

Abstract
Background
Propofol can be associated with delayed awakening after prolonged infusion. The aim of this study was to characterize the preclinical pharmacology of AZD-3043, a positive allosteric modulator of the γ-aminobutyric acidA (GABAA) receptor containing a metabolically-labile ester moiety. We postulated that its metabolic pathway would result in a short acting clinical profile.
Methods
The effects of AZD-3043, propofol and propanidid were studied on GABAA receptor-mediated chloride currents in embryonic rat cortical neurons. Radioligand binding studies were also performed. The in vitro stability of AZD-3043 in whole blood and liver microsomes was evaluated. The duration of the loss of righting reflex and effects on the electroencephalograph evoked by bolus or infusion intravenous (IV) administration were assessed in rats. A mixed-effects kinetic-dynamic model using minipigs permitted exploration of the clinical pharmacology of AZD-3043.
Results
AZD-3043 potentiated GABAA receptor-mediated chloride currents and inhibited [35S]tert-butylbicyclophosphorothionate binding to GABAA receptors. AZD-3043 was rapidly hydrolyzed in liver microsomes from humans and animals. AZD-3043 produced hypnosis and electroencephalograph depression in rats. Compared to propofol, AZD-3043 was shorter acting in rats and pigs. Computer simulation using the porcine kinetic-dynamic model demonstrated that AZD-3043 has very short 50 and 80% decrement times independent of infusion duration.
Conclusions
AZD-3043 is a positive allosteric modulator of the GABAA receptor in vitro and a sedative/hypnotic agent in vivo. The esterase dependent metabolic pathway results in rapid clearance and short duration of action even for long infusions. AZD-3043 may have clinical potential as a sedative/hypnotic agent with rapid and predictable recovery.
Introduction
The sedative/hypnotic activity of barbiturates, benzodiazepines, neurosteroids and propofol (2,6-diisopropylphenol in lipid emulsion) results from potentiation of γ-aminobutyric acid (GABA)-mediated inhibition of synaptic activity within the central nervus system (CNS) via an allosteric interaction at the GABAA receptors. Thus, propofol produces a marked increase in the affinity of [3H]GABA binding to rat cortical membrane preparations, and potentiates radiolabeled chloride uptake evoked by muscimol, a GABAA receptor agonist.1 Acting at a binding site distinct from that of the barbiturates, benzodiazepines or neurosteroid sedative/hypnotics, propofol increases the open probability of GABAA receptor chloride channels.2 Propofol has gained considerable use for the induction and maintenance of sedation/hypnosis as patient awakening is relatively rapid. However, the rapidity of emergence from propofol-mediated sedation/hypnosis is dose-dependent.3 In order to avoid tissue accumulation and delayed or unpredictable patient emergence, dose titration is necessary, particularly with prolonged infusion.
Propanidid ((4-diethylcarbamoylmethoxy-3-methoxy-phenyl)-acetic acid propyl ester) is a short-acting sedative/hypnotic agent containing an ester moiety that was available in some countries in the 1960’s and 70’s.4 As was the case with propofol, propanidid was introduced as a cremophor-containing solution; both were associated with histamine release and adverse hemodynamic effects.5,6 Unlike the situation with propofol, propanidid was not re-introduced commercially in an alternative, more acceptable, lipid-based emulsion.
AZD-3043 ([4-[(N,N-Diethylcarbamoyl)methoxy]-3-ethoxyphenyl]acetic acid propyl ester), like propanidid, contains a metabolically-labile ester (Figure 1). Formerly known as TD-4756, AZD-3043 is formulated in a lipid emulsion similar to that used for propofol. It was postulated that emergence from hypnosis with AZD-3043 would be more rapid and predictable than that following propofol. This study characterized the nature of the interaction of AZD-3043 with the GABAA receptor in vitro, and examined the hypnotic profile of the compound in vivo. The stability of AZD-3043 was also evaluated in vitro, in liver microsomes and whole blood from several species, including man. The onset, duration of hypnosis, and recovery profile after intravenous bolus and infusion administration of AZD-3043 was evaluated in rats. The electroencephalogram of the rat was recorded during AZD-3043 administration. The pharmacokinetics and pharmacodynamics of AZD-3043 were investigated in a porcine model; pharmacokinetic simulations based on a combined kinetic-dynamic model were used to explore the clinical behavior of AZD-3043.
Figure 1.

The chemical structures of AZ-3043, its major, inactive, carboxylate metabolite THRX-108893, and propanidid are shown. AZ-3043 is an analog of propanidid that is substituted with an aryl ethoxy rather than an aryl methoxy group. In rodents, THRX-108893, accounts for more than 90% of the total radioactivity in urine, feces, plasma, liver and kidney following dosing with radiolabeled AZ-3043. GABA = γ-aminobutyric acid
Materials and Methods
All experiments were conducted according to guidelines established by the ‘Institutional Animal Care and Use Committee’ at Theravance, Inc., Brigham Young University, Provo, Utah (rat electroencephalography studies), or at the University of Utah, Salt Lake City, Utah (porcine study). The procedures described complied with the Animal Welfare Act and Public Health Service Policy, 1999. Human blood was collected under a voluntary blood donor program at Theravance, Inc., the policy for which was set up by our occupational health physician, and included volunteer consent and Institutional Biosafety Committee approval.
In Vitro Electrophysiology
Cultures of cortical neurons were prepared from embryos of untimed (approximately E-18) pregnant dam rats.7 Cells were seeded onto poly-D-lysine (100 μg/mL) coated glass coverslips in Dulbecco’s Modified Eagles Medium, supplemented with 10 % fetal bovine serum and 200 μg/mL penicillin/streptomycin. One day after plating, cells were maintained in serum-free Dulbecco’s Modified Eagles Medium, supplemented with B27 (GibcoBRL). Cultures were maintained in a humidified, 5% CO2 environment at 37°C until use. The coverslips were transferred to the recording chamber and perfused continuously (0.5 mL/min) with bathing solution at room temperature. The bathing solution (pH 7.4) contained (mM): NaCl (140), KCl (5.4), CaCl2 (1.3), glucose (33), 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES) (25), and tetrodotoxin (0.3 μM). Gigaohm seals were formed between the cell and a patch electrode (initial resistance 1 – 2 MΩ). The patch electrode contained (mM): CsCl (140), CaCl2 (1), ethylene glycol-bis(beta-aminoethyl ether) N,N,N′,N′-tetra acetic acid salt (EGTA) (11), HEPES (10), pH 7.2 with CsOH. The membrane beneath the electrode tip was ruptured to establish a whole cell patch-clamp configuration. Recording commenced in the voltage clamp mode, with an initial holding potential of −60 mV.
GABA (5 μM, the EC20 concentration) was applied once every 2 minutes (BioLogic RSC-200 rapid solution exchanger; Bio-Logic SAS, Claix, France) for a total of 20 minutes per cell in the absence, or presence, of increasing concentrations of AZD-3043, propofol or propanidid. The EC20 for GABA was determined on a separate pool of primary cortical neurons. Within this control pool, the GABA-evoked responses were similar in potency (though varying in magnitude).
Ionic currents were recorded using an AXON Instruments AxoPatch 200B amplifier (Axon Instruments, Inc., Union City, CA). Peak current amplitudes were measured using pCLAMP® software (Molecular Devices Corporation, Sunnyvale, CA), and data were then exported to Origin® (OriginLab Corp., Northampton, MA) where the individual current amplitudes were normalized to the initial current amplitude in the absence of AZD-3043, propofol or propanidid.
Radioligand Binding Studies
In order to investigate their potential off-target activities, radioligand binding experiments were performed in duplicate with AZD-3043 (50 μM), propanidid (50 μM), and their primary carboxylate metabolites at 30 different receptors. With the exception of the human β1- and β2-adrenoceptor, and the muscarinic M1–5 receptor subtype binding studies performed at Theravance, Inc., investigations were carried out under contract at Cerep (Le Bois l’Evêque – B.P. 1 – 86600 Celle l’Evescault, France; see catalog for assay conditions). Non-specific radioligand binding was determined in the presence of an excess of unlabeled ligand, and specific binding was defined as the difference between total and non-specific binding. Data (individual values and means) were expressed as a percentage inhibition of specific binding.
Stability in Blood and Liver Microsomes
Human whole blood stability experiments were conducted in whole blood from seven separate human donors. Single experiments were conducted with pooled blood (in duplicate) from rats (n=5), cats (n=4), beagle dogs(n=4) and minipigs(n=4). Blood was collected in vacutainer tubes containing sodium heparin. Single experiments were conductedwith pooled liver microsomes (in duplicate) from beagle dogs (n=4), cynomolgus monkeys (n=8), humans(n=10) and minipigs(n = 3), purchased from Xenotech, Inc. (Kansas City, KS). In vitro stability of AZD-3043 (50 μM) was evaluated following incubation (for 10 seconds to 60 minutes) with whole blood or liver microsomes (1 mg of protein/mL, 37°C)in polypropylene test tubes. In experiments using blood, incubation samples (0.3 mL) were withdrawn and mixed with 0.6 mL of ice-cold ethanol to terminate metabolism. The samples were centrifuged, and the supernatants dried under a stream of nitrogen at room temperature. The residue was reconstituted in 200 μL of 5% acetonitrile with 0.1% trifluroacetic acid, and then centrifuged. For microsomal studies, incubation samples (0.1 mL) were withdrawn and combined with 50 μL acetonitrile with 1% trifluoracetic acid to terminate the reaction. The precipitated microsomal protein was pelleted by centrifugation at 10,000 rpm for 10 minutes at 4°C. The supernatants (40 μL from the blood studies or 50 μL from microsomal experiments) were injected on to an Agilent 1100 high pressure liquid chromography system equipped with a Luna C18 analytical column (5 μm, 150 × 2.0 mm; Phenomenex, Torrance, CA) at a flow rate of 0.5 mL/min. The mobile phases were 5% acetonitrile in 0.1% trifluroacetic acid (Mobile Phase A), and 95% acetonitrile in 0.1% trifluroacetic acid (Mobile Phase B), and a wavelength of 214 nm was used. The mobile phase gradient started at 94% A/6% B and ramped to 35% A/65% B over 15 minutes before reverting to an isocratic hold at 94% A/6% B over the final 5 minutes. The substrate remaining at each sampling time point was monitored by peak area on the chromatogram and expressed as a percentage of that at time 0. The observed rate constant (kobs) for substrate disappearance was determined as the slope of the line from the plot of the natural log of the percentage of substrate remaining versus time of incubation. The half-life (t1/2; i.e., the time required for 50% of the substrate to be metabolized) was calculated according to the following equation: T1/2 = 0.693/kobs.
Rodent Pharmacology
Adult male and female Sprague Dawley rats (Harlan, Indianapolis, IN), weighing 120 to 750 g, were used. Food and water were available ad libitum.
Hypnosis in response to bolus administration
A rodent model8 was used to provide a measure of onset and duration of hypnosis, and to evaluate the recovery profile following return of consciousness. AZD-3043 (6.5 to 27 mg/kg), propofol (5 to 20 mg/kg), propanidid (15 to 45 mg/kg), and their respective vehicles were administered to rats via the tail vein (1 mL/kg over 5 seconds). The hypnotic activities of THRX-108893 (0.2 to 0.9 g/kg) and 1-propanol (0.1 to 0.5 g/kg), the primary metabolites of AZD-3043, were also assessed. In one study, remifentanil (2.5 and 10 μg/kg) or fentanyl (10 μg/kg) was co-administered with AZD-3043, propofol or propanidid. Following dosing, rats were placed in a supine position on a heating blanket to maintain body temperature at 37 to 38°C (monitored rectally with a sensor (Physitemp BAT-12, Physitemp Instruments, Inc., Clifton, NJ). The duration of the loss of the righting reflex was recorded. Upon regaining their righting reflex, the time that lapsed until the rats were able to grip and climb a steel frame, and ambulate normally was determined (the “time to behavioral recovery”). To provide a measure of relative potencies, the dose estimated to produce a mean loss of righting reflex of 2 minutes (i.e. the “2-minute bolus dose”) was determined from the dose-response curves. In one study, the ED50 values for the loss of righting reflex produced by AZD-3043 and propofol were determined (i.e., the dose at which 50% of rats lost their righting reflex, regardless of duration). The ED50 values were derived using Prism graphics software 3.0 (GraphPad, Inc., San Diego, CA).
Hypnosis in response to IV infusion
Induction of hypnosis in rats was achieved using the approximate 2-minute bolus dose of each compound, and immediately following induction, infusion, via the tail vein, was commenced at half that dose per minute. In some experiments, the infusion rate was maintained for 20 minutes. In other studies, the infusion rate was modified to ensure that the depth of hypnosis remained constant, as monitored by the magnitude of a withdrawal reflex to intermittent paw pinch provided by a pair of forceps. The degree of closure of the tips of the forceps was fixed to ensure that a consistent paw pressure was applied. Following completion of the infusion (20 minutes, or 3 or 5 hours later), the duration of the loss of righting reflex and time to behavioral recovery were determined.
Rodent Electroencephalography
Under anesthesia (pentobarbital: 40 mg/kg intraperitoneally, and ketamine 20 to 27 mg/kg intramuscularly), a longitudinal incision was made in the rat’s neck, and a jugular vein was catheterized. Following placement of the rat in a stereotaxic frame, a midline incision (2 cm in length) was made in the scalp. Holes (1 to 2 mm in diameter) were drilled in seven perimeter skull locations for insertion of grounding and support screws. A silver grounding wire was wrapped around the seven supporting skull screws. Holes (1 mm in diameter) were drilled in four skull locations, corresponding to the bilateral parietal and frontal cortices. Four stainless steel skull screws, wrapped with 120 μm HML-coated stainless steel wire, served as the electroencephalography electrodes. The ground and electroencephalography electrodes were connected to a subminiature Amphenol 9-pin D-connector (Amphenol Corporation, Cedar Grove, NJ), cemented to the cranium with dental acrylate.
At least one week after surgery, each rat was placed in a sound-proof box. Electroencephalography activity was amplified (100 to 10,000 X) and filtered (0.1 to 100 Hz) using a multichannel signal conditioner (National Instruments, Austin, TX). Following collection of 20 minutes of baseline data, rats were dosed via the jugular catheter (1 mL/kg over 5 seconds) with vehicle, followed by increasing doses of AZD-3043 (5, 10, 20 and 30 mg/kg) or propofol (1, 3, 6 and 10 mg/kg) at 30-minute intervals (n = 6 rats in a cross-over design, 5 day washout between treatments). The electroencephalography data were digitized at 500 Hz, and analysis performed as described previously.9 Epochs, consisting of 2 seconds of averaged electroencephalography data, were classified based on an “interpretation map”, that was constructed from a learning algorithm of the electroencephalography data.10 Classification values represent the degree of presence (>0) or absence (<0) of a pattern of features (e.g., phase-weighted frequencies) inherent in the raw spontaneous electroencephalography. The learning algorithm identified feature patterns that discriminated optimally between drug and non-drug states. Five sets of frequencies were used to classify the electroencephalography epochs (Gaussian center frequency ± Gaussian half-width = 3.5±3.5 Hz, 5±5 Hz, 10.0±10.0 Hz, 14.0±14.0 Hz and 17.0±31.0 Hz). Using commercial signal processing software (NeuroInsight LLC, Bountiful, UT), statistical analysis (pattern discovery, recognition and verification) was performed. Pre- and post-injection electroencephalography patterns were compared to detect drug effects, and electroencephalography activation plots, representing 2 seconds of filtered electroencephalography activity, were created. The values in the electroencephalography activation plots ranged from −1 (fully inactivated) to 1 (fully activated).
Large Mammal Pharmacology
Minipigs were used to model the clinical pharmacokinetics and pharmacodynamics of AZD-3043. All animals were obtained from a single breeding source and were approximately the same age.
The anesthetic induction was performed using intramuscular injection of telazol, xylazine and ketamine.10 Anesthesia was maintained with inhaled isoflurane. The pig’s trachea was intubated and mechanically ventilated to maintain arterial pCO2 at approximately 35 to 40 mmHg. Physiologic monitoring included the electrocardiogram, a pulmonary artery catheter, femoral arterial catheters, pulse oximetry (SpO2), and a urinary catheter.
AZD-3043 (1.5 or 3.0 mg·kg−1·min−1, 20 min) was administered via the peripheral IV catheter. Arterial blood samples (1 mL each) were collected at 0 (control sample), 2, 4,8, 12, 16, 20, 21, 22, 23, 24, 25, 27.5, 30, 35, 40, 45, 60, 90, 120 and 180 minutes after the start of the infusion. Immediately after collection, methanol (2.5 mL) was added to each blood sample (1 mL) and the mixture vortexed. Samples were then centrifuged (3,000 × g) for 15 minutes at 5°C and 2 mL of the supernatant transferred to a new tube for analysis.
Blood concentrations of AZD-3043 and the acid metabolite were determined in minipigs by LC-MS/MS (Sciex API 3000, Applied Biosystems, Foster City, CA). Samples (25 μL) were injected on a Higgins Analytical C18 Targa column (30 × 0.5 mm; 5 μM) with a flow rate of 0.125 mL/min (Higgins Analytical Inc., Mountain View, CA). Mobile phase A consisted of 0.25% formic acid in water, and mobile phase B consisted of 0.25% formic acid in acetonitrile. The gradient elution started with a 1-minute loading step at 30% B followed by a linear gradient up to 80% B over 2 minutes, a 95% B wash for 30 seconds, and a 1-minute re-equilibration at 10% B. The mass spectrometer was operated in positive ion multiple reaction monitoring mode. The calibration range of the assay was from 0.01 μg/mL (limit of quantification) to 1000 μg/mL in whole blood.
The raw electroencephalography was collected using bipolar, frontal, low impedence surface electrodes and processed into the Bispectral Index (BIS; Aspect Medical Systems, Natick, MA) parameter. The BIS was used as the primary pharmacodynamic signal.
Pharmacokinetic-Pharmacodynamic analysis
The raw concentration versus time data were plotted and inspected. The pharmacokinetic parameters for a three compartment model were estimated using a mixed-effects population approach based on the NONMEM program (University of California, San Francisco, CA) with a log-normal error model applied to interindividual error on each parameter. Model performance was assessed by visual inspection of residual plots and by computing the accuracy and bias of the model predictions as described by Varvel et al.11
The raw BIS versus time data were plotted and inspected. The pharmacodynamic parameters for an inhibitory sigmoidal Emax model with an effect compartment linking the plasma and the effect site12 were also estimated using a naïve pooled approach implemented in NONMEM.
Computer Simulations
Computer simulations using the combined population pharmacokinetic-pharmacodynamic models were performed to provide an illustration of the predicted time-course of AZD-3043 effect-site concentrations and BIS effect after the administration of clinically relevant doses of the drug. For comparison, similar simulations were performed for propofol using pharmacokinetic and pharacodynamic parameters from the literature.13 Furthermore, we performed 80% and 50% decrement time simulations,14 predicting the time necessary to achieve an 80% or 50% decrease in effect site concentration after termination of a variable length, continuous, steady state infusions. The simulations were implemented using PKPD Tools.ψ
Drug Formulations
AZD-3043, THRX-108893 and propanidid were synthesized at Theravance, Inc., while propofol (for in vitro studies) was purchased from Sigma Aldrich (St. Louis, MO). For in vitro electrophysiology, radioligand binding and blood/liver microsome stability studies, test compounds were formulated in dimethyl sulfoxide to provide stock solutions of 10, 1 or 20 mM respectively, which were then diluted with either perfusion buffer or distilled water. For in vivostudies, propofol (Diprivan®, 1%, Astra Zeneca, Caponago, Italy), 1-propanol (Aldrich, HPLC grade), remifentanil (Ultiva®, Abbott Labs, North Chicago, IL) and fentanyl (Fentanyl citrate, Baxter, Deerfield, IL) were purchased. AZD-3043 and propanidid were formulated in lipid emulsions, while THRX-108893 and 1-propanol were formulated in distilled water. Dilutions of AZD-3043, propanidid, propofol, THRX-108893 and 1-propanol were made in their respective vehicles. Dilutions of fentanyl and remifentanil were made in 5% dextrose in distilled water (D5W).
Statistical Analysis
In the rat hypnotic infusion experiments, two types of statistical analyses were performed. For the 20 minute infusion studies, to compare the emergence times of propofol, AZD-3043 and propanidid, a one-way ANOVA with Dunnett’s post-hoc test (statistical significance at p<0.05) was used. In a subsequent study in which the emergence times were analyzed at different infusion durations (i.e., 20 minutes, or 3 or 5 hours), a Student’s unpaired t-test (two-tailed hypothesis testing, with statistical significance at p<0.05) was used to compare the effects of propofol and AZD-3043 at each of the timepoints. Data were analyzed using Prism™ (GraphPad, Inc.) software.
Results
In Vitro Electrophysiology
The vehicle, DMSO, had no effect on the chloride currents recorded in embryonic rat cortical neurons. GABA (3 μM to 1 mM) and the selective GABAA receptor agonist, muscimol (0.3 μM to 1 mM), evoked a concentration-dependent increase in chloride currents. The GABAA receptor selective antagonist, bicuculline (10 μM) inhibited the GABA-induced chloride currents which were therefore concluded to be GABAA receptor-mediated. AZD-3043, propanidid and propofol potentiated the GABA (5 μM; EC20 concentration)-mediated current (EC50 values of 36, 26 and 6 μM respectively; Figures 2A and 2B). The maximum potentiation achieved by AZD-3043 was approximately 65% of that produced by propofol, and 115% of that of propanidid. THRX-108893 (300 μM) had no effect on GABA-evoked current (data not shown).
Figure 2.

Potentiation of GABA (5 μM)-evoked chloride currents in embryonic rat cortical neurons by (A) AZD-3043 (n = 6 cells), propanidid (n = 6 cells) and propofol (n = 4 cells). Data are expressed as the mean (+/− SD) percentage increases in the current evoked by GABA, at its EC50 concentration. A representative, raw trace for AZD-3043 is also shown (B). EC50 is the concentration producing 50% of maximal effect.
Radioligand Binding Studies
Of the 30 binding sites at which they were tested, AZD-3043 and propanidid (each 50 μM) only produced >50% inhibition of specific binding at GABAA chloride channels (83 and 65% respectively); see AZD-3043 data in Supplemental Digital Content 1, Figure 1. Propanidid (50 μM) produced 49% inhibition of strychnine-sensitive glycine receptor binding, while AZD-3043 (50 μM) was inactive. The carboxylate metabolites of AZD-3043 and propanidid produced <50% inhibition of specific binding in all the radioligand binding assays at 200 μM; see THRX-108893 data in Supplemental Digital Content 1, Figure 2.
Stability in Blood and Liver Microsomes
AZD-3043 was rapidly metabolized in human, minipig, dog and cynomolgus monkey liver microsomes (t1/2 values in Table 1). There was no difference in the metabolic stability of AZD-3043 in human liver microsomes in the absence or presence of an NADPH-regenerating system suggesting that the metabolism of AZD-3043 is not mediated by cytochrome P450. Metabolism of AZD-3043 resulted in the formation of the carboxylate metabolite, THRX-108893, in all species evaluated.
Table 1.
The in vitro stability of AZD-3043 in various spieces.
| Preparation | Species | Mean t1/2 value (minutes) |
|---|---|---|
| Liver microsomes | Human | 4 |
| Dog | 7 | |
| Minipig | 0.4 | |
| Cynomolgus monkey | 3 | |
| Whole blood | Human | 27 ± 8* |
| Cat | >60 | |
| Dog | >60 | |
| Minipig | >60 | |
| Rat | 0.6 | |
| Guinea pig | 0.1 |
Data are expressed with respect to the mean t1/2 values from at least two separate studies.
Human whole blood stability experiments were conducted in whole blood from seven separate human donors.
AZD-3043 was rapidly metabolized in rat and guinea pig whole blood (mean t1/2 values of 0.6 and 0.1 minutes respectively), but more slowly metabolized in human blood (mean t1/2 = 27 minutes). In cat, dog orminipig blood, AZD-3043 was stable (Table 1). The rank order of stability of AZD-3043 in blood from the various species tested was cat = dog = minipig> human > rat > guinea pig. When AZD-3043 was metabolized, the formation of THRX-108893 was observed.
Rodent Pharmacology
Hypnosis in response to bolus administration
AZD-3043 (6.5, 13 and 27 mg/kg; n = 4 per dose), propanidid (15, 30 and 45 mg/kg; n = 4, 5 and 3, respectively) and propofol (5, 10 and 20 mg/kg; n = 9, 7 and 7, respectively), but not their vehicles, which were inactive (data not shown), produced a dose-dependent loss of the righting reflex following IV bolus administration (Figure 3A). For each compound, onset of hypnosis was equally rapid (<10 seconds from completion of injection). The slope of the AZD-3043 and propanidid dose-response curves was shallower than that of propofol. In addition, the time to behavioral recovery for AZD-3043 and propanidid, but not propofol, changed little with increasing doses (Figure 3B). THRX-108893 and 1-propanol, the primary metabolites of AZD-3043, were also evaluated to determine if they possessed hypnotic activity. THRX-108893 (200 to 900 mg/kg; n = 2 per dose) failed to produce hypnosis while 1-propanol (200 to 500 mg/kg; n = 3 or 4 per dose) was found to be only weakly active. With respect to the dose of each compound calculated to produce a mean loss of righting reflex of 2 minutes, AZD-3043 was approximately 2.5-fold less potent than propofol, and 2.5-fold more potent than propanidid (potency ratios estimated from the mean doses associated with a 2-minute loss of the righting reflex). The ED50 values for AZD-3043 and propofol (i.e., the doses at which 50% of rats lost their righting reflex, regardless of duration) were 3.3 and 3.0 mg/kg respectively (n=4).
Figure 3.
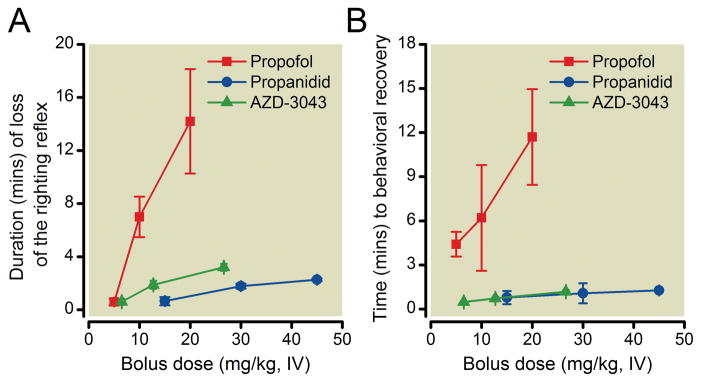
Duration (mean +/− SD) of the loss of righting reflex (A) and time to behavioral recovery (B) produced by AZD-3043 (6.5, 13 and 27 mg/kg; n = 4), propanidid (15, 30 and 45 mg/kg; n = 4, 5 and 3, respectively) and propofol (5, 10 and 20 mg/kg; n = 9, 7 and 7, respectively) following bolus IV administration (1 mL/kg over 5 seconds) in rats.
A study was performed to determine the degree of synergy between opioids and AZD-3043. Remifentanil alone (2.5 μg/kg; n=6) produced no loss of righting reflex and lacked obvious analgesic activity (i.e., a robust withdrawal response to noxious paw pinch was present), while at a higher dose (10 μg/kg) there was a loss of righting reflex in two of six rats and demonstrable analgesia in all animals. Upon co-administration of remifentanil (2.5 or 10 μg/kg), lower doses of AZD-3043, propanidid and propofol were required for hypnosis compared to co-administration with vehicle (Figure 4). The remifentanil-induced leftward shift in the hypnoisis dose-response curve was greater for AZD-3043 than for propofol. Indeed, upon co-administration of 10 μg/kg remifentanil, the dose of AZD-3043 producing 2 minutes loss of righting reflex was lower than that of propofol (approximately 1.5 and 3 mg/kg, respectively; Figure 4). Fentanyl (10 μg/kg) alone produced no loss of righting reflex although analgesia was evident. Upon co-administration of fentanyl (10 μg/kg), the leftward shifts in the hypnosis dose-response curves of AZD-3043 and propofol were similar (Figure 4).
Figure 4.

Duration (mean +/− SD) of the loss of righting reflex produced by AZD-3043 (A; n = 3, 4 or 5) and propofol (B; n = 3, 4 or 5), upon co-administration with remifentanil (2.5 or 10 μg/kg), fentanyl (10 μg/kg) or vehicle (D5W; 1 mL/kg) in rats. D5W = 5% dextrose in water.
Hypnosis in response to IV infusion
Following induction of hypnosis with the approximate 2-minute bolus doses of AZD-3043, propanidid or propofol (15, 30 and 6 mg/kg respectively), hypnosis was maintained by a 20-minute infusion at half that dose·kg−1·min−1. Upon constant infusion of AZD-3043 or propanidid, the depth of hypnosis remained at a similar level, as indicated by a consistent, and small, withdrawal response to intermittent noxious paw pinch with a pair of forceps. In contrast, the depth of hypnosis increased during the 20-minute propofol infusion. Following completion of the infusion, recovery of the righting reflex was rapid in rats treated with AZD-3043 or propanidid, but prolonged and more variable in those that had received propofol (mean durations (±SD) of 1.7±0.2, 2.1±0.9 and 72.1±23.7 minutes respectively; Figure 5A). Similarly, time to behavioral recovery (Figure 5B) was more rapid following AZD-3043 or propanidid infusion (mean durations ((±SD) of 1.1±0.2 and 1.8±0.7 minutes, respectively, compared to propofol (mean duration of 33.1±10.7 minutes).
Figure 5.
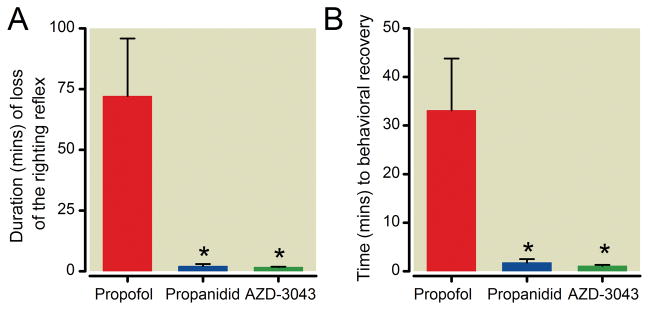
Duration (mean +/− SD) of the loss of righting reflex (A) and time to behavioral recovery (B) following a 20-minute infusion of AZD-3043 (7.5 mg·kg−1·min−1; n = 4), propanidid (20 mg·kg−1·min−1; n = 3) or propofol (3 mg·kg−1·min−1; n = 5) in rats (*p<0.05 compared to propofol, one-way ANOVA and Dunnett’s post-hoc test).
To evaluate further the hypnotic profile of AZD-3043, infusions of 20 minutes, or 3 or 5 hours duration were carried out, and the infusion rate was adjusted, when necessary, to maintain a consistent depth of hypnosis (a small withdrawal reflex to noxious paw pinch). The return of the righting reflex and time to behavioral recovery following termination of the infusion of AZD-3043, but not propofol, occurred rapidly and was relatively unaffected by the duration of infusion (Figure 6).
Figure 6.

Duration (mean +/− SD) of the loss of righting reflex (A) and time to behavioral recovery (B) following a 20-minute, or 3 or 5-hour IV infusion of AZD-3043 (n = 4, 4 or 3, respectively) or propofol (n = 5, 4 or 5, respectively) in rats (*p<0.05 compared to propofol at the same time-point, Student’s unpaired t-test). The infused dose was adjusted as necessary to maintain a small withdrawal reflex to noxious paw pinch.
Rat Electroencephalography
The electroencephalography was recorded in rats to compare the effects of AZD-3043 and propofol on brain activity, and their duration of action following bolus IV dosing. The electroencephalography analysis was able to detect modulation of electrocortical activity produced by AZD-3043 and propofol within 5 to 10 seconds following their administration. Administration of AZD-3043 (5 to 30 mg/kg) and propofol (1 to 10 mg/kg), but not their respective vehicles, produced a rapid, dose-dependent suppression of the rat electroencephalogram; see AZD-3043 and propofol processed data and raw electroencephalogram tracings in Supplemental Digital Content 1, Figures 3–16. Suppression of the electroencephalogram by AZD-3043 (5 to 30 mg/kg) was short-lived (recovery within approximately 5 minutes). Propofol produced a greater, dose-dependent increase in the duration of electroencephalographic suppression and decrease in the slope of the electroencephalogram recovery relative to AZD-3043 (see Supplemental Digital Content 1, Figures 3–16).
Large Mammal Pharmacology
All fiveminipigs completed the experiments. One male and four female swine, weighing 27.4 to 35.0 kg (average = 30.5±3.3 kg) were used. No infusion was terminated early because of an adverse event, such as severe bradycardia, tachycardia, or hypotension.
Pharmacokinetic-Pharmacodynamic Analysis
Four datapoints out of 105 were removed from the pharmacokinetic analysis because they were considered to be outliers due to a sample processing problem. Figure 7A shows the raw concentration-time curves for all animals.
Figure 7.

(A) The dose normalized raw concentration versus time data from the porcine model. AZD-3043 concentration is shown on a linear scale. The black bar represents the 20 min infusion of AZD-3043. The concentrations from the single animal receiving 3.0 mg·kg−1·min−1 were divided by 2 in order to dose normalize the raw data for ease of plotting.(B) The raw effect versus AZD-3043 effect site concentration (“collapsed” hysteresis loops) and the estimated concentration-effect relationship. Dots and solid lines represent the individual data. The bold solid line represents the population prediction based on a naïve pooled analysis. BIS = Bispectral Index Scale
The three compartment model adequately described the pharmacokinetics of AZD-3043. The best model parameters along with the numerical measures of model performance are shown in Table 2. In general terms, these models have excellent performance in terms of bias and accuracy.
Table 2.
Population compartmental pharmacokinetic parameters by mixed effect modeling and pharmacodynamic parameters by naïve pooled analysis.
| Estimate | CV (%) | 95%CI | |
|---|---|---|---|
| Pharmacokinetic parameters | |||
| Volumes (l) | |||
| Central | 2.84 | 24.7 | 2.17, 3.39 |
| Peripheral1 | 5.6 | 32.4 | 4.24, 7.62 |
| Peripheral2 | 5.37 | 34.1 | 3.31, 7.09 |
| Clearances (L/min) | |||
| Central | 1.63 | 13.8 | 1.42, 1.86 |
| Intercompartmental1 | 0.15 | 36.7 | 0.10, 0.28 |
| Intercompartmental2 | 0.486 | 46.3 | 0.33, 0.69 |
| MDPE | 0.062 | ||
| MDAPE | 0.181 | ||
| Pharmacodynamic parameters | |||
| E0 | 84.6 | ||
| Emax | 0.705 | ||
| Ce50 (μ/mL) | 16.7 | ||
| γ | 4.42 | ||
| keo (/min) | 0.242 | ||
CV = coefficient of variation (Omega from NONMEM); 95%CI = the 95% confidence intervals (computed from a bootstrap technique with 1000 replications); MDPE = median prediction error; MDAPE = median absolute prediction error; E0 = predrug effect; Emax = maximal effect; Ce50 = 50% effective concentration; γ = the steepness of the concentration-effect relationship; keo = first-order rate constant characterizing effect site equilibration kinetics.
where n is the total number of samples in the study population. The median prediction error (MDPE), a measure of model bias, was also computed for each model.
Data from one animal was excluded from the pharmacodynamic analysis because of an inadequate electroencephalographic signal. Figure 7B shows the shapes of the raw concentration-effect relationships and the population estimate. The pharmacodynamics were well described by the model (model parameters displayed in Table 2).
Computer simulations
The simulations revealed substantial differences in the predicted clinical pharmacology behavior of AZD-3043 and propofol. The simulations of a combined bolus and infusion dosing scheme for the two drugs are displayed in Figure 8A; these simulations predict a substantially faster drug off-set for AZD-3043 compared to propofol. The decrement time simulations displayed in Figure 8B illustrate several important points. First, the 50% decrement times showed moderate difference between two drugs for long infusions. Second, AZD-3043 exhibits a substantially shorter 80% decrement time than propofol especially after long infusion.
Figure 8.

(A) A simulation of a total body weight based dosage of a bolus and infusion for 30 kg pig. Propofol’s kinetic parameters are from Johnson et al.13 The propofol dosing scheme was designed to achieve peak concentrations near the effect site concentration estiamted to produce 50% of maximal effect (i.e., the Ce50). AZD-3043 dosing scheme (3-fold higher than propofol) was based on the current experiment design (1.5 mg·kg−1·min−1 for AZD-3043 vs. 0.5 mg·kg−1·min−1 for propofol). (B) A simulation of the time necessary to achieve a 80% or 50% decrease in effect site concentration after the termination of a continuous infusion targeting a constant concentration.
Discussion
Propofol is a positive allosteric modulator of the GABAA receptor and produces sedation/hypnosis by potentiating GABAA-mediated neuronal inhibition within the CNS.1,2,15 Clinically, dose titration of propofol is common, particularly upon prolonged infusion, to ensure acceptable emergence from sedation or hypnosis. AZD-3043 is a novel sedative/hypnotic agent, that was designed to be rapidly hydrolyzed by esterases in liver or blood. The in vitro stability data in the present study demonstrated that AZD-3043 is metabolized rapidly in liver microsomes from a variety of species, including man. Interestingly, a species-dependent rate of metabolism of AZD-3043 was evident in whole blood. Upon metabolism of AZD-3043, the formation of the corresponding carboxylic acid metabolite, THRX-108893, was observed, suggesting that AZD-3043 is metabolized by esterase present in the blood. A more complete description of the in vitro and in vivo pharmacokinetic profile of AZD-3043 will be the subject of a future publication.
Whole cell patch clamp data confirmed that like propofol and propanidid, AZD-3043 potentiated GABAA-mediated chloride currents. In the embryonic rat cortical neuron preparation, AZD-3043 had a similar potency to propanidid but was less potent than propofol. The significance of the apparent lower intrinsic activity of AZD-3043 and propanidid relative to propofol in these rat neurons is unclear. It is possible that AZD-3043 and propofol possess different efficacies and/or affinities for the many subtypes of GABAA receptors in the CNS. Pentameric in structure, the GABAA receptor consists of numerous combinations of different subunits (i.e. α, β, γ, δ, ε, θ and π), and although more than 10,000 pentameric subunit combinations are possible, it is postulated that fewer than 10 subtypes contribute to the major physiological responses to GABA in the adult mammalian brain.16 It is also possible that the low aqueous solubilities of each test agent, despite the absence of any visible compound precipitation, contributed to the apparent differences noted in their intrinsic activities. Notwithstanding, in vivo, AZD-3043 and propofol produced a similar degree of electroencephalographic suppression in pigs, cats (unpublished observations; contemporaneous cat experiment data on file at Theravance, South San Francisco; the studies were conducted between September and December 2004, supervised by David T Beattie Ph.D., San Francisco, CA; Cat electroencephalogram, cardiovascular parameters and blood chemistries were assessed) and rats.
AZD-3043, but not its carboxylate metabolite, THRX-108893, inhibited GABAA ([35S]tert-butylbicyclophosphorothionate) binding in the in vitro radioligand binding studies. AZD-3043 had little or no affinity for a variety of other CNS-expressed receptors and ion channels. The binding and electrophysiological data are consistent with the proposal that AZD-3043 is a positive allosteric modulator of GABAA activity, and as such, should potentiate GABAA-mediated inhibitory neurotransmission in the CNS.
AZD-3043 produced hypnosis in rats, with rapid onset, following bolus IV administration. Despite similar potencies in experiments with the embryonic rat cortical neurons, AZD-3043 was approximately 2.5-fold more potent than propanidid following bolus IV dosing. Importantly, THRX-108893 and 1-propanol, the primary metabolites of AZD-3043, were either inactive or only very weak hypnotic agents. Upon return of the righting reflex, the time to behavioral recovery was rapid, and remained so, with increasing doses of AZD-3043 or propanidid, in contrast to propofol. Following bolus IV administration to rats, AZD-3043 produced a transient and dose-dependent suppression of electroencephalographic activity with rapid onset of action. Such activity is consistent with a sedative/hypnotic agent possessing rapid CNS penetration and clearance. The recovery of the electroencephalogram, following suppression with AZD-3043, was more rapid and less influenced by increasing bolus dosage than with propofol. In general, the electroencephalographic (BIS) data obtained in the porcine model paralleled the findings from the rodent studies, although a clear limitation of the pig experiments was the concomitant administration of isoflurane (for ethical reasons).
In the majority of general anesthetic procedures, opioids are co-administered with hypnotic agents. Synergy between a hypnotic agent and an opioid is commonly observed, the analgesia conferred by the opioids allowing the dose of hypnotic to be reduced.17 Interestingly, in this study the leftward shift in the hypnosis dose-response curve of AZD-3043 was greater than that of propofol when remifentanil was co-administered, although similar upon fentanyl co-administration. Upon bolus dosing of remifentanil and AZD-3043, esterases may have been saturated transiently and the synergy with these agents was thus more marked than the remifentanil/propofol combination. The similarly large reduction in the hypnotic dose of propanidid, upon co-administration of remifentanil, supports such an hypothesis. The data are consistent with a reduction of the required clinical dose of AZD-3043 when used in combination with opioids.
The most important finding in this study was that the time to emergence from AZD-3043-induced hypnosis was rapid, and relatively unaffected by the duration of infusion, in contrast to findings with propofol. It was also apparent that the infused dose of AZD-3043 required less alteration, in comparison to that of propofol, to maintain a consistent depth of hypnosis throughout (as monitored by the, admittedly subjective, observation of noxious stimulus-induced paw withdrawal). The infusion data for AZD-3043 are consistent with a short, and constant, 80% or 50% decrement time. These results suggest that the use of AZD-3043 in the clinic may allow more rapid and predictable patient emergence, more precise control of hypnotic depth and less requirement for dose titration upon prolonged infusion, in comparison to propofol. Of course it is very important to emphasize that the pharmacokinetic simulations from the animal models may not be predictive of human pharmacology and require confirmation in man.
From a pharmaceutics perspective, AZD-3043 is a “soft drug.” Soft drugs are molecules that are purposefully designed to be rapidly metabolized (metabolically labile).18 In anesthesia, the soft drug concept is useful because it enables precise titration to effect and rapid recovery. The short acting opioid remifentanil and beta-blocker esmolol are familiar applications of this pharmaceutic approach in perioperative practice. AZD-3043, CNS7056 and MOC-etomidate (and others) are all more recent examples of the soft drug trend in anesthesia drug discovery.19–21 The first-in-man studies of AZD-3043 have now been completed and the data analysis is ongoing. Given the unique requirements of anesthesia therapeutics, we can expect to see more soft drugs in anesthesiology in the future.
Supplementary Material
Final Boxed Summary Statement.
What we already know about this topic
AZD-3043 is a chemical analog of propanidid that was designed to be a rapidly metabolized hypnotic (“soft drug”) because of its ester moiety
What this article tells us that is new
AZD-3043 is metabolized rapidly by liver microsomes
It is a positive allosteric modulator of γ-aminobutyric acidA-mediated chloride currents
It produces rapid onset, dose-dependent electroencephalograph activity suppression in rats and pigs
Emergence from AZD-3043-induced hypnosis is rapid and relatively unaffected by dose and infusion duration
Acknowledgments
Funding Statement: All studies were by supported by Theravance, Inc., 901 Gateway Boulevard, South San Francisco, CA 94080 USA
Footnotes
Minto and Schnider, URL; http://www.pkpdtools.com/doku.php?id=start, last accessed December 14, 2011
References
- 1.Peduto VA, Concas A, Santoro G, Biggio G, Gessa GL. Biochemical and electrophysiologic evidence that propofol enhances GABAergic transmission in the rat brain. Anesthesiology. 1991;75:1000–9. doi: 10.1097/00000542-199112000-00012. [DOI] [PubMed] [Google Scholar]
- 2.Lambert JJ, Belelli D, Pistis M, Hill-Venning C, Peters JA. The Interaction of Intravenous Anesthetic Agents with Native and Recombinant GABAA Receptors. In: Enna SJ, Bowery NG, editors. The GABA Receptors. Totowa, NJ: Humana Press Inc; 1997. pp. 121–56. [Google Scholar]
- 3.Shafer A, Doze VA, Shafer SL, White PF. Pharmacokinetics and pharmacodynamics of propofol infusions during general anesthesia. Anesthesiology. 1988;69:348–56. doi: 10.1097/00000542-198809000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Gunner BW, Harrison GA, Walker WD, Jenkinson IS. Propanidid, a new short-acting anaesthetic. Med J Aust. 1965;2:327–9. doi: 10.5694/j.1326-5377.1965.tb25278.x. [DOI] [PubMed] [Google Scholar]
- 5.Habazettl H, Vollmar B, Rohrich F, Conzen P, Doenicke A, Baethmann A. Anesthesiologic efficacy of propanidid as a liposome dispersion. An experimental study with rats. Anaesthesist. 1992;41:448–56. [PubMed] [Google Scholar]
- 6.Glen JB, Hunter SC. Pharmacology of an emulsion formulation of ICI 35 868. Br J Anaesth. 1984;56:617–26. doi: 10.1093/bja/56.6.617. [DOI] [PubMed] [Google Scholar]
- 7.Lindsay RM, Evison CJ, Winter J. A Practical Approach. New York: Oxford University Press; 1991. Culture of adult mammalian peripheral neurons, Cellular Neurobiology; pp. 3–17. [Google Scholar]
- 8.Lingamaneni R, Krasowski MD, Jenkins A, Truong T, Giunta AL, Blackbeer J, MacIver MB, Harrison NL, Hemmings HC., Jr Anesthetic properties of 4-iodopropofol: Implications for mechanisms of anesthesia. Anesthesiology. 2001;94:1050–7. doi: 10.1097/00000542-200106000-00020. [DOI] [PubMed] [Google Scholar]
- 9.Steffensen SC, Lee RS, Henriksen SJ, Packer TL, Cook DR. A novel electroencephalographic analysis method discriminates alcohol effects from those of other sedative/hypnotics. J Neurosci Methods. 2002;115:145–56. doi: 10.1016/s0165-0270(02)00009-2. [DOI] [PubMed] [Google Scholar]
- 10.Ko JC, Williams BL, Smith VL, McGrath CJ, Jacobson JD. Comparison of Telazol, Telazol-ketamine, Telazol-xylazine, and Telazol-ketamine-xylazine as chemical restraint and anesthetic induction combination in swine. Lab Anim Sci. 1993;43:476–80. [PubMed] [Google Scholar]
- 11.Varvel JR, Donoho DL, Shafer SL. Measuring the predictive performance of computer-controlled infusion pumps. J Pharmacokinet Biopharm. 1992;20:63–94. doi: 10.1007/BF01143186. [DOI] [PubMed] [Google Scholar]
- 12.Holford NH, Sheiner LB. Understanding the dose-effect relationship: clinical application of pharmacokinetic-pharmacodynamic models. Clin Pharmacokinet. 1981;6:429–53. doi: 10.2165/00003088-198106060-00002. [DOI] [PubMed] [Google Scholar]
- 13.Johnson KB, Egan TD, Kern SE, White JL, McJames SW, Syroid N, Whiddon D, Church T. The influence of hemorrhagic shock on propofol: A pharmacokinetic and pharmacodynamic analysis. Anesthesiology. 2003;99:409–20. doi: 10.1097/00000542-200308000-00023. [DOI] [PubMed] [Google Scholar]
- 14.Youngs EJ, Shafer SL. Pharmacokinetic parameters relevant to recovery from opioids. Anesthesiology. 1994;81:833–42. doi: 10.1097/00000542-199410000-00010. [DOI] [PubMed] [Google Scholar]
- 15.Lin LH, Chen LL, Zirrolli JA, Harris RA. General anesthetics potentiate gamma-aminobutyric acid actions on gamma-aminobutyric acidA receptors expressed by Xenopus oocytes: Lack of involvement of intracellular calcium. J Pharmacol Exp Ther. 1992;263:569–78. [PubMed] [Google Scholar]
- 16.McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–43. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- 17.Avramov MN, White PF. Use of alfentanil and propofol for outpatient monitored anesthesia care: determining the optimal dosing regimen. Anesth Analg. 1997;85:566–72. doi: 10.1097/00000539-199709000-00015. [DOI] [PubMed] [Google Scholar]
- 18.Egan TD. Is anesthesiology going soft?: Trends in fragile pharmacology. Anesthesiology. 2009;111:229–30. doi: 10.1097/ALN.0b013e3181ae8460. [DOI] [PubMed] [Google Scholar]
- 19.Sneyd JR, Rigby-Jones AE. New drugs and technologies, intravenous anaesthesia is on the move (again) Br J Anaesth. 2010;105:246–54. doi: 10.1093/bja/aeq190. [DOI] [PubMed] [Google Scholar]
- 20.Kilpatrick GJ, McIntyre MS, Cox RF, Stafford JA, Pacofsky GJ, Lovell GG, Wiard RP, Feldman PL, Collins H, Waszczak BL, Tilbrook GS. CNS 7056: A novel ultra-short-acting Benzodiazepine. Anesthesiology. 2007;107:60–6. doi: 10.1097/01.anes.0000267503.85085.c0. [DOI] [PubMed] [Google Scholar]
- 21.Cotten JF, Husain SS, Forman SA, Miller KW, Kelly EW, Nguyen HH, Raines DE. Methoxycarbonyl-etomidate: A novel rapidly metabolized and ultra-short-acting etomidate analogue that does not produce prolonged adrenocortical suppression. Anesthesiology. 2009;111:240–9. doi: 10.1097/ALN.0b013e3181ae63d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.