

Abstract
The endoplasmic reticulum (ER) senses both extracellular and intracellular stresses that can disrupt its ability to facilitate the maturation of proteins destined for secretory pathways. The accumulation of misfolded proteins within the ER triggers an adaptive signaling pathway coined the Unfolded Protein Response (UPR). UPR activation contributes to cell adaptation by reducing the rate of protein translation, while increasing the synthesis of chaperones. Although we have gained considerable insight into the mechanisms that regulate gene expression and certain aspects of protein translation, the contribution of micro-RNAs (miRNAs) to UPR-dependent activities has only recently been investigated. Here, we highlight recent insights into the contribution of miRNAs to UPR-dependent cellular adaptive responses.
Keywords: PERK, IRE1α, ATF6, ER stress, UPR, microRNA, apoptosis
The Unfolded Protein Response (UPR) signal transduction pathway
The UPR constitutes a signal transduction pathway that responds to perturbations in the folding and maturation of proteins within the endoplasmic reticulum (ER). The oxidative, calcium rich lumen of the ER is sensitive to a variety of stresses such as viral infection, oncogene activation, and metabolic deprivation (a frequent consequence of neoplastic growth or metabolic disorders such as diabetes or obesity). Such stresses compromise protein folding within the ER, thereby triggering the UPR as a mechanism to regulate cell homeostasis and ultimately cell fate. The UPR consists of three upstream regulators: protein kinase RNA-like endoplasmic reticulum kinase (PERK), Inositol regulated enzyme 1 alpha (IRE1α/β; there are two isoforms, alpha and beta with alpha being the ubiquitous isoform), and Activating Transcription Factor 6 (ATF6; Figure 1). Both PERK and IRE1 are type 1 single pass protein kinases. Upon activation, PERK phosphorylates eukaryotic initiation factor 2 alpha (eIF2α), which results in reduced translation initiation of many mRNA transcripts. Phosphorylation of eIF2α concurrently increases translation efficiency of select transcripts, such as that of Activating Transcription Factor 4 (ATF4). Increased translation of ATF4 is a consequence of overlapping short open reading frames in the ATF4 5’UTR. ATF4 regulates a transcriptional program that contributes, paradoxically, to both pro-survival and pro-death cellular programs [1]. Pro-survival transcripts include amino acid transporters and regulators of redox control [2]. A key pro-apoptotic transcript directly activated by ATF4 is chop/gadd153, a C/EBP family member linked to cellular apoptosis. Proper regulation of chop expression appears to play a key role in cellular commitment to apoptosis [3]. IRE1α, although a protein kinase, also harbors nuclease activity that is essential for regulated splicing of the X-box binding protein 1 (Xbp1) transcription factor and for limiting ER-bound transcripts during cellular stress [4] [5, 6]. ATF6, a transmembrane transcription factor, undergoes stress-dependent translocation to the Golgi apparatus, where it is cleaved thereby allowing nuclear accumulation of the N-terminal transcriptional regulatory domain [7]. ATF6 regulates transcriptional activation of genes, such as chaperones, whose products facilitate adaptation to stress [8].
Figure 1. Signal transducers of the UPR.
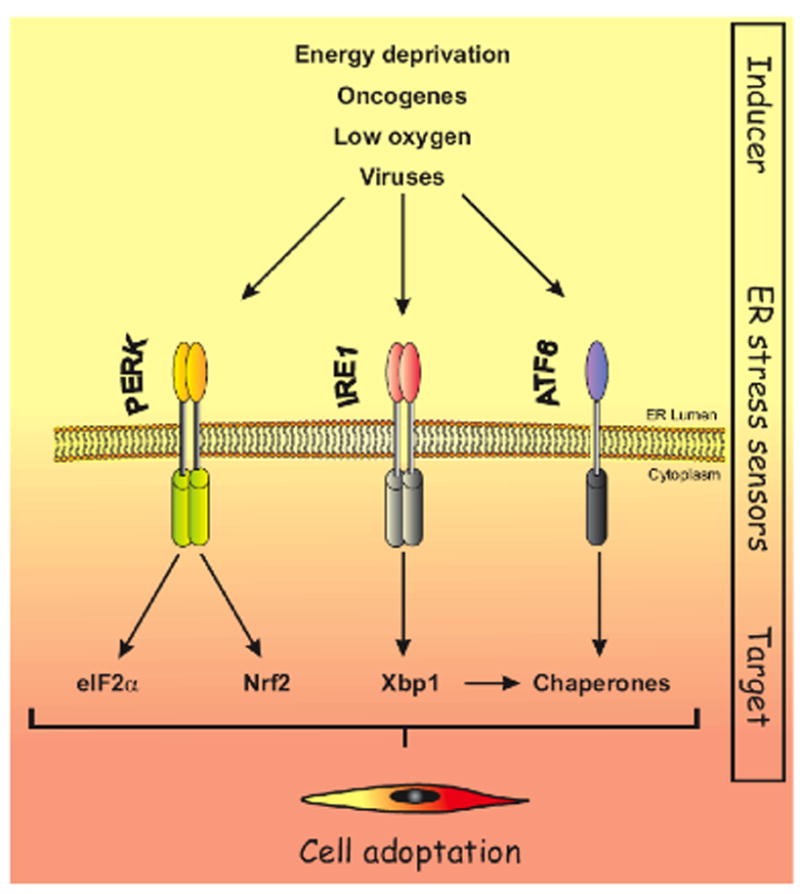
The Unfolded Protein Response (UPR) is triggered under conditions of cellular stress such as energy deprivation, oncogene activation, low oxygen (hypoxia), and viral challenge. Signals from the stressed ER trigger the activation of two transmembrane protein kinases, IRE1α and PERK as well the transmembrane transcription factor ATF6. PERK phosphorylates eIF2α which regulates global protein translation and Nrf2 which regulates expression of anti-oxidant enzymes. IRE1α activates transcription factor Xbp1 via RNase-dependent removal of an intron that prevents efficient translation; ATF6 regulates transcription of chaperone genes. Collectively, PERK, Ire1 and ATF6 signaling modulates cellular adaptation in response to ER stress.
Although we understand the basic activation and general function of UPR components, there remain significant gaps in our understanding of the molecular mechanisms whereby they regulate cell fate. Indeed, recent work has implicated small 20-22 nucleotide RNAs, commonly referred to as microRNAs/miRs (also known as miRNAs or miRs), in the regulation of life and death decisions following ER stress, and all three UPR signal transducers contribute to miRNA regulation. We now discuss emerging data describing UPR-dependent regulation of key miRNAs (Table 1) and their contribution to cell survival.
Table 1.
ER stress dependent microRNAs
| miRNA | Target(s) | UPR Pathway | Function | Ref |
|---|---|---|---|---|
| miR-211 | CHOP | PERK/ATF4 | Pro-survival | Chitnis et al., 2012 |
| miR-204 | CHOP | PERK/ATF4 | Pro-survival | Chitnis et al., 2012 |
| miR-708 | Rhodopsin/Neurotanin | ?/CHOP | Homeostasis | Behrman et al., 2011 |
| miR-30c-2p | Xbp-1 | PERK/NF-κB | Pro-apoptotic | Byrd et al., 2012 |
| miR-106 | Bim | PERK/ATF4 | Pro-survival | Gupta et al., 2012 |
| miR-17 | Caspase-2 | IRE1α | Pro-survival | Upton et al., 2012 |
| miR-34a | Caspase-2 | IRE1α | Pro-survival | Upton et al., 2012 |
| miR-96 | Caspase-2 | IRE1α | Pro-survival | Upton et al., 2012 |
| miR-125b | Caspase-2 | IRE1α | Pro-survival | Upton et al., 2012 |
| miR -346 | TAP1 | XBP1 | Immunomodulation | Bartoszewski et al., 2011 |
MiRNAs contribute to PERK-dependent regulation of cell fate
Although there is significant data to support a pro-survival function for PERK, PERK-dependent activation of ATF4 also regulates expression of chop and thereby establishes a pro-apoptotic pathway for cells experiencing stress. Because maximal accumulation of the CHOP protein requires extended or chronic stress, mechanisms that contribute to its temporal regulation could conceivably contribute to a switch from a pro-survival to a pro-death fate. Until recently, our understanding of the mechanisms that antagonize CHOP accumulation has been limited. New insights regarding the regulation of chop expression have stemmed from an unbiased search for miRNAs that respond to PERK activation. This approach led to the identification of miR-211 as a PERK-inducible miRNA (Figure 2) [9]. Although the expression of miRNAs can be directed by their own unique promoter, other miRNAs are physically located in introns of protein coding genes resulting in their expression being somewhat dependent upon host gene expression. MiR-211 is embedded in an intron of trpm1, which encodes a calcium channel. MiR-211 lacks its own regulatory sequences, and its induction depends on PERK-dependent activation of ATF4. A striking feature of miR-211 regulation is not only its rapid accumulation, with expression peaking 5 hours post-stress induction, but also its precipitous decline, reaching baseline levels by 8 hours post-stress induction. Although the mechanism underlying its downregulation remains to be discerned, the transient nature of miR-211 accumulation suggests that its key target must be repressed during early stages of ER stress, but at later time points target expression would be observed as miR-211-dependent silencing is relieved. This indeed appears to be the case, as chop was identified as a direct and critical target of miR-211. Antagonizing miR-211 expression resulted in increased CHOP accumulation and premature commitment to apoptosis, whereas overexpression of miR-211 antagonized CHOP induction and cell death.
Figure 2. Multiple miRNAs contribute to cell adaptation following UPR activation.
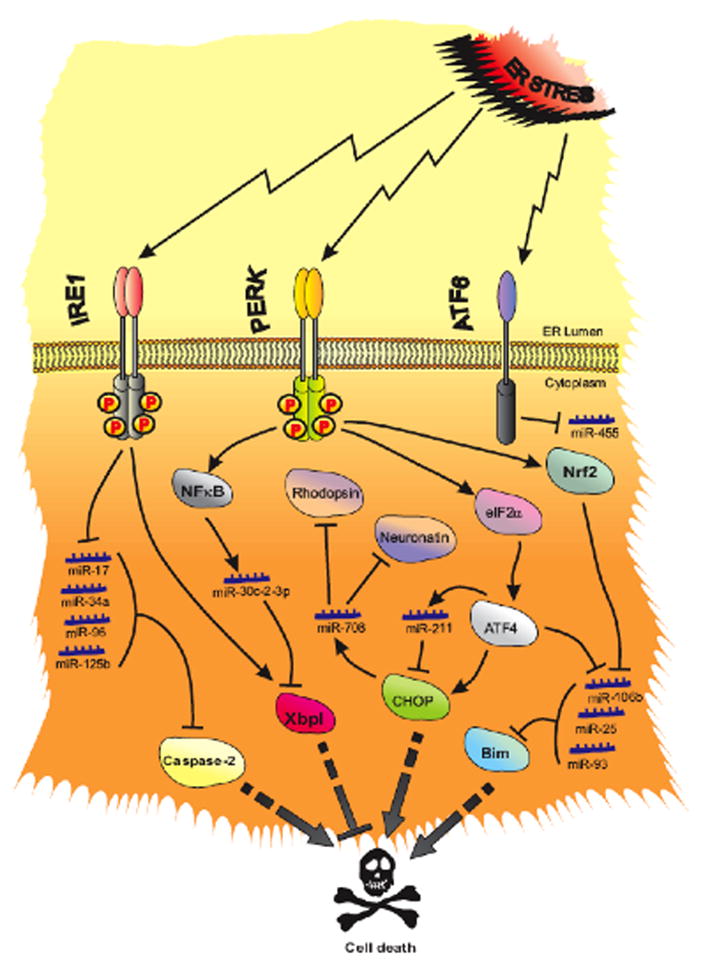
ER stress induces IRE1α, PERK, and ATF6. IRE1α degrades precursors of miR-17, miR-34a, miR-96 and miR-125b leading to elevation of their target, caspase-2, and sets the stage for apoptosis. PERK phosphorylates eIF2α, induces ATF4, CHOP and NF-κB. ATF4 induces miR-211, which in turn attenuates CHOP expression and prevents premature apoptosis. Upon expression, CHOP induces miR-708, which in turn targets Rhodopsin and Neuronatin. NF-κB activation is necessary for expression of miR-30-2-3p and moderation of IRE1α-dependent induction of the pro-survival transcription factor, XBP1. PERK also induces Nrf2 (Nuclear Factor-like-2), which along with ATF4, suppresses the miR-106b-25 cluster (miR-106b, miR-25, and miR-93) enabling induction of pro-apoptotic Bim. The third upstream component of ER stress signaling, ATF6, suppresses miR-455, and induces calreticulin which in turn contributes to proper protein folding in heart.
An additional striking feature of miR-211 function stems from its mechanism of action. Most miR targets are regulated through mRNA degradation or translational silencing, which is determined by base-pairing between the miR and seed sequences in the target 3’UTR. This mechanism depends upon incorporation of the processed, mature miR into RNA-induced silencing complex (RISC). The key components of RISC are the miR and one of four Argonaute (Ago) proteins that harbor RNA slicing activity; the miR functions as the specificity factor that guides Ago to a target mRNA. Chop, however, lacks any potential regulatory seed sequence in its 3’UTR; rather, the chop promoter harbors two seed sequence matches for miR-211, and these sequences mediate miR-211-dependent transcriptional repression of chop. Thus, in contrast to the canonical mode of miR action, which occurs in the cytoplasm, this mode requires nuclear import of a miR-containing complex (RITS, RNA-Induced Transcriptional Silencing) that. like RISC. harbors an Ago protein, but in addition also contains a histone methyltransferase that catalyzes marks associated with transcriptional repression (e.g. EZH2). Target specificity is determined by base pairing between the miR and seed sequences in a target promoter [9]. Critically, the transient accumulation of miR-211 is mirrored precisely by a transient accumulation of the repressive histone modification of H3K27me3 in the chop promoter [9].
Intriguingly, although CHOP is subject to regulation by miR-211, it also regulates the induction of miR-708 [10]. The induction of miR-708, and consequentially its host gene odz4, is a feature of CHOP transcriptional activation. One confirmed target of miR-708 is rhodopsin, a highly synthesized and key component of the developing retina. MiR-708 induction limits or “fine-tunes” rhodopsin synthesis in the developing eye, thereby preventing translational stress and accumulation of misfolded rhodopsin protein within the ER. Though miR-708 expression has not been formally shown to be dependent on PERK, given its dependence on CHOP one would anticipate that PERK induction might also mediate ER stress-dependent induction of miR-708. In addition to regulation of rhodopsin, miR-708 has also been implicated as a putative regulator of cancer metastasis [11] through regulation of the resident ER protein, neuronatin (Figure 2). Neuronatin is a regulator of cellular calcium levels and through this, contributes to mitogen activated protein kinase (MAPK) and focal adhesion kinase (FAK) activity. The downregulation of miR-708 correlated with increased neuronatin and increased metastatic spread, as would be expected with increased MAPK and FAK activity. Whether PERK and/or CHOP contribute to miR-708 expression in this context remains to be ascertained.
Are there additional PERK-regulated miRs that contribute to the switch between adaptation and commitment to cell death? To address this issue, Byrd et al. used a computational approach to search for miRs that potentially regulate the accumulation of Xbp1. Xbp1 is induced downstream of Ire1, and it activates transcription of genes that encode enzymes involved in protein folding and maturation within the ER[12]. Using algorithms that predict miR targets, miR-30c-2-3p was identified as a likely regulator of Xbp1. miR-30c-2-3p was subsequently found to be transcriptionally induced by ER stress via PERK. Although many of the transcriptional targets of PERK are regulated by ATF4, the induction of miR-30c-2-3p is dependent upon activation of Nuclear Factor kappa B (NF-κB). The activation of NF-κB following ER stress is triggered by PERK-dependent translational inhibition of its inhibitor, IκB.
Because miR-30c-2-3p antagonizes accumulation of Xbp1, which in turn elicits a proadaptive, pro-survival transcriptional program, one would anticipate that miR-30c-2-3p antagonizes cell survival under conditions of ER stress. Consistent with this notion, inhibition of endogenous miR-30c-2-3p decreased UPR-mediated cell death. Thus, miR-30c-2-3p is engaged in cross-talk between the PERK and Ire1 branches of the UPR and, like miR-211, regulates the capacity of a cell to adapt to cell stress [13]. Based upon our current molecular understanding, miR-211 and miR-30c-2-3p function to moderate an excessive pro-death and pro-survival response, respectively, thus maintaining homeostasis.
PERK not only induces key miRNAs, but it has also been implicated in the downregulation of certain miRNAs [9, 14] (Table 1). One class of miRNAs whose repression is dependent upon PERK is the miR-106b-25 cluster, composed of miR-106b, miR-25, and miR-93 [14]. Downregulation of miR-106b-25 is dependent on ATF4 and Nrf2, the latter being a direct PERK substrate [15]. Unlike miR-30c-2-3p and miR-211, which regulate factors that indirectly regulate cell death, the miR-106b-25 cluster targets the pro-apoptotic BH3 only protein, BIM. Thus, regulation of the miR-106b-25 cluster, like miR-211 and miR-30c-2-3p, may also function to establish a point of no return wherein cells commit to an apoptotic fate.
PERK activity can also be attributed to repression of miR-199a/214 through its capacity to induce NF-κB (Saito et al., 2011). MiR-214 down-regulation contributes to UPR signaling through de-repression of XBP-1 and ATF4 [16]. Overexpression of miR-214 can reduce proliferation, colony formation and induced apoptosis. Moreover, interaction of miR-214 with ATF4 mRNA was implicated in reduced bone formation [17]. Additional miRNAs that are subject to repression include miR-221/222 [18]. The ability of a miR-221 and 222 mimetics to accelerate ER-stress-induced apoptosis supports the importance of this regulatory process.
The accumulating data reveal a role for miRNAs in the mediation of PERK function. The majority of the miRNAs have pro-survival activity that is consistent with their demonstrated target specificity. Understanding the broader significance of PERK-dependent miRNAs requires continued investigation.
IRE1α antagonizes anti-apoptotic miRNA accumulation
Aspects of cell commitment to and ultimately execution of apoptosis depend upon the activation of cellular caspases. Mitochondrial execution caspase-3 contributes to cell death; however, mechanisms linking UPR signal transducers with caspase activation remain to be firmly established. Current data suggest that activation of caspase-2 during ER stress is required for activation of caspase-3 [19]. How, then, is caspase-2 regulated? Insights into caspase-2 regulation emerged from work focusing on IRE1α associated pro-apoptotic activities. Like PERK, IRE1α mediates both pro-apoptotic and pro-survival responses. IRE1α -mediated prosurvival functions are tightly linked with its inherent RNase activity. IRE1α activates Xbp1 by mediating a splicing event that results in generation of an alternate Xbp1 mRNA that is translated more efficiently; Xbp1 in turn induces a pro-survival gene expression program that facilitates cellular adaptation. In addition, Ire1 can directly target and degrade mRNAs that cotranslationally traffic into the ER [20]. Through this mechanism of mRNA degradation, IRE1α cooperates with PERK to limit protein load in the ER during cellular stress. Conversely, the proapoptotic function of IRE1α has been linked to its capacity to phosphorylate and activate c-Jun N-terminal kinase (JNK) [21]. Collectively, these observations suggest a model wherein IRE1α -RNase function is pro-survival, whereas its protein kinase activity regulates apoptotic signaling. However, recently published work refutes such a simplistic model. Through experimental dissection of the mechanism of apoptosis induction, Upton et al. elucidated a requisite role for IRE1α in brefeldin A-dependent induction of caspase-2 protein accumulation and activation [22]. Strikingly, caspase-2 protein accumulation was strictly dependent upon IRE1α RNase activity, but independent of its kinase function. However, cleavage and thus activation of caspase-2 depended on both IRE1 activities, revealing the need for a second IRE1-dependent signal.
Concerning the mechanism of IRE1α-mediated caspase-2 accumulation, a bioinformatic search revealed high confidence binding sites for miRs-17, 34a, 96 and 125b (Table 1) in the caspase-2 3’UTR [22]. Importantly, IRE1 activation reduces the levels of RNA precursors of these miRNAs in cells, and specifically cleaves precursor-miRNAs at sites distinct from those targeted by the miRNA maturation RNase, Dicer. IRE1α-dependent suppression of miR-17 contributes to cell death by increasing thioredoxin-interacting protein (TXNIP), which in turn causes procaspase-1 cleavage and interleukin 1β secretion [23]. Thus, through its ability to selectively impair the biogenesis of these select miRNAs, IRE1α facilitates the accumulation of caspase-2, thereby setting the stage for cellular commitment to apoptosis.
ER stress, micro-RNAs and the immune response
ER stress can modulate immune responses through regulation of MHC class I surface expression [24]. Peptides degraded by proteasome are translocated by ATP-binding cassette transporter (TAP1) into ER lumen. These peptides are then presented on cell surface with MHCI. ER stress reduces MHC-class I antigen presentation and suppresses TAP1 levels. XBP-1-dependent miR-346 links immunomodulation with ER stress by suppressing TAP1 mRNA [25]. A detailed analysis of how miR-346 regulates immune response will offer further insights into how ER stress regulates immune response.
An ATF6-dependent miRNA contributes to cardioprotection
Analogous to PERK and IRE1, ATF6 activation also contributes to differential expression of miRNAs. Our understanding of ATF6 function in miRNA regulation is perhaps best understood in the context of cardiovascular stress. ATF6 protects the heart from ischemic cell death [26]. Activation of ATF6 in mouse heart contributes to increased calreticulin expression and coordinate suppression of miR-455 [27]. Calreticulin helps fold native proteins and is required for cardiac development [28]. Overexpression of miR-455 abrogated calreticulin expression, whereas anti-miR-455 elevated it. Analysis of miR-455 function in other organs is required to reveal different facets of ATF6 dependent regulation of microRNAs.
MiRNAs, cell fate and neoplastic growth
The pro-survival function of the UPR pathway has recently become a key point of interest with regard to its potential pro-neoplastic activities. Gene targeting and shRNA strategies have collectively revealed that PERK, IRE1α, and Xbp1 contribute to tumor progression [29-32]. The pro-neoplastic functions of the UPR have stimulated efforts to develop strategies for targeting key UPR activities or downstream effectors as novel therapeutic modalities. Our ability to fully understand or interpret the success or failure of such strategies will depend on a detailed understanding of the signaling pathways and molecular mechanisms triggered by UPR signal transducers in this context. An obvious question that arises is whether UPR-inducible miRNAs are differentially expressed according to UPR activation status in tumors, and in turn whether they contribute to adaptive responses in tumors. Of the miRNAs discussed, both miR-708 and miR-211 have been implicated in tumorigenesis. MiR-708 has also been associated with breast cancer metastasis [11]. In the retina, induction of miR-708 limits over-production of rhodopsin and thus preserves cell fate. However, miR-708 expression is epigenetically silenced during the course of breast cancer metastasis, leading to increased expression of pro-metastatic neuronatin and deregulation of calcium homeostasis. The loss of miR-708 in the context of metastasis is correlated with increased MAPK and FAK signaling, both of which could contribute to cell growth and migration. However, dysregulation of calcium in the ER is also associated with UPR activation. This raises two points of interest. First, reduced expression of miR-708 was associated with an increase in H3K27me3; although this mark would likely override the pro-transcriptional activity of CHOP towards miR-708, CHOP status in these tumors is unknown. Might CHOP be coordinately silenced? Second, because UPR activation is associated with dysregulation of calcium homeostasis, it is anticipated that loss of miR-708 might trigger increased UPR signaling. Thus, metastatic growth and survival could potentially be synergistically enhanced through down-regulation of CHOP and repression of miR-708.
MiR-211 expression has been linked with PERK activation in both mouse models of mammary cancer and human lymphoma [9]. PERK activity has also been associated with several models of human cancer including breast cancer [30]. Deletion of PERK in a HER2/Neu-driven model of mammary cancer significantly reduced tumor growth; importantly, in this model PERK activity was associated with high miR-211 and low CHOP levels, whereas tumors lacking PERK exhibited reduced miR-211 expression. Increased PERK signaling has also been reported in lymphomas harboring activated c-myc and PERK signaling was found to be essential for survival of c-myc-transformed lymphocytes [32]. Analogous to HER2/Neudriven mammary tumors, miR-211 levels were high in PERK- and c-myc-expressing transformed lymphocytes consistent with PERK functioning as an upstream regulator of lymphomagenesis. Although these data would argue that the pro-survival activity of miR-211 also provides a pro-neoplastic activity, this may be tissue-specific as reduced miR-211 levels correlate with increased metastasis in melanoma [33]. Understanding the functional contribution of miRNAs to UPR-triggered pathways, as well as their role in regulating cell fate, will provide critical insight necessary for therapeutic advances. In addition, with the development of mechanisms for the delivery of nucleic acids as a therapy, targeting pro-survival miRs might prove to be a viable therapeutic modality.
Concluding remarks
The complex relationship between miRNAs and stress pathways is only beginning to be experimentally dissected. The UPR regulators PERK, Ire1α and ATF6 induce or suppress miRNAs, which has implications for cell fate. The model emerging from these studies is that miRNAs fine-tune ER stress machinery and modulate cellular adaptation to stress. However, regulation of expression of miRNAs is multifaceted; in some instances expression is temporally regulated, as witnessed with miR-211 and miR-30c-2-3p. In addition, some miRNAs are pro- adaptive (miR-211) whereas others are pro-apoptotic (miR-30c-2-3p), and the relative levels of expression will likely determine cell fate. Because miRNAs can target many distinct mRNA targets, the physiological consequence of expression is likely to be context and/or tissue-dependent. Therefore, it will be important to determine which miRNAs form essential nodes in this network and which are redundant. Apart from microRNAs, it seems likely that other noncoding RNAs such as long non-coding RNAs may also be regulated by and contribute to the biological effects of the UPR. In the coming years the development of a complete catalogue of ER-stress dependent non-coding RNAs, along with their targets, would prove an invaluable resource in our efforts to understand their contributions to UPR signaling.
Highlights.
miRNAs function as pro-adaptive molecules during ER stress.
miRNAs may provide a critical link between the UPR and tumorigenesis.
miRNAs are regulated positively and negatively by the UPR
Acknowledgments
This work was supported by National Institutes of Health grants P01 CA104838 (JAD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pavitt GD, Ron D. New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harbor perspectives in biology. 2012:4. doi: 10.1101/cshperspect.a012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 3.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nature cell biology. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He Y, et al. Emerging roles for XBP1, a sUPeR transcription factor. Gene expression. 2010;15:13–25. doi: 10.3727/105221610x12819686555051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 6.Han D, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nature reviews Molecular cell biology. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 8.Parmar VM, Schroder M. Sensing endoplasmic reticulum stress. Advances in experimental medicine and biology. 2012;738:153–168. doi: 10.1007/978-1-4614-1680-7_10. [DOI] [PubMed] [Google Scholar]
- 9.Chitnis NS, et al. miR-211 Is a Prosurvival MicroRNA that Regulates chop Expression in a PERK-Dependent Manner. Mol Cell. 2012;48:353–364. doi: 10.1016/j.molcel.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behrman S, et al. A CHOP-regulated microRNA controls rhodopsin expression. The Journal of cell biology. 2011;192:919–927. doi: 10.1083/jcb.201010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryu S, et al. Suppression of miRNA-708 by polycomb group promotes metastases by calcium-induced cell migration. Cancer Cell. 2013;23:63–76. doi: 10.1016/j.ccr.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 12.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. The Journal of cell biology. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byrd AE, et al. MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. The Journal of cell biology. 2012;196:689–698. doi: 10.1083/jcb.201201077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta S, et al. Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell death & disease. 2012;3:e333. doi: 10.1038/cddis.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cullinan SB, et al. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duan Q, et al. ER stress negatively modulates the expression of the miR-199a/214 cluster to regulates tumor survival and progression in human hepatocellular cancer. PLoS ONE. 2012;7:e31518. doi: 10.1371/journal.pone.0031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, et al. miR-214 targets ATF4 to inhibit bone formation. Nature medicine. 2013;19:93–100. doi: 10.1038/nm.3026. [DOI] [PubMed] [Google Scholar]
- 18.Dai R, et al. miR-221/222 suppression protects against endoplasmic reticulum stress-induced apoptosis via p27(Kip1)- and MEK/ERK-mediated cell cycle regulation. Biological chemistry. 2010;391:791–801. doi: 10.1515/BC.2010.072. [DOI] [PubMed] [Google Scholar]
- 19.Cheung HH, et al. Involvement of caspase-2 and caspase-9 in endoplasmic reticulum stress-induced apoptosis: a role for the IAPs. Experimental cell research. 2006;312:2347–2357. doi: 10.1016/j.yexcr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 20.Gaddam D, et al. Comparison of mRNA localization and regulation during endoplasmic reticulum stress in Drosophila cells. Molecular biology of the cell. 2013;24:14–20. doi: 10.1091/mbc.E12-06-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urano F, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 22.Upton JP, et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oslowski CM, et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell metabolism. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ulianich L, et al. ER stress impairs MHC Class I surface expression and increases susceptibility of thyroid cells to NK-mediated cytotoxicity. Biochimica et biophysica acta. 2011;1812:431–438. doi: 10.1016/j.bbadis.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 25.Bartoszewski R, et al. The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J Biol Chem. 2011;286:41862–41870. doi: 10.1074/jbc.M111.304956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martindale JJ, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circulation research. 2006;98:1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 27.Belmont PJ, et al. Regulation of microRNA expression in the heart by the ATF6 branch of the ER stress response. Journal of molecular and cellular cardiology. 2012;52:1176–1182. doi: 10.1016/j.yjmcc.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mesaeli N, et al. Calreticulin is essential for cardiac development. The Journal of cell biology. 1999;144:857–868. doi: 10.1083/jcb.144.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mimura N, et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood. 2012;119:5772–5781. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bobrovnikova-Marjon E, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29:3881–3895. doi: 10.1038/onc.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blais JD, et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol. 2004;24:7469–7482. doi: 10.1128/MCB.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hart LS, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. The Journal of clinical investigation. 2012;122:4621–4634. doi: 10.1172/JCI62973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levy C, et al. Intronic miR-211 assumes the tumor suppressive function of its host gene in melanoma. Mol Cell. 2010;40:841–849. doi: 10.1016/j.molcel.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]