assessed by flow cytometry. Data represent the average percentage of neutrophils containing intracellular beads (three mice for each strain). Error bars represent standard errors of the means. (B) Following 30 min of infection, LDH release was assessed as a measure of neutrophil lysis. Cell lysis was normalized using a Triton X-100-lysed control. Background lysis was measured in uninfected neutrophils incubated without bacteria and subtracted from all experimental values. Data represent the average percentage of neutrophil lysis (three mice for each strain). Error bars represent standard errors of the means.
It has been shown that the ADPRT domain has a cytotoxic effect on mammalian cells (23, 28). Another explanation for the decreased internalization of latex beads observed in the presence of intact ADPRT domain is that the ADPRT domain caused neutrophil lysis and subsequent release of internalized beads back into the medium. To exclude neutrophil lysis as a mechanism accounting for decreased bead internalization, we quantified lactate dehydrogenase (LDH) release from bone marrow neutrophils after infection with ExoS-secreting strains. In comparison to uninfected neutrophils, we found no LDH release at 30 min postinfection (Fig. 3B), despite a difference in ExoS-mediated phagocytic uptake (Fig. 3A). Therefore, the decreased numbers of internalized beads associated with infection by ExoS+ bacteria were not the result of neutrophil lysis. Rather, ExoS inhibited phagocytic uptake of beads by neutrophils in an ADPRT-dependent but GAP-independent manner.
A fluorescence-based internalization assay indicates that ExoS inhibits phagocytic uptake in vitro and in vivo.
To examine the impact of ExoS on phagocytosis in vivo, we developed a fluorescence-based internalization assay. In this assay, intracellular and extracellular bacteria are distinguished by differential fluorescent-antibody staining (29). Of note, morphologically intact internalized bacteria are quantified whether they are viable or dead. Unpermeabilized cells are exposed to antibodies against pseudomonal bacteria followed by green fluorescent secondary antibodies, which stain extracellular bacteria. Cells are then washed, permeabilized, and reexposed to the antibodies against pseudomonal bacteria, but this time a red fluorescent secondary antibody is used, which results in both intracellular and extracellular bacteria fluorescing red. The net result is that intracellular bacteria fluoresce red, whereas extracellular bacteria fluoresce red and green. We first tested this technique in vitro by using J774 macrophage-like cells infected for 3 h with PA99S or PA99null bacteria (Fig. 4A). Fewer PA99S bacteria than PA99null bacteria were observed inside J774 cells (Fig. 4B). This validated our fluorescence-based approach and further supported previous data showing that ExoS has an antiphagocytic phenotype in vitro.
FIG 4 .
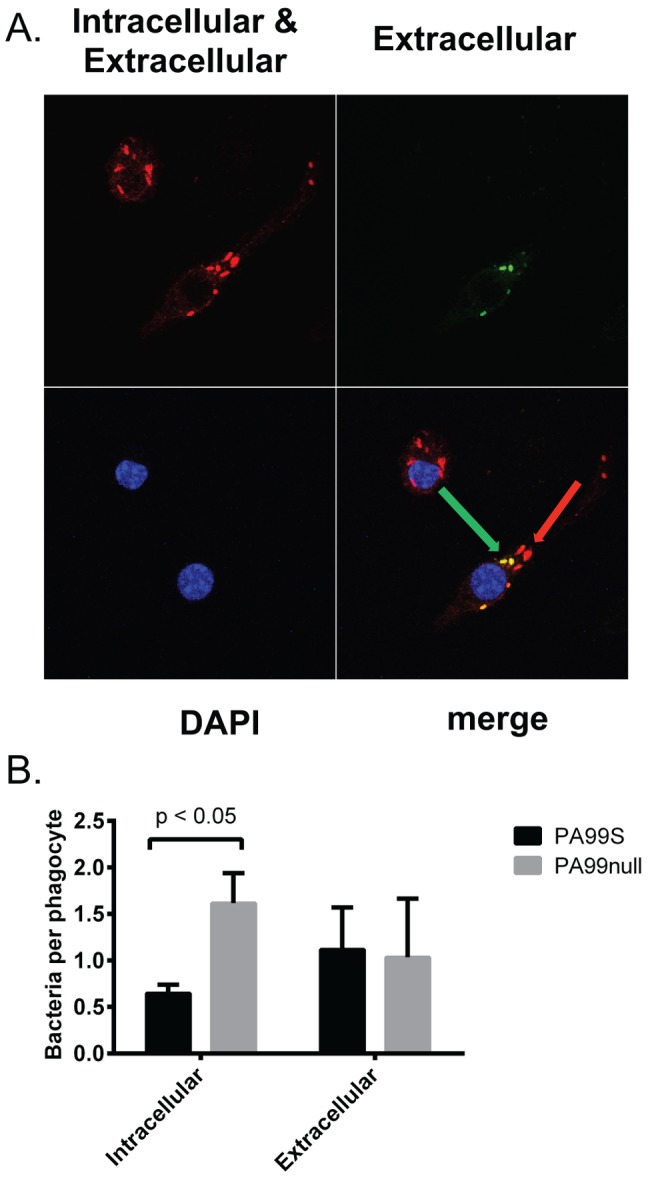
ExoS contributes to decreased bacterial uptake by phagocytes in vitro. (A) J774 macrophage-like cells were infected for 3 h with strain PA99S or PA99null and stained differentially to distinguish intracellular bacteria (red; red arrow) from extracellular bacteria (red and green; green arrow). DAPI, 4′,6′-diamidino-2-phenylindole. (B) Average numbers of intracellular and extracellular bacteria per phagocyte were counted (300 phagocytes pooled from four separate experiments). Error bars represent standard errors of the means between four separate experiments.
We next applied the fluorescence approach to phagocytes from mice with pneumonia. Leukocytes (CD45+ cells) were isolated from the lungs of mice previously infected with strain PA99null+S, PA99null, PA99null+S(R146A), or PA99null+S(E379A/E381A) for 24 h. The cells were stained with the fluorescent antibodies and quantified by flow cytometry. Cells with at least one extracellular bacterium (red and green) or intracellular bacterium (red) were categorized into the respective group. A higher proportion of leukocytes isolated from PA99null-infected mice contained intracellular bacteria than leukocytes from PA99null+S-infected mice, although this difference did not reach statistical significance (P = 0.17; Fig. 5A). Furthermore, the phenotype of the ADPRT-deficient PA99null+S(E379A/E381A) strain was similar to that of the PA99null strain, suggesting that the ADPRT activity of ExoS was responsible for inhibiting phagocytosis. Consistent with this conclusion, phagocytes from mice infected with the GAP-defective mutant PA99null+S(R146A) had similar numbers of intracellular bacteria as phagocytes from mice infected with strain PA99null+S. One potential problem with this technique is that leukocytes sorted into the extracellular bacteria pool (red+ green+) could have both intracellular and extracellular bacteria, causing an underestimation of the number of leukocytes containing intracellular bacteria. To determine whether this was the case, 20 leukocytes from the sorted population associated with each strain were examined by confocal microscopy. Visualization of cells and bacteria by microscopy confirmed that cell sorting had indeed appropriately binned the leukocyte populations with respect to associated intracellular and extracellular bacteria (Fig. 5B). Approximately 25% of the leukocytes from the extracellular bacterial population contained both extracellular and intracellular bacteria, but this proportion was independent of the ExoS strain used for infection (data not shown) and therefore did not affect the differences observed in Fig. 5A. Since cell sorting also could not distinguish leukocytes with one bacterium or multiple associated bacteria, we next visually examined the pools of sorted cells to manually determine the number of bacteria per leukocyte. On average, leukocytes sorted into the intracellular (red+ green−) population had 3 bacteria per cell (see Fig. S5A in the supplemental material), and leukocytes sorted into the extracellular (red+ green+) bacterial population had two adherent bacteria per cell (Fig. S5B). The average number of bacteria associated with each leukocyte did not vary with ExoS strains. Together, these results suggest that ExoS inhibits internalization by phagocytes during pneumonia through an ADPRT-dependent mechanism.
FIG 5 .
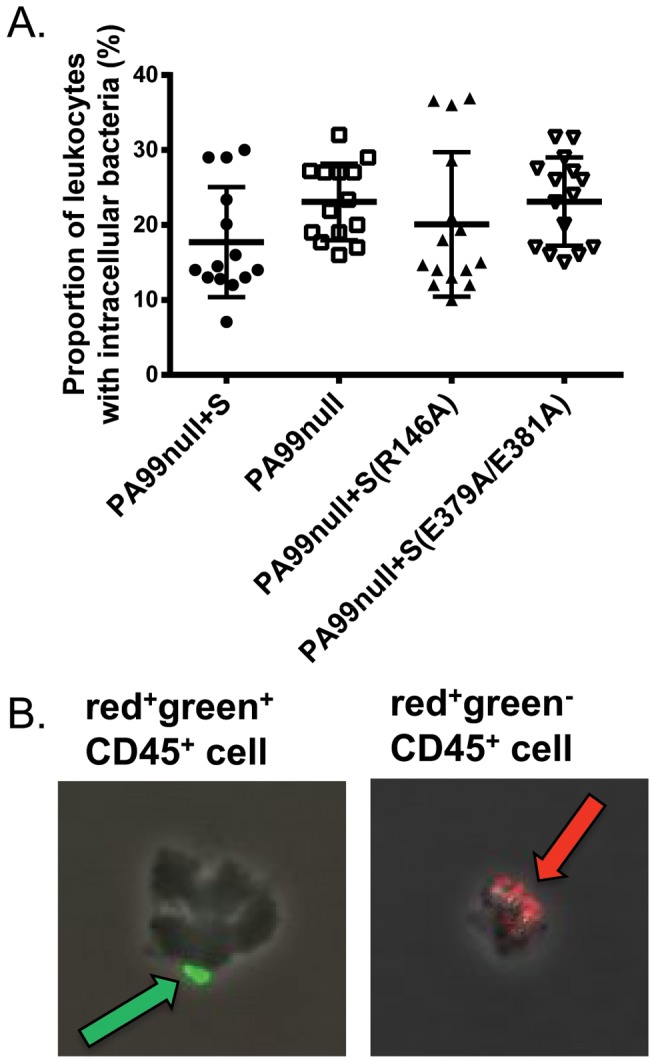
A fluorescent-antibody internalization assay indicates that ExoS inhibits phagocytosis through an ADPRT-dependent mechanism. (A) Mice were infected with strain PA99null+S, PA99null, PA99null+S(R146A), or PA99null+S(E379A/E381A) for 24 h. Leukocytes were isolated from whole lungs and analyzed by flow cytometry to identify those with intracellular bacteria (CD45+ red+ green−). Each symbol represents the value for an individual mouse (15 mice per strain). Black bars indicate the medians for the groups of mice. The confidence intervals (CIs) of the medians (confidence levels shown in the parentheses) are as follows: 12.8 to 29.0 (98.71) for strain PA99null+S, 17.7 to 27.2 (98.71) for PA99null, 13.0 to 28.6 (96.48) for PA99null+S(R146A), and 17.0 to 27.5 (96.48) for PA99null+S(E379A/E381A). P = 0.05 (overall ANOVA). (B) Intracellular/extracellular bacteria were confirmed by viewing cell populations with a fluorescence microscope. The green arrow indicates an extracellular bacterium associated with a CD45+ cell sorted in the red+ green+ cell population; the red arrow indicates intracellular bacteria associated with a CD45+ cell sorted in the red+ green− cell population.
Confirmation that ExoS inhibits phagocytic uptake in mice using an amikacin protection internalization assay.
Because the results of the fluorescence-based internalization assay showed only a trend toward less internalization of an ExoS-secreting strain, we used an amikacin protection internalization assay to additionally confirm this phenotype. In this method, BALF samples were collected from infected mice and split into two fractions. The murine cells in the first fraction were lysed immediately with saponin, and the cell lysate plus BALF was plated to enumerate total viable bacteria (intracellular and extracellular) within the airways. Phagocytic cells were isolated from the second fraction by centrifugation and incubated with amikacin to kill any extracellular bacteria. Phagocytic cells were then washed, lysed, and plated for enumeration of viable intracellular bacteria. At early time points, no difference in the percentage of internalized bacteria was observed between the PA99S and PA99null strains (Fig. 6A). At 18 and 24 h postinfection, however, a lower proportion of ExoS-secreting bacteria was observed within mouse phagocytes from the lavage fluid in comparison to a nonsecreting strain (Fig. 6A). Additionally, significantly more PA99null+S(E379A/E381A) bacteria (ADPRT−) were inside phagocytes than PA99null+S(R146A) bacteria (GAP−) at 24 h postinfection (Fig. 6B). These results confirmed that ExoS inhibits phagocytosis during pneumonia and that the ADPRT domain of ExoS is primarily responsible for this phenotype.
FIG 6 .
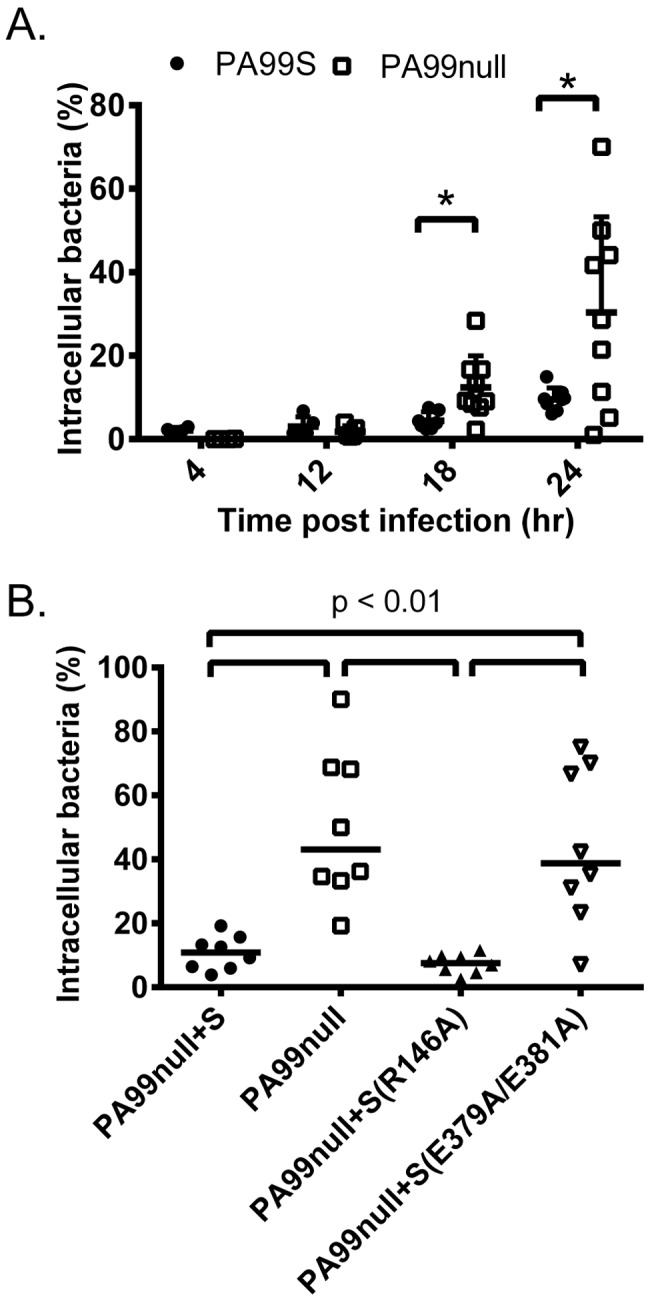
An amikacin protection internalization assay indicates that ExoS inhibits bacterial uptake by phagocytes in vivo. (A) Mice were infected with strain PA99S or PA99null for 4, 12, 18, and 24 h. The percentage of intracellular bacteria was calculated by plating total BALF viable bacteria and viable bacteria within phagocytes after incubation with amikacin for 1 h. Each symbol represents the value for an individual mouse (at least four mice for each strain at each time point) (*, P < 0.05). Error bars represent the standard errors of the mean. (B) The ADPRT domain of ExoS contributes to inhibition of phagocytosis in vivo. Mice were infected with strain PA99null+S, PA99null, PA99null+S(R146A), or PA99null+S(E379A/E381A) for 24 h. The percentage of intracellular bacteria was calculated by plating total BALF viable bacteria and viable bacteria within BALF phagocytes after incubation with amikacin for 1 h. Each symbol represents the value for an individual mouse (eight mice for each strain). Black bars represent medians for the groups of mice. The confidence intervals (CIs) of the medians (confidence levels shown in the parentheses) are as follows: 3.9 to 19.19 (99.22) for strain PA99null+S, 19.32 to 90.0 (99.22) for PA99null, 2.52 to 11.58 (99.22) for PA99null+S(R146A), and 7.051 to 75.0 (99.22) for PA99null+S(E379A/E381A).
DISCUSSION
ExoS is a major virulence determinant of P. aeruginosa, but how it functions during infections to cause severe disease remains unclear. In vitro assays had suggested a role for ExoS in inhibiting phagocytosis of P. aeruginosa. In the present study, we show that ExoS does indeed limit the ability of phagocytes to internalize P. aeruginosa during pneumonia. In contrast to in vitro results, which had suggested that both the GAP and ADPRT activities of ExoS contribute to inhibition of phagocytosis (18–20), we did not observe an appreciable role for the GAP activity in this inhibition. Rather, the ADPRT domain of ExoS was the main contributor in preventing phagocytic uptake of bacteria.
Our results indicate that the in vivo antiphagocytic activity of ExoS is attributable to its ADPRT domain. The ADPRT domain of ExoS has a wide range of targets, including Ras, ezrin/radixin/moesin (ERM) proteins, and Rab5. ExoS ADP-ribosylates Ras, a small GTPase involved in host cell actin rearrangement, at Arg41 (30). This modification results in inhibition of Ras by locking it in its inactive GDP-bound state, which disrupts downstream interactions with Raf1 and prevents further signaling for actin polymerization (30). ADP-ribosylation of the host ERM protein moesin at Arg553, Arg560, and Arg563 prevents its phosphorylation (31). This in turn may disrupt ERM interaction with Rho signaling by preventing the exchange of Rho-GDI (guanine nucleotide dissociation inhibitor) to Rho-GEF (guanine nucleotide exchange factor), consequently causing RhoGTPases to remain in the inactive GDP-bound state. Additionally, ADP-ribosylation of ERM proteins may disrupt the interactions of focal adhesions with the host cell actin cytoskeleton and block phagocytosis by this mechanism. Recently, it was reported that ADP-ribosylation of Rab5, which is known to play a role in phagosome maturation (32), caused an inhibition of bacterial uptake into J774 macrophage-like cells only minutes after infection (16). ExoS modification of Rab5 disrupted its interaction with downstream early endosome antigen 1 (EEA1) and inhibited endosome recycling (33). In this manner, ExoS may interfere with phagocytosis by targeting multiple distinct signaling pathways, including Ras- and ERM-mediated cytoskeletal rearrangements necessary for bacterial engulfment and Rab5-mediated phagosome maturation.
Since the GAP domain of ExoS targets the small GTPases Rho, Rac, and CDC42, it was thought to block bacterial uptake by host cells. The ExoS GAP domain has functional and structural similarity to YopE of Yersinia spp., which inhibits phagocytosis (34). Structural analysis of the GAP domain bound to Rac1 illustrated that ExoS uses an arginine finger at residue Arg146 to lock Rac1 into its inactive GDP-bound state (35). This in turn disrupts Rac signaling and the actin cytoskeleton (27, 36), leading to increased cell rounding and decreased bacterial internalization in vitro (18, 20). Surprisingly, we found that inactivation of the ExoS GAP domain did not lead to an appreciable difference in bacterial uptake by neutrophils in vitro or by phagocytic cells during pneumonia. This raises the question of what role if any is played by the GAP domain during infection. It is conceivable that the GAP and ADPRT activities work synergistically and that the GAP activity manifests an antiphagocytic phenotype only in the presence of an active ADPRT domain. Alternatively, the GAP domain may be important in an aspect of pathogenicity other than phagocytosis. However, the fact that the GAP− mutant had internalization and virulence phenotypes similar to those of wild-type ExoS argues against these interpretations. Perhaps more likely is that GAP activity does not play an appreciable role in mammalian infections. Rather, the GAP activity of ExoS may have evolved to exert an effect against other organisms that P. aeruginosa encounters in the environment, such as amoebae (37).
As we have mentioned, some (17, 27) but not all (16) previous in vitro studies had suggested a role for the ExoS GAP domain in preventing bacterial internalization and therefore in pathogenesis. Our results, like those of Rucks and colleagues (38), suggest that some of this variability occurs because ExoS phenotypes are cell type specific. Since neutrophils are the predominant immune cell present in the lungs during in vivo infection, we performed experiments examining the effect of ExoS on primary neutrophils in vitro. We confirmed that it is the ADPRT domain of ExoS, not the GAP domain, that hinders the phagocytosis of P. aeruginosa bacteria by neutrophils (Fig. 3A). Our results underscore the importance of studying virulence factor functions in the context of animal models or with cell lines that best represent infection conditions.
We used two different in vivo methods to investigate the effects of ExoS on phagocytosis: an amikacin protection assay and a fluorescence-based flow cytometric assay. While the fluorescence-based assay did not show significant differences between strains, there was a trend in the data similar to that of the amikacin assay, namely, that ExoS, specifically its ADPRT domain, inhibits phagocytosis. These assays differed in experimental readouts, allowing us to address slightly different aspects of bacterial internalization. The fluorescence-based assay quantified the proportion of lung leukocytes containing intracellular bacteria. The amikacin protection assay quantified the proportion of bacteria within lung phagocytes. Furthermore, the amikacin assay measured only viable intracellular bacteria, whereas the fluorescence assay measured all morphologically intact intracellular bacteria regardless of viability. The fact that two methodologically distinct assays gave similar results strengthens our conclusions regarding ExoS and phagocytosis.
Previously, the ADPRT domain of ExoS was shown to promote survival of intracellular P. aeruginosa bacteria within corneal epithelial cells (39). Bacterial cells formed plasma membrane bleb structures that allowed for bacterial replication and protection from non-cell-permeable antibiotic treatment. These bleb structures, which contained actively swimming bacteria, were capable of detaching and traveling away from the host cell surface (39). In these studies, ExoS facilitated intracellular survival of P. aeruginosa, an observation that at first glance appears inconsistent with our results. One possibility is that ExoS prevents overall bacterial internalization but promotes the viability of the few bacteria that are successfully internalized. Perhaps more likely is the possibility that bleb formation is specific to epithelial cells or even corneal epithelial cells and does not occur in leukocytes. Of note, we did not observe bleb structures within the leukocytes in our assays (data not shown).
ExoS-mediated inhibition of phagocytosis was associated with decreased survival in the mouse model of pneumonia and increased bacterial persistence. While we demonstrate an inhibition in phagocytosis by two different in vivo assays, the <2-fold difference in internalized bacteria in Fig. 5A suggests that other phenotypes of the ADPRT domain may also contribute to its virulence effects. The ADPRT activity of ExoS leads to disruption of the actin cytoskeleton, focal adhesions, and tight junctions (27, 28), which may allow P. aeruginosa penetration through epithelial barriers and dissemination (7). The ExoS ADPRT domain also has cytotoxic activity with characteristics of apoptosis, including chromatin condensation and caspase-3 activation (23). However, other aspects of cell death, such as loss of cell membrane integrity, are more characteristic of necrosis or pyroptosis (20, 28). ExoS also inhibits DNA synthesis in fibroblast-like cells, although it is unclear whether this effect is due to the ADPRT domain (40). Given its many targets and associated cellular phenotypes, it is likely that ExoS contributes to pathogenicity not simply by inhibiting phagocytosis but in multiple ways. Alternatively, it is conceivable that a small difference in phagocytosis may be compounded over time to yield a large cumulative difference in viable bacteria, as we observed. This difference may in turn directly correlate with the overall survival of mice.
Our findings suggest the following model for the role of ExoS in P. aeruginosa pathogenesis. Upon entering the lungs, bacteria inject ExoS into phagocytic cells. This toxin then ADP-ribosylates host cell proteins to block phagocytosis, which allows large numbers of bacteria to persist in the lungs despite the robust recruitment of phagocytic cells. These bacteria may utilize ExoS or other virulence factors to cause subsequent injury to pulmonary tissues and septic shock. P. aeruginosa may also take advantage of the collateral damage caused by the exuberant but ineffectual inflammatory response to cause more severe pneumonia. Together, these effects lead to the poor clinical outcomes associated with ExoS+ P. aeruginosa strains.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
PA99 is a P. aeruginosa clinical isolate that naturally secretes ExoU, ExoS, and ExoT (11). Previously, the exoU and exoT genes were disrupted to generate a strain, designated PA99S, that secretes only ExoS (7). As previously described, the PA99null strain, which produces an intact type III secretion apparatus but no known effector proteins, was generated by disrupting all three effector genes (exoU, exoS, and exoT) (7). A strain complemented for ExoS secretion (designated PA99null+S) was previously generated by integrating the exoS gene into a neutral site in the chromosome (7). Complementation was confirmed by measuring mouse mortality (see Fig. S3A in the supplemental material). A strain secreting an enzymatic ExoS variant defective in ADPRT activity [PA99null+S(E379A/E381A)] was previously generated (41). These substitutions have been previously shown to disrupt only ADPRT activity (25). A strain secreting an enzymatic ExoS variant defective in GAP activity [PA99null+S(R146A)] was generated for this work as previously described (7). This substitution has been previously shown to disrupt only GAP activity (12). Protein secretion was confirmed by immunoblot analysis (Fig. S1). None of the mutants displayed a growth defect in vitro (7). Note that these variant forms of ExoS have been previously shown to be translocated into mammalian cells by the P. aeruginosa type III secretion system and to retain functional activity of the nonmutated domain (42).
Bacterial strains were streaked from frozen cultures onto Vogel-Bonner minimal (VBM) agar (43). Overnight cultures were grown in 5 ml MINS medium (44) at 37°C. Cultures were diluted into fresh medium the next day and regrown to exponential phase prior to infections.
Mouse model of acute pneumonia.
In vivo infections used the mouse model of pneumonia of Comolli et al. (45). Briefly, 6- to 8-week-old female BALB/c mice were anesthetized by intraperitoneal injection of a mixture of ketamine (75 mg/kg of body weight) and xylazine (5 mg/kg). Mice were intranasally inoculated with either 1.2 × 106 (persistence, inflammatory, in vivo phagocytosis experiments) or 9.2 × 106 (survival experiments) CFU bacteria in phosphate-buffered saline (PBS). Inocula were confirmed by plating serial dilutions on VBM agar. At appropriate times postinfection, the mice were anesthetized and sacrificed by cervical dislocation. The lungs were excised and homogenized in PBS. Viable bacteria were enumerated by plating serial dilutions on VBM agar. For survival experiments, mice were infected and survival was monitored for 65 h after infection. Mice that became severely ill, as defined by ruffled fur and decreased respiratory rate and activity, were euthanized and scored as dead.
Animals were purchased from Harlan Laboratories, Inc. (Indianapolis, IN) and housed in the containment ward of the Center of Comparative Medicine at Northwestern University. All experiments were approved by the Northwestern University Animal Care and Use Committee.
Analysis of inflammatory response by flow cytometry.
Immune cells were quantified as previously described (46). Briefly, lungs were perfused, excised, pressed through 40-µm filters (BD Falcon; Becton, Dickinson and Company, Franklin Lakes, NJ), and rinsed repeatedly with PBS. The recovered cells were pelleted, and red blood cells were lysed by incubation with water. The remaining cells were pelleted, resuspended in 1 ml PBS, and quantified using trypan blue exclusion and a hemocytometer. A total of 1 × 106 cells were pelleted and blocked for nonspecific antibody binding. Individual cell types were identified using cell-discriminatory antibodies at appropriate dilutions in a total of 125 µl fluorescence-activated cell sorting (FACS) buffer (1% bovine serum albumin, 1 mM EDTA in 1× PBS) and incubated for 30 min on ice protected from light. Cells were pelleted, fixed in 3.7% paraformaldehyde in PBS for 2 min, and washed twice with FACS buffer. Final cell suspensions were filtered through 80-µm nylon mesh (Small Parts Inc., Miami Lakes, FL) into 12- by 75-mm round-bottom tubes (BD Falcon).
Analysis was performed on a BD FACSCanto II flow cytometer (Becton, Dickinson and Company) and analyzed using FlowJo version 8.8.6 software (Tree Star, Inc., Ashland, OR). Immune cells were gated as follows: total inflammatory cells, CD45+; neutrophils, CD45+ Gr1hi; recruited monocytes, CD45+ Gr1lo; resident macrophages, CD45+ Gr1− F4/80+ (see Fig. S6 in the supplemental material). The total number of inflammatory cells per mouse lung was determined by equating the number of viable inflammatory cells measured by flow cytometry to the number of trypan blue-negative cells counted on the hemocytometer.
Bone marrow neutrophil phagocytosis and cell death assays.
Marrow cavities of the tibias and femurs of female BALB/c mice were flushed with 1× Hanks’ balanced salt solution (HBSS). Cells were pelleted at 500 × g for 10 min, resuspended in 1× HBSS, and placed over a 62% Percoll gradient in 1× HBSS (GE Healthcare Biosciences, Piscataway, NJ). Gradients were spun at 1,500 × g for 30 min. The neutrophil pellet was washed and quantified using trypan blue exclusion and a hemocytometer.
For both assays, a total of 1 × 106 neutrophils were infected at a multiplicity of infection (MOI) of 10 for 30 min. For cell lysis assessment, a 50-µl aliquot of the infection medium was pelleted. The supernatant was removed and assayed for release of LDH using the CytoTox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI). Cell lysis was quantified by measuring absorbance at 490 nm and normalizing to total cell lysis achieved by adding 0.05% Triton X-100. Background lysis was measured in uninfected neutrophils incubated without bacteria and subtracted from all experimental values. For phagocytosis assessment, neutrophils were pelleted at 400 × g for 5 min after 30 min of infection. Neutrophils were then incubated with latex beads for 30 min at 37°C using the phagocytosis assay kit (IgG FITC [fluorescein isothiocyanate]) (Cayman Chemical Company, Ann Arbor, MI) per the manufacturer’s instructions. Cells were washed twice, and bead uptake was quantified using a BD FACSCanto II flow cytometer (Becton, Dickinson and Company).
Fluorescence internalization assay.
For in vivo experiments, white blood cells were isolated from whole mouse lungs using the method outlined above for inflammatory response experiments. A total of 1 × 106 cells were blocked with 10% rat serum (Sigma-Aldrich) and anti-CD16/32 (eBioscience) in FACS buffer for 10 min at 4°C. Leukocytes were stained by adding CD45 PE (phycoerythrin) antibody (eBioscience) at a 1:1,250 final dilution in FACS buffer for 15 min at 4°C. Cells were fixed as previously described. For staining of extracellular bacteria, primary antibody to P. aeruginosa strain PA99 (47) was diluted 1:1,000 in FACS buffer and added to cells for 1 h at 4°C. For staining of extracellular bacteria, cells were washed with FACS buffer and incubated with FITC-conjugated goat anti-rabbit secondary antibody (Molecular Probes, Eugene, OR) at 1:1,000 dilution in FACS buffer for 1 h at 4°C. For staining intracellular bacteria, cells were washed, permeabilized with 0.5% saponin for 20 min, and then incubated again with primary antibody to strain PA99 at 1:1,000 dilution in FACS buffer with 0.5% saponin for 1 h at 4°C. Cells were washed with FACS buffer and incubated with Alexa Fluor 647-conjugated goat anti-rabbit secondary antibody (Molecular Probes) at 1:1,000 dilution in FACS buffer with 0.5% saponin for 1 h at 4°C. This stained all intracellular and extracellular bacteria with red fluorescence. Cells were resuspended and filtered as described above. Cells were analyzed by using a Becton Dickinson FACSAria 4 cell sorting system or BD FACSCanto II flow cytometer (Becton, Dickinson and Company) (see Fig. S7 in the supplemental material). Leukocytes (CD45+ cells) associated with intracellular (red+ green−) and extracellular (red+ green+) bacteria were sorted and viewed with a Zeiss UV LSM510 confocal microscope in the Nikon Cell Imaging Facility at Northwestern University. A minimum of 20 cells within each sorted population per strain were analyzed for manual quantification of intracellular/extracellular bacteria per cell.
For in vitro cell culture experiments, J774 macrophage-like cells were infected with an MOI of 10 for 3 h. Cells were stained using the antibodies described above and imaged on a Zeiss UV LSM510 confocal microscope in the Nikon Cell Imaging Facility at Northwestern University. The number of intracellular/extracellular bacteria associated with 300 macrophages was quantified manually using ImageJ 1.47 analysis software (pooled from four separate experiments).
Amikacin protection assay.
Mice were anesthetized and sacrificed by cervical dislocation, and BALF was collected by instilling and withdrawing 1 ml of PBS into the trachea three times. The BALF was split into two aliquots. One aliquot was incubated with 1% saponin for 5 min for lysis of all murine cells and plated on VBM agar for enumeration of total bacteria in the lungs. Histopaque 1083 (Sigma-Aldrich) was added to the second aliquot, and phagocytic cells were pelleted by centrifugation at 500 × g for 30 min at 4°C. Residual red blood cells were lysed with cold, sterile H2O as described above. The remaining cells were quantified using trypan blue exclusion and a hemocytometer. The cells were incubated for 1 h with amikacin at a final concentration of 100 µg/ml in PBS to kill extracellular bacteria. Cells were pelleted, washed with PBS, lysed with 1% saponin, and plated on VBM agar for enumeration of viable intracellular bacteria. The percentage of intracellular bacteria was calculated by dividing the number of intracellular bacteria by total BALF bacteria.
Statistical methods.
Statistical analysis was performed using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA). An analysis of variance (ANOVA) followed by Bonferroni’s correction for multiple comparisons was performed for persistence, inflammatory response, and amikacin/in vitro phagocytosis experiments. The Kruskal-Wallis comparison of medians followed by Dunn’s test for multiple comparisons was performed on flow cytometric phagocytosis experiments. A log rank (Mantel-Cox) test was performed on survival curves. A P value of 0.05 was used as the threshold for significance for all tests.
SUPPLEMENTAL MATERIAL
Supplemental methods. Download
Confirmation of ExoS secretion by immunoblot analysis. Isogenic mutants of clinical isolate PA99 were grown in 5 ml MINS medium at 37°C overnight to induce type III secretion. Culture supernatants were harvested, purified, and subjected to immunoblot analysis with polyclonal rabbit antisera against ExoS/ExoT. Parental strain PA99 secreted ExoS and ExoT. Each of the complemented PA99null strains secreted ExoS. Download
ExoS enhances persistence of P. aeruginosa in the lungs of a mouse model of pneumonia through an ADPRT-dependent mechanism. (A and B) Mice were inoculated with ExoS variants lacking either GAP or ADPRT activity, and BALF was collected at 18 h (A) or 24 h (B) postinfection. The ADPRT domain, but not the GAP domain, of ExoS contributed to bacterial persistence in vivo. Each symbol represents the bacterial load in BALF from an individual mouse (at least seven mice for each strain). The bars represent the medians for the groups of mice. Each experiment was performed twice with cumulative results shown. Download
ExoS causes increased mortality in a mouse model of pneumonia. (A) Mice were intranasally infected with strain PA99S, PA99null, or complemented strain PA99null+S. There was no significant difference between the values for strain PA99S and complemented strain PA99null+S (*, P < 0.001 compared to strains PA99S and PA99null+S). (B) Mice were intranasally infected with strain PA99null+S, PA99null+S(R146A), or PA99null+S(E379A/E381A). The ADPRT domain of ExoS was primarily responsible for the higher mortality. There was no significant difference between the values for strains PA99null+S and PA99null+S(R146A) [*, P < 0.001 compared to PA99null+S and PA99null+S(R146A)]. All mice received a dose of 9.2 × 106 CFU. Survival was monitored for 65 h postinfection. At least four mice for each strain were used. Download
ExoS causes greater inflammatory infiltrate within the lungs. Histology from mice inoculated with strain PA99S, PA99null, or PBS alone at 24 h postinfection. The lungs from three mice for each strain were sectioned and stained with hematoxylin and eosin. Three slides per mouse were imaged; representative images are shown. Download
Quantification of bacteria associated with leukocytes for fluorescence internalization assay. (A and B) Leukocytes (CD45+ cells) associated with intracellular (A) and extracellular (B) bacteria were sorted and viewed with a Zeiss UV LSM510 confocal microscope, since flow cytometry could not distinguish between cells with one bacterium or multiple bacteria. A minimum of 20 cells within each sorted population for each strain were analyzed for manual quantification of intracellular/extracellular bacteria per cell. The values are the mean number of bacteria per leukocyte. Error bars represent the standard deviations. Values are not significantly different. The means (standard deviations shown in the parentheses) for the different strains are as follows: in panel A, 3.15 (1.93) for strain PA99null+S, 2.8 (1.15) for PA99null, 3.75 (1.86) for PA99null+S(R146A), and 2.7 (1.26) for PA99null+S(E379A/E381A); for panel B, 1.95 (0.94) for PA99null+S, 2.5 (1.0) for PA99null, 1.8 (0.77) for PA99null+S(R146A), and 1.9 (0.79) for PA99null+S(E379A/E381A). Download
Gating strategy for flow cytometric analysis of immune cell populations. Immune cells were gated on the basis of CD45 allophycocyanin (APC) antibody staining to determine the total number of leukocytes isolated from whole lungs. CD45+ cells were further analyzed for Gr1 FITC staining to determine Gr1+ and Gr1− cells. Neutrophils (PMNs) and recruited monocytes (monos) were distinguished on the basis of Gr1hi and Gr1lo staining, respectively. CD45+ Gr1− cells were further analyzed for F4/80 PE staining to distinguish resident macrophages. The image shows a representative gating strategy from a mouse infected with strain PA99S at 18 h postinfection. Download
Gating strategy for flow cytometric fluorescence internalization assay. Total leukocytes were gated from cells isolated from whole lungs on the basis of CD45 PE antibody staining. CD45+ leukocytes were further analyzed to quantify cells associated with intracellular (red+ green−) and extracellular (red+ green+) bacteria. The image shows a representative gating strategy from a mouse infected with strain PA99null+S at 24 h postinfection. SSC-A, side scatter area. Download
ACKNOWLEDGMENTS
This work was supported by the Northwestern University Pathology Core Facility, the Northwestern University Interdepartmental ImmunoBiology Flow Cytometry Core Facility, the Northwestern University RHLCCC Flow Cytometry Facility, and a Cancer Center Support Grant (NCI CA060553). Imaging work was performed at the Northwestern University Cell Imaging Facility generously supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. Additional support was provided by the National Institutes of Health: AI089015 (S.M.R.), AI053674, AI075191, AI099269, AI04831, and AI088286 (A.R.H.).
Footnotes
Citation Rangel SM, Logan LK, Hauser AR. 2014. The ADP-ribosyltransferase domain of the effector protein ExoS inhibits phagocytosis of Pseudomonas aeruginosa during pneumonia. mBio 5(3):e01080-14. doi:10.1128/mBio.01080-14.
REFERENCES
- 1. Centers for Disease Control and Prevention 2013. Antibiotic resistance threats in the United States, 2013. Centers for Disease Control and Prevention, Atlanta, GA. http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. [Google Scholar]
- 2. Fagon JY, Chastre J, Domart Y, Trouillet JL, Pierre J, Darne C, Gibert C. 1989. Nosocomial pneumonia in patients receiving continuous mechanical ventilation. Prospective analysis of 52 episodes with use of a protected specimen brush and quantitative culture techniques. Am. Rev. Respir. Dis. 139:877–884. 10.1164/ajrccm/139.4.877 [DOI] [PubMed] [Google Scholar]
- 3. George DL, Falk PS, Wunderink RG, Leeper KV, Jr, Meduri GU, Steere EL, Corbett CE, Mayhall CG. 1998. Epidemiology of ventilator-acquired pneumonia based on protected bronchoscopic sampling. Am. J. Respir. Crit. Care Med. 158:1839–1847. 10.1164/ajrccm.158.6.9610069 [DOI] [PubMed] [Google Scholar]
- 4. Rello J, Gallego M, Mariscal D, Soñora R, Valles J. 1997. The value of routine microbial investigation in ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 156:196–200. 10.1164/ajrccm.156.1.9607030 [DOI] [PubMed] [Google Scholar]
- 5. Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7:654–665. 10.1038/nrmicro2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wiener-Kronish JP, Sakuma T, Kudoh I, Pittet JF, Frank D, Dobbs L, Vasil ML, Matthay MA. 1993. Alveolar epithelial injury and pleural empyema in acute Pseudomonas aeruginosa pneumonia in anesthetized rabbits. J. Appl. Physiol. 75:1661–1669 [DOI] [PubMed] [Google Scholar]
- 7. Shaver CM, Hauser AR. 2004. Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect. Immun. 72:6969–6977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vance RE, Rietsch A, Mekalanos JJ. 2005. Role of the type III secreted exoenzymes S, T, and Y in systemic spread of Pseudomonas aeruginosa PAO1 in vivo. Infect. Immun. 73:1706–1713. 10.1128/IAI.73.3.1706-1713.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hauser AR, Cobb E, Bodí M, Mariscal D, Vallés J, Engel JN, Rello J. 2002. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit. Care Med. 30:521–528. 10.1097/00003246-200203000-00005 [DOI] [PubMed] [Google Scholar]
- 10. Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. 2001. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J. Infect. Dis. 183:1767–1774. 10.1086/320737 [DOI] [PubMed] [Google Scholar]
- 11. Feltman H, Schulert G, Khan S, Jain M, Peterson L, Hauser AR. 2001. Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology (Reading, Engl.) 147:2659–2669 [DOI] [PubMed] [Google Scholar]
- 12. Goehring U-M, Schmidt G, Pederson KJ, Aktories K, Barbieri JT. 1999. The N-terminal domain of Pseudomonas aeruginosa exoenzyme S is a GTPase-activating protein for Rho GTPases. J. Biol. Chem. 274:36369–36372. 10.1074/jbc.274.51.36369 [DOI] [PubMed] [Google Scholar]
- 13. DiNovo AA, Schey KL, Vachon WS, McGuffie EM, Olson JC, Vincent TS. 2006. ADP-ribosylation of cyclophilin A by Pseudomonas aeruginosa exoenzyme S. Biochemistry 45:4664–4673. 10.1021/bi0513554 [DOI] [PubMed] [Google Scholar]
- 14. Maresso AW, Baldwin MR, Barbieri JT. 2004. Ezrin/radixin/moesin proteins are high affinity targets for ADP-ribosylation by Pseudomonas aeruginosa ExoS. J. Biol. Chem. 279:38402–38408. 10.1074/jbc.M405707200 [DOI] [PubMed] [Google Scholar]
- 15. Vincent TS, Fraylick JE, McGuffie EM, Olson JC. 1999. ADP-ribosylation of oncogenic Ras proteins by Pseudomonas aeruginosa exoenzyme S in vivo. Mol. Microbiol. 32:1054–1064. 10.1046/j.1365-2958.1999.01420.x [DOI] [PubMed] [Google Scholar]
- 16. Mustafi S, Rivero N, Olson JC, Stahl PD, Barbieri MA. 2013. Regulation of Rab5 function during phagocytosis of live Pseudomonas aeruginosa in macrophages. Infect. Immun. 81:2426–2436. 10.1128/IAI.00387-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cowell BA, Chen DY, Frank DW, Vallis AJ, Fleiszig SMJ. 2000. ExoT of cytotoxic Pseudomonas aeruginosa prevents uptake by corneal epithelial cells. Infect. Immun. 68:403–406. 10.1128/IAI.68.1.403-406.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garrity-Ryan L, Kazmierczak B, Kowal R, Comolli J, Hauser A, Engel JN. 2000. The arginine finger domain of ExoT contributes to actin cytoskeleton disruption and inhibition of internalization of Pseudomonas aeruginosa by epithelial cells and macrophages. Infect. Immun. 68:7100–7113. 10.1128/IAI.68.12.7100-7113.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fleiszig SM, Wiener-Kronish JP, Miyazaki H, Vallas V, Mostov KE, Kanada D, Sawa T, Yen TS, Frank DW. 1997. Pseudomonas aeruginosa-mediated cytotoxicity and invasion correlate with distinct genotypes at the loci encoding exoenzyme S. Infect. Immun. 65:579–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frithz-Lindsten E, Du Y, Rosqvist R, Forsberg A. 1997. Intracellular targeting of exoenzyme S of Pseudomonas aeruginosa via type III-dependent translocation induces phagocytosis resistance, cytotoxicity and disruption of actin microfilaments. Mol. Microbiol. 25:1125–1139. 10.1046/j.1365-2958.1997.5411905.x [DOI] [PubMed] [Google Scholar]
- 21. Barclay NG, Spurrell JC, Bruno TF, Storey DG, Woods DE, Mody CH. 1999. Pseudomonas aeruginosa exoenzyme S stimulates murine lymphocyte proliferation in vitro. Infect. Immun. 67:4613–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jia J, Wang Y, Zhou L, Jin S. 2006. Expression of Pseudomonas aeruginosa toxin ExoS effectively induces apoptosis in host cells. Infect. Immun. 74:6557–6570. 10.1128/IAI.00591-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaufman MR, Jia J, Zeng L, Ha U, Chow M, Jin S. 2000. Pseudomonas aeruginosa mediated apoptosis requires the ADP-ribosylating activity of ExoS. Microbiology (Reading, Engl.) 146:2531–2541 [DOI] [PubMed] [Google Scholar]
- 24. Sun Y, Karmakar M, Taylor PR, Rietsch A, Pearlman E. 2012. ExoS and ExoT ADP ribosyltransferase activities mediate Pseudomonas aeruginosa keratitis by promoting neutrophil apoptosis and bacterial survival. J. Immunol. 188:1884–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Radke J, Pederson KJ, Barbieri JT. 1999. Pseudomonas aeruginosa exoenzyme S is a biglutamic acid ADP-ribosyltransferase. Infect. Immun. 67:1508–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DiMango E, Zar HJ, Bryan R, Prince A. 1995. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J. Clin. Invest. 96:2204–2210. 10.1172/JCI118275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rocha CL, Coburn J, Rucks EA, Olson JC. 2003. Characterization of Pseudomonas aeruginosa exoenzyme S as a bifunctional enzyme in J774A.1 macrophages. Infect. Immun. 71:5296–5305. 10.1128/IAI.71.9.5296-5305.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pederson KJ, Barbieri JT. 1998. Intracellular expression of the ADP-ribosyltransferase domain of Pseudomonas aeruginosa exoenzyme S is cytotoxic to eukaryotic cells. Mol. Microbiol. 30:751–759. 10.1046/j.1365-2958.1998.01106.x [DOI] [PubMed] [Google Scholar]
- 29. Criss AK, Katz BZ, Seifert HS. 2009. Resistance of Neisseria gonorrhoeae to non-oxidative killing by adherent human polymorphonuclear leucocytes. Cell. Microbiol. 11:1074–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ganesan AK, Vincent TS, Olson JC, Barbieri JT. 1999. Pseudomonas aeruginosa exoenzyme S disrupts Ras-mediated signal transduction by inhibiting guanine nucleotide exchange factor-catalyzed nucleotide exchange. J. Biol. Chem. 274:21823–21829. 10.1074/jbc.274.31.21823 [DOI] [PubMed] [Google Scholar]
- 31. Maresso AW, Deng Q, Pereckas MS, Wakim BT, Barbieri JT. 2007. Pseudomonas aeruginosa ExoS ADP-ribosyltransferase inhibits ERM phosphorylation. Cell. Microbiol. 9:97–105. 10.1111/j.1462-5822.2006.00770.x [DOI] [PubMed] [Google Scholar]
- 32. Alvarez-Dominguez C, Stahl PD. 1999. Increased expression of Rab5a correlates directly with accelerated maturation of Listeria monocytogenes phagosomes. J. Biol. Chem. 274:11459–11462. 10.1074/jbc.274.17.11459 [DOI] [PubMed] [Google Scholar]
- 33. Barbieri AM, Sha Q, Bette-Bobillo P, Stahl PD, Vidal M. 2001. ADP-ribosylation of Rab5 by ExoS of Pseudomonas aeruginosa affects endocytosis. Infect. Immun. 69:5329–5334. 10.1128/IAI.69.9.5329-5334.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Evdokimov AG, Tropea JE, Routzahn KM, Waugh DS. 2002. Crystal structure of the Yersinia pestis GTPase activator YopE. Protein Sci. 11:401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Würtele M, Wolf E, Pederson KJ, Buchwald G, Ahmadian MR, Barbieri JT, Wittinghofer A. 2001. How the Pseudomonas aeruginosa ExoS toxin downregulates Rac. Nat. Struct. Biol. 8:23–26. 10.1038/83007 [DOI] [PubMed] [Google Scholar]
- 36. Pederson KJ, Vallis AJ, Aktories K, Frank DW, Barbieri JT. 1999. The amino-terminal domain of Pseudomonas aeruginosa ExoS disrupts actin filaments via small-molecular-weight GTP-binding proteins. Mol. Microbiol. 32:393–401. 10.1046/j.1365-2958.1999.01359.x [DOI] [PubMed] [Google Scholar]
- 37. Matz C, Moreno AM, Alhede M, Manefield M, Hauser AR, Givskov M, Kjelleberg S. 2008. Pseudomonas aeruginosa uses type III secretion system to kill biofilm-associated amoebae. ISME J 2:843–852. 10.1038/ismej.2008.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rucks EA, Fraylick JE, Brandt LM, Vincent TS, Olson JC. 2003. Cell line differences in bacterially translocated ExoS ADP-ribosyltransferase substrate specificity. Microbiology (Reading, Engl.) 149:319–331. 10.1099/mic.0.25985-0 [DOI] [PubMed] [Google Scholar]
- 39. Angus AA, Evans DJ, Barbieri JT, Fleiszig SMJ. 2010. The ADP-ribosylation domain of Pseudomonas aeruginosa ExoS is required for membrane bleb niche formation and bacterial survival within epithelial cells. Infect. Immun. 78:4500–4510. 10.1128/IAI.00417-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Olson JC, McGuffie EM, Frank DW. 1997. Effects of differential expression of the 49-kilodalton exoenzyme S by Pseudomonas aeruginosa on cultured eukaryotic cells. Infect. Immun. 65:248–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ottmann C, Yasmin L, Weyand M, Veesenmeyer JL, Diaz MH, Palmer RH, Francis MS, Hauser AR, Wittinghofer A, Hallberg B. 2007. Phosphorylation-independent interaction between 14-3-3 and exoenzyme S: from structure to pathogenesis. EMBO J. 26:902–913. 10.1038/sj.emboj.7601530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fraylick JE, La Rocque JR, Vincent TS, Olson JC. 2001. Independent and coordinate effects of ADP-ribosyltransferase and GTPase-activating activities of exoenzyme S on HT-29 epithelial cell function. Infect. Immun. 69:5318–5328. 10.1128/IAI.69.9.5318-5328.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vogel HJ, Bonner DM. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97–106 [PubMed] [Google Scholar]
- 44. Nicas TI, Iglewski BH. 1984. Isolation and characterization of transposon-induced mutants of Pseudomonas aeruginosa deficient in production of exoenzyme S. Infect. Immun. 45:470–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Comolli JC, Hauser AR, Waite L, Whitchurch CB, Mattick JS, Engel JN. 1999. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect. Immun. 67:3625–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Diaz MH, Shaver CM, King JD, Musunuri S, Kazzaz JA, Hauser AR. 2008. Pseudomonas aeruginosa induces localized immunosuppression during pneumonia. Infect. Immun. 76:4414–4421. 10.1128/IAI.00012-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Howell HA, Logan LK, Hauser AR. 2013. Type III secretion of ExoU is critical during early Pseudomonas aeruginosa pneumonia. mBio 4(2):e00032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Download
Confirmation of ExoS secretion by immunoblot analysis. Isogenic mutants of clinical isolate PA99 were grown in 5 ml MINS medium at 37°C overnight to induce type III secretion. Culture supernatants were harvested, purified, and subjected to immunoblot analysis with polyclonal rabbit antisera against ExoS/ExoT. Parental strain PA99 secreted ExoS and ExoT. Each of the complemented PA99null strains secreted ExoS. Download
ExoS enhances persistence of P. aeruginosa in the lungs of a mouse model of pneumonia through an ADPRT-dependent mechanism. (A and B) Mice were inoculated with ExoS variants lacking either GAP or ADPRT activity, and BALF was collected at 18 h (A) or 24 h (B) postinfection. The ADPRT domain, but not the GAP domain, of ExoS contributed to bacterial persistence in vivo. Each symbol represents the bacterial load in BALF from an individual mouse (at least seven mice for each strain). The bars represent the medians for the groups of mice. Each experiment was performed twice with cumulative results shown. Download
ExoS causes increased mortality in a mouse model of pneumonia. (A) Mice were intranasally infected with strain PA99S, PA99null, or complemented strain PA99null+S. There was no significant difference between the values for strain PA99S and complemented strain PA99null+S (*, P < 0.001 compared to strains PA99S and PA99null+S). (B) Mice were intranasally infected with strain PA99null+S, PA99null+S(R146A), or PA99null+S(E379A/E381A). The ADPRT domain of ExoS was primarily responsible for the higher mortality. There was no significant difference between the values for strains PA99null+S and PA99null+S(R146A) [*, P < 0.001 compared to PA99null+S and PA99null+S(R146A)]. All mice received a dose of 9.2 × 106 CFU. Survival was monitored for 65 h postinfection. At least four mice for each strain were used. Download
ExoS causes greater inflammatory infiltrate within the lungs. Histology from mice inoculated with strain PA99S, PA99null, or PBS alone at 24 h postinfection. The lungs from three mice for each strain were sectioned and stained with hematoxylin and eosin. Three slides per mouse were imaged; representative images are shown. Download
Quantification of bacteria associated with leukocytes for fluorescence internalization assay. (A and B) Leukocytes (CD45+ cells) associated with intracellular (A) and extracellular (B) bacteria were sorted and viewed with a Zeiss UV LSM510 confocal microscope, since flow cytometry could not distinguish between cells with one bacterium or multiple bacteria. A minimum of 20 cells within each sorted population for each strain were analyzed for manual quantification of intracellular/extracellular bacteria per cell. The values are the mean number of bacteria per leukocyte. Error bars represent the standard deviations. Values are not significantly different. The means (standard deviations shown in the parentheses) for the different strains are as follows: in panel A, 3.15 (1.93) for strain PA99null+S, 2.8 (1.15) for PA99null, 3.75 (1.86) for PA99null+S(R146A), and 2.7 (1.26) for PA99null+S(E379A/E381A); for panel B, 1.95 (0.94) for PA99null+S, 2.5 (1.0) for PA99null, 1.8 (0.77) for PA99null+S(R146A), and 1.9 (0.79) for PA99null+S(E379A/E381A). Download
Gating strategy for flow cytometric analysis of immune cell populations. Immune cells were gated on the basis of CD45 allophycocyanin (APC) antibody staining to determine the total number of leukocytes isolated from whole lungs. CD45+ cells were further analyzed for Gr1 FITC staining to determine Gr1+ and Gr1− cells. Neutrophils (PMNs) and recruited monocytes (monos) were distinguished on the basis of Gr1hi and Gr1lo staining, respectively. CD45+ Gr1− cells were further analyzed for F4/80 PE staining to distinguish resident macrophages. The image shows a representative gating strategy from a mouse infected with strain PA99S at 18 h postinfection. Download
Gating strategy for flow cytometric fluorescence internalization assay. Total leukocytes were gated from cells isolated from whole lungs on the basis of CD45 PE antibody staining. CD45+ leukocytes were further analyzed to quantify cells associated with intracellular (red+ green−) and extracellular (red+ green+) bacteria. The image shows a representative gating strategy from a mouse infected with strain PA99null+S at 24 h postinfection. SSC-A, side scatter area. Download