

Abstract
To achieve permanent correction of Wilson’s disease by a cell therapy approach, replacement of healthy hepatocytes will be most desirable. There is a physiological need for hepatic ATP7B-dependent copper transport in bile, which is deficient in Wilson’s disease, producing progressive copper accumulation in the liver or brain with organ damage. The ability to repopulate the liver with healthy hepatocytes raised possibilities for cell therapy in Wilson’s disease. Therapeutic principles included reconstitution of bile canalicular network as well as proliferation in transplanted hepatocytes, despite toxic amounts of copper in the liver. Nonetheless, cell therapy studies in animal models elicited major differences in the mechanisms driving liver repopulation with transplanted hepatocytes in Wilson’s disease versus nondiseased settings. Recently, noninvasive imaging was developed to demonstrate copper removal from the liver, including after cell therapy in Wilson’s disease. Such developments will help advance cell/gene therapy approaches, particularly by offering roadmaps for clinical trials in people with Wilson’s disease.
Keywords: liver, bile, stem cell, copper, excretion, transplantation
Considerable experience has been gained over the nearly 100 years since the original description of Wilson‘s disease (WD) in terms of clinical presentations, diagnostic challenges, and drug treatments (e.g., copper (Cu) chelators) for this complex condition.1 As drug treatment for WD is a lifelong requirement that can be challenging owing to drug side effects or compliance issues, alternative therapeutic approaches, particularly those capable of offering a permanent cure, are of considerable interest. For instance, transplantation of healthy cells (cell therapy approach), introduction of healthy copies of the WD gene, ATP7B (gene therapy approach), or a combination of these approaches (cell/gene therapy) offer opportunities for permanently altering disease progression in WD.
The following discussion will succinctly outline critical principles for cell therapy in WD, especially by contrasting outcomes of cell transplantation in WD with outcomes in the nondiseased liver. It should be noted that, as cell therapy has not yet been undertaken in people with WD, this discussion focuses on preclinical animal studies. Also, it should be noted that allogeneic hepatocytes are subject to rejection, which will require immunosuppression of individuals similar to orthotopic liver transplantation (OLT), although rejection mechanisms are different in these situations. Therefore, the following discussion explores studies where transplanted cells could engraft, proliferate, and survive indefinitely without confounding by rejection-related issues.
Relevant molecular mechanisms
Copper is obligatorily required for biochemical processes in cells throughout the body. The mechanisms regulating cellular Cu uptake, trafficking, utilization, and disposal are evolutionarily conserved, with extensive complexities that are incompletely understood. 2 Nonetheless, the most significant problem related to excessive Cu accumulation in the body concerns inadequate excretion of Cu into the hepatic bile canaliculus by ATP7B. Physiologically, Cu is mostly, but not exclusively, recognized at the cell membrane by Ctr1, which forms a membrane pore to permit entry into the cell. Subsequently, intracellular routing, secretion, or excretion of Cu involves chaperoning by copper chaperone to superoxide dismutase-1 (CCS), by unknown ligands to mitochondria, and by Atox1 to ATP7B, which is expressed largely in hepatocytes, and serves to excrete Cu ions into the bile, or to ATP7A, which is expressed in cells other than hepatocytes, and serves to secrete Cu ions into blood.
The function of ATP7B may be impaired by genetic mutations that are mostly sporadic but may travel through families and may affect multiple regions of the gene, including Cu-binding domains or other parts of the gene.3,4 Over 300 disease-causing ATP7B mutations have been identified in WD with differences related to individual families, which poses technical difficulties for the gene therapy approach since it must be customized for individuals. Moreover, the ATP7B gene is very large, which makes it difficult to package therapeutic constructs into gene transfer vectors. Also, mutations may affect intracellular processing of ATP7B transcripts.5 Therefore, proposed gene therapy constructs must be prospectively validated for Cu binding and transport capacity in suitable cell culture and intact animal systems, as further considered below.
A common problem related to ATP7B mutations in WD is progressive Cu accumulation with hepatocellular injury, hepatic fibrosis, and chronic liver disease. Hepatic injury may manifest with acute liver failure, which may involve mitochondrial damage,6 but many underlying pathophysiological aspects of this liver injury need to be better understood at the molecular level. On the other hand, in the setting of impaired hepatic Cu excretion due to ATP7B mutations, Cu may also accumulate in the brain, resulting in neurological damage. Early and rapid mobilization of Cu from affected parts of the brain is critical for avoiding or reversing further neurological damage. The major physiological pathway for elimination of Cu from the brain involves ATP7A-mediated secretion via the choroid plexus into the cerebrospinal fluid followed by entry into the blood and eventually excretion by hepatocytes into the bile.
Therefore, the fundamental purpose of cell/gene therapy in WD is to restore ATP7B-mediated hepatobiliary Cu excretion. This could be achieved by transplanting healthy hepatocytes, although these must come from another donor. If person-specific cells are to be utilized from individuals with WD (e.g., human inducible pluripotent stem cells (hiPS) or another stem cell type), these must meet requirements for hepatic differentiation, genetic modification with healthy ATP7B gene copies, and the ability to survive and function after transplantation. These requirements constitute unmet challenges at present.
Liver is the optimal target for curative cell/gene therapy in WD
The specific need for cell therapy in WD is to restore hepatobiliary Cu excretion. This could be accomplished by reconstituting the liver with suitable numbers or masses of healthy transplanted cells. An alternative possibility is to correct the defect in native hepatocytes with healthy copies of an ATP7B transgene introduced by vectors capable of integrating and/or persisting indefinitely in cells. For cell or gene therapy in WD, the rationale for targeting the liver first and foremost is based on the physiological restriction of ATP7B expression to hepatocytes as well as the availability of mechanisms required for the transfer of Cu ions by ATP7B into the bile canaliculus, the entry of Cu into bile ducts and intestines, and the elimination of Cu from the body.2
Besides transplantation of hepatocytes into the liver itself, which is further discussed below, transplantation of liver tissue into extrahepatic locations has been examined for tissue engineering applications (e.g., by creating an auxilary liver in small intestinal segments or by transplantation of tissue in the abdominal cavity).7,8 However, mobilization and removal of excess Cu still requires an intact hepatobiliary apparatus that will excrete bile outside of the body. As transplantation of tissues in locations other than the liver does not typically produce hepatobiliary excretory apparatus, this approach will be insufficient for cell therapy in WD.
By contrast, provision of liver support from extrahepatic liver tissue or cells could potentially help in tiding over an acute crisis in WD (e.g., rescue from acute liver failure), as established by studies of drug-induced hepatotoxicity.9 In this situation, transplanted liver tissue or cells are thought to contribute metabolic functions for liver support as well as paracrine factors that may promote regeneration of the injured liver. Through this mechanism, people could potentially be bridged to OLT or even recover spontaneously.
Organs other than the liver are either less attractive or constitute inadequate targets for cell or gene therapy in WD. For instance, ATP7B is also normally expressed in glomeruli, medulla, and proximal tubular epithelial cells in the kidneys.10 However, ATP7A is also expressed in glomeruli, which adds complexity to Cu transport owing to the simultaneous existence of excretory and absorptive mechanisms in kidneys. Therefore, glomerular filtration of Cu could be countered by Cu reabsorption in the loops of Henle, unless Cu was unavailable for reabsoption, as will be the situation when urinary Cu excretion is induced by various chelators. As a result, the possibility that expression of introduced ATP7B transgene in suitable renal tubular epithelial cell subsets might increase urinary Cu excretion in WD has been of far less interest. Alternative luminal sites (e.g., pancreatic ducts and colonic mucosa) are also of less interest owing to either limited accessibility or roles in Cu excretion for these locations for cell or gene therapy.
Cell type–specific mechanisms
Exciting advances have recently been made in understanding how healthy hepatocytes or other liver cell types may be isolated, characterized, manipulated, and transplanted to repopulate the liver.11 Moreover, advances have been made in stem cell biology, including insights into the nature and biology of stem/progenitor cells from various donor sources, at least in part, for cell therapy applications.12,13 Although the potential of stem/progenitor cells has begun to be uncovered, far more needs to be understood about mechanisms in hepatic differentiation and whether stem cell–derived hepatocytes could repopulate the liver. These aspects converge into basic stem cell biology, including mechanisms of hepatic endoderm lineage specification and lineage advancement through outside-in cell signaling, transcriptional regulation, and related intracellular processes, which are beyond the scope of this discussion. Moreover, cells must acquire appropriate adhesion factors to engraft in the liver. The current state of the art suggests that iPS-derived hepatocyte-like cells may express some hepatic functions, but these are largely restricted to fetal-like stages. The determination of whether pluripotent stem cell–derived hepatocytes could reconstitute the liver after transplantation is in an early experimenal state.14
The context of healthy versus diseased liver
Hepatocyte transplantation studies in a variety of animal models, including with unmanipuated healthy cells and genetically modified cells, established an excellent basis for the requirements of transplanted cell engraftment, proliferation, and function for correcting various disorders, including WD.15–19 Here, important early insights included delineation of substantial differences in cell and gene therapy for WD, where the liver has undergone acute, chronic, or both types of injury, compared to other genetic conditions where the liver is uninjured and other target organs are affected (e.g., congenital enzymatic deficiency states with hyperbilribunemia and neurotoxicity or hypercholesterolemia with cardiovascular diseases).20,21 In part, these differences revolve around alterations in organ architecture, which may lead to redistributions of transplanted cells; the presence of inflammatory changes may impair engraftment of transplanted cells or various processes and events related to hepatic injury may impair proliferation of transplanted cells to the required extent.
Similarly, when ATP7B is expressed in native hepatocytes as part of gene therapy protocols, it remains unresolved whether disease processes or issues related to gene transfer vectors may limit either transgene expression or survival of transduced cells, yielding only temporary benefits.22,23 Therefore, further studies are required investigatingthe fate of genetically modified native cells within the liver in WD. It has yet to be determined whether targeting will be effective for subsets of undamaged native cells or putative endogenous stem/progenitor cells that have not been affected by Cu-induced damage.
Cell therapy in WD requires the bile canalicular network for hepatic Cu excretion
To excrete Cu, transplanted hepatocytes must integrate in the liver parenchyma and reconstitute the hepatobiliary excretory apparatus. This represents a seminal requirement for cell therapy in WD, because without the ability in transplanted hepatocytes to appropriately process and transfer Cu to the bile canaliculus,2 excess amounts of the metal will not be removed. This critical requirement determines whether alternative cell types expressing some hepatic functions (e.g., stem cell–derived cells, immature hepatocyte-like cells) will be of therapeutic value in WD, as further discussed below.
Fortunately, transplantation studies using donor cells with suitable reporters, such as those introducting the bile canalicular marker dipeptidyl peptidase 4 (Dpp4) into recipients lacking Dpp4 have allowed visualization of bile canalicular reconstitution in liver parenchyma (Fig. 1). 24 In Dpp4-deficient rats, which arose through a spontaneous point mutation in Dpp4, no other function is lost, and the liver, as well as other organs, remains healthy throughout life. However, cell transplantation models using Dpp4-deficient rats provided convenient and very effective ways to localize transplanted cells and to study the fate of transplanted cells throughout the life span of animals.11 Studies in Dpp4-deficient rats confirmed that reconstituted bile canaliculi in transplanted and adjacent native hepatocytes were functionally intact by demonstrating biliary excretion of a fluorescently tagged bile salt.24 This ability for biliary excretion in transplanted cells provided the first definitive clue that biliary transport of Cu, or for that matter, other toxic substances, will be feasible for cell therapy in WD and additional conditions characterized by biliary transport defects.25 Subsequently, the challenges in repopulating the liver with transplanted cells were systematically addressed.
Figure 1.
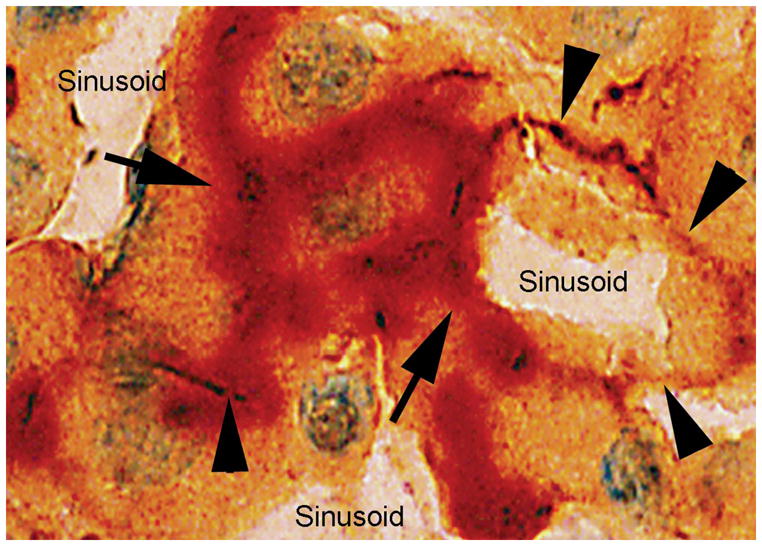
Reconstitution of bile canalicular apparatus in transplanted cells. Dpp4+ bile canalicular domains (red color, arrows) in transplanted hepatocytes adjacent to ATPase+ bile canalicular domains (brown color, arrowheads) in adjacent native hepatocytes in liver of Dpp4-deficient recipient rat. Overlapping areas with both Dpp4 and ATPase activities are also seen. This confirmed that transplanted cells were integrated within the liver parenchyma and plasma membrane structures were reconstituted. Original magnification 1000×; methylgreen counterstain. Modified from Ref. 15.
Mechanisms of transplanted cell engraftment and proliferation
It became clear that, for transplanted cells to enter the liver parenchyma, the cells had to be deposited into hepatic sinusoids, because injection of cells into the hepatic arterial bed or other vascular beds with high blood flow rapidly led to the destruction of transplanted cells, presumably due to shear stresses or lack of suitable adhesion factors or extracellular matrix components.26 Similarly, transplantion of cells into the splenic artery was ineffective, due to entrapment and loss of transplanted cells in the arterial circulation.27 By contrast, injection of cells into the portal venous system, including direct injection into the splenic pulp or portal vein radicles, was effective, with instantaneous migration of cells into the portal vein and hepatic sinusoids.28 However, this became different in the setting of portal hypertension, particularly when portasystemic collaterals were present, as might be found in WD with liver fibrosis, because substantial fractions of transplanted cells would enter the pulmonary circulation, with the potential for serious cardiorespiratory complications.29
Therefore, suitable precautions will be necessary for evaluating and establishing the safety of cell transplantation in the presence of chronic liver disease, as in WD. On the other hand, integration of transplanted hepatocytes or of other cell types, such as liver sinusoidal endothelial cells (LSEC), took place over several days, with gradual fusion of plasma membrane structures in transplanted cells and adjacent native cells, whereas the vast majority of transplanted cells (approximately 80–90% of cells) was rapidly cleared within the first day or two.30,31
This clearance of transplanted cells was multifactorial and included mechanisms like instantaneous ischemia/reperfusion-type events because transplanted cells served as emboli within hepatic sinusoids (Fig. 2). Although mechanical consequences of sinusoidal blood flow occlusion were relevant for the subsequent fate of transplanted cells, additional changes due to cell–cell signaling between transplanted and native cells occurred with the recruitment of inflammatory cells (i.e., neutrophils (PMN) and Kupffer cells (KC) or monocytes).32,33 These inflammatory cells expressed numerous inflammatory cytokines/chemokines/receptors capable of clearing transplanted cells, as prior depletion of neutrophils or Kupffer cells had substantial benefits for transplanted cell engraftment. Adhesion of transplanted cells to the hepatic sinusoidal endothelium was helpful in the activation of LSEC and the separation of the endothelial barrier for transplanted cells to enter the space of Disse and to insinuate into the liver parenchyma between native cells.34 Therefore, endothelial separation was particularly beneficial for transplanted cell engraftment, which was substantiated by studies showing superior engraftment of transplanted cells when the hepatic endothelium was disrupted before cell transplantation. During the entry of transplanted cells into the liver parenchyma, benefits accrued from the activation of hepatic stellate cells (HSC) in the space of Disse and the release of various matrix-type metalloproteinases, which produced remodeling of the liver parenchyma.35
Figure 2.
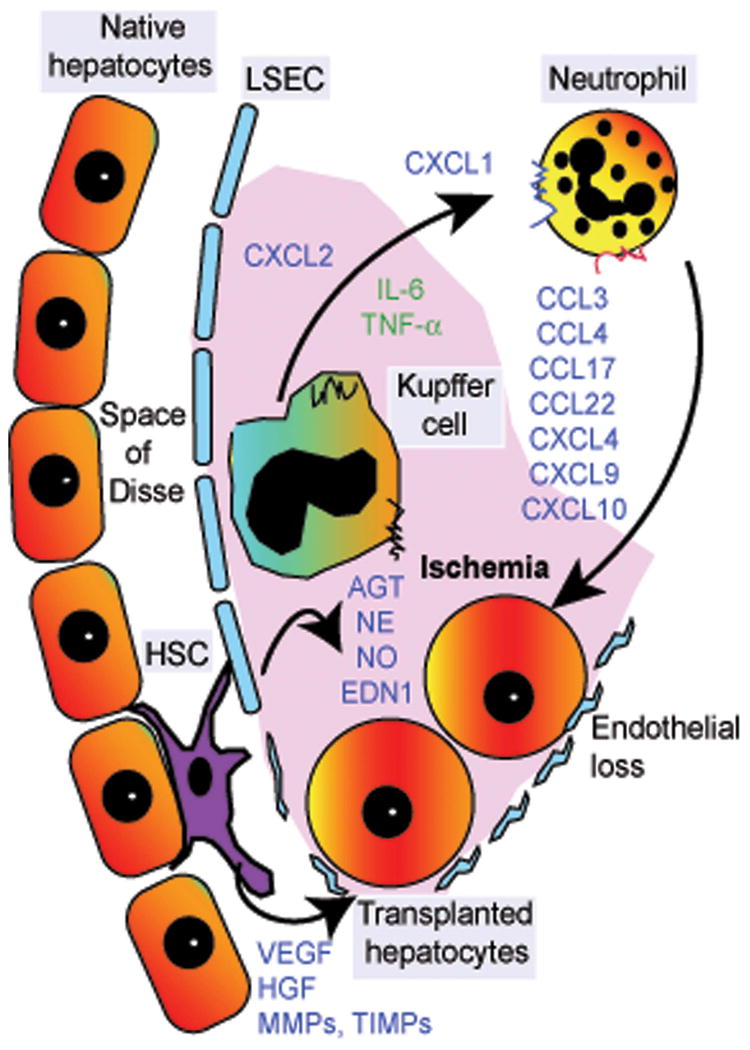
Schematic of early hepatic sinusoidal events following cell transplantation. Arrival of transplanted hepatocytes in hepatic sinusoids triggers ischemia-type processes, which lead to the release of locally acting vascular substances (e.g., angiotensin, norepinephrine, nitrous oxide, endothelin-1) as well as activation of neutrophils and Kupffer cells. This, in turn, leads to the release of multiple cytokines, chemokines, and receptors, including those with cytotoxic properties, which leads to transplanted cell clearance. Simultaneously, ischemic damage to liver sinusoidal endothelial cells (LSECs), along with cell–cell interactions resulting from adhesion of transplanted hepatocytes to LSECs, advances the entry of transplanted cells into the space of Disse. Activation of hepatic stellate cells (HSCs) causes expression of matrix-type metalloproteinases (MMPs), tissue inhibitors of MMPs (TIMPs), etc., which facilitates tissue remodeling during the integration of transplanted cells into the liver parenchyma. Release of additional substances (e.g., VEGF, HGF) from native hepatocytes, LSECs, and HSCs also assists transplanted cell engraftment. These mechanisms provide opportunities for drug-based approaches to control deleterious inflammatory events and to promote beneficial cell engraftment processes. The mechanisms are different in the setting of normal liver and are less well defined in the setting of chronic liver disease, including WD.
Therefore, while the process of transplanted cell integration was complex, the central elements in cell engraftment were identifiable and seemed to be well organized. Moreover, candidate vascular, inflammatory, and cytoprotective mechanisms were discovered that were amenable to nonoverlapping drug-based approaches for improving engraftment of transplanted cells without interfering with their ability to proliferate during subsequent liver repopulation.36 In these ways, transplanted cell engraftment was improved by several fold, and the kinetics of liver repopulation were also accelerated by several fold in animals without liver disease.
Chronic liver disease alters engraftment of transplanted cells
In contrast, in chronic liver disesase, ongoing inflammation and activation of PMN or KC; capillarization of sinusoidal endothelium; biological alterations in HSC, along with fibrosis and portasystemic shunting; and engraftment of transplanted cells became less efficient, and many transplanted cells were additionally lost due to onward translocation into the pulmonary circulation.37 However, transplanted cells were able to overcome augmented endothelial barriers in animals with liver cirrhosis and enter the liver parenchyma. Subsequently, transplanted cells showed variable proliferation in liver lobules that likely corresponded with the underlying survival/proliferation activity in adjacent native hepatocytes.
These findings are relevant for liver repopulation in WD, although major questions remain to be addressed in cell transplantation mechanisms in this setting, particularly to achieve liver repopulation in an efficient and expeditious fashion.
Long-term fate of transplanted cells in liver
After integration of transplanted cells into the liver parenchyma, transplanted hepatocytes survived for the entire life span of the animals.38 Moroever, hepatic gene expression was well maintained in transplanted cells, which was similar to adjacent hepatocytes. Indeed, gene expression in transplanted hepatocytes was better preserved in the liver compared with extrahepatic sites (e.g., the peritoneal cavity or dorsal fat pad).39
Studies in animal models of WD have been informative. The Long–Evans cinnamon (LEC) rat model of WD arose spontaneously in an inbred Japanese colony of inbred Long–Evans agouti (LEA) rats and was eventually used for cell/gene therapy studies.15 The LEC rat was later found to suffer extensive liver damage owing to Cu accumulation, which resulted from a deletion in the 3′ region of Atp7b causing loss of hepatobiliary Cu-excretion capacity. Surprisingly, LEC rats did not exhibit a susceptibility for neurological disease. A mouse model of WD was generated by knocking out a portion of Atp7b.40 These Atp7b–.– mice develop significant liver disease, although not neurological disease, similar to LEC rats. Over time, some Atp7b–/– mice may exhibit healthy livers, presumably due to genetic recombination, as has been observed in other instances of induced mutations. On the other hand, the Atp7b mutation in LEC rats has been stable. The toxic milk mouse represents another mouse model for excessive Cu accumulation similar to WD.41 These animal models have been useful for defining whether cell/gene therapy approaches could be effective in WD.
The ability of transplanted hepatocytes to express ATP7B was verified in LEC rats,15–19 Atp7b–/– knockout mice (Bahde R, Gupta S et al. Unpublished observation), and toxic milk mice.41 However, the number of transplanted cells did not increase over many months or even thee life span of recipients with normal healthy liver.17 Studies in a variety of animal models have now conclusively established that transplanted cells can proliferate in the liver when an imabalance is created in survival/proliferation between native and transplanted cells. For instance, in response to DNA damage in native hepatocytes by adduct-forming drugs, radiation, or toxins, along with additional events (e.g., partial hepatectomy, ischemia/reperfusion, thyroid hormone) the liver may be extensively, virtually completely, replaced by transplanted cells.11
In LEC rats with copper toxicosis, similar findings were observed: transplanted cells did not proliferate over a period of several months, except when substantial damage began to occur in native hepatocytes, which eventually resulted in compensatory proliferation of transplanted cells.17,18 This was likely the result of ongoing lipid peroxidative damage in the liver because of Cu toxicosis, including depletion of cellular antioxidant defences, as well as perturbations in cell cycling events. These possibilities were substantiated by significant acceleration of transplanted cell proliferation following additional DNA damage (e.g., by the DNA adduct–forming chemical retrorsine or radiation), along with partial hepatectomy, ischemia/reperfusion, and even administration of toxic bile salts.16–19 However, the kinetics of liver repopulation with transplanted cells in LEC rats were quite different from healthy animals (e.g., after radiation and ischemia/reperfusion),11 which probably reflected the effects of the liver microenvironment in the setting of Cu toxicosis, as well as preconditioning-related mechanisms and shorter or longer duration of survival in native hepatocytes. This had direct effects on the extent of liver repopulation and therapeutic benefits in animals with copper toxicosis. Also, it was necessary to repopulate at least 30–40%% of the liver with healthy hepatocytes to achieve sufficient Cu clearance and therapeutic benefits. Not every animal subjected to cell therapy showed equivalent benefits. It was most remarkable, however, that when liver was extensively repopulated and excessive Cu was cleared, even established liver disease and hepatic fibrosis resolved, suggesting that presence of significant liver damage in WD will not be a restriction for cell therapy (Fig. 3). These mechanisms in transplanted cell engraftment and proliferation will be of paramount significance in achieving suitable liver repopulation for WD as follows: (1) despite adequate engraftment in liver of transplanted cells, the gradual onset of liver repopulation over a very long period implies that therapeutic correction in WD will require significant time, and (2) the nature of preconditioning regimens suitable for clinical applications in people will require substantial additional work because chemicals (e.g., retorsine) and ablative procedures (e.g., partial hepatectomy) will be undesirable in the setting of existing liver injury in WD.
Figure 3.

Benefits of cell therapy in LEC rats with copper toxicosis. (A) Healthy donor liver of LEA rats showing normal morphology. (B) Liver of LEC rat with extensive injury, including cholangiofibrosis and other morphological abnormalities. (C) Liver of LEC rat several months after transplantation of healthy LEA rat hepatocytes with reversal of hepatic injury and return of normal morphology. The liver of these animals showed corresponding normal, increased, and near-normal Cu content, respectively. Original magnification 100×. H&E counterstain.
Therefore, additional mechanisms to repopulate the liver with healthy cells constitute another major need for cell therapy in WD.
The role of stem cells in cell therapy for WD
The interest in endogenous stem cells, including from within the liver or from other organs, and in transplantation of exogenous stem cell–derived cells has progressively risen over recent years (Table 1).8,12–14 In part, isolation of pluripotent stem cells (e.g., human embryonic stem cells (hESC) or hiPS cells) that could be manipulated to generate hepatocyte-like cells is responsible for some of the excitement.8,14 As indicated above, mechanisms to achieve extensive hepatic differention in cases of hESC or hiPS cells are incompletely understood at present. Similarly, whether appropriate hepatocyte-like cells could be generated from hematopoietic, mesenchymal, adipocyte, or placental stem cells that will engraft and repopulate the liver with capacity for disease correction is under active study but is at an early stage.13 Relevant issues include whether hepatocyte-like cells that may express liver genes in vitro will be capable of engrafting in sufficient numbers and whether such cells will proliferate with sufficient functionality for therapeutic benefits in vivo. This has generally not been the case with hematopoeitic or mesenchymal stem cells. Although adipocyte- or placenta-derived liver-like cells have been reported to proliferate after transplantation into the liver, the value of such cells has not been determined for WD.
Table 1.
Potential sources of donor cells for treating WD
| Hepatocytes derived from pluripotent stem cellsa | Stem cells from adult liverb | Hepatocytes derived from extrahepatic stem cellsc |
|---|---|---|
| Embryonic stem cells | Hepatocyte subpopulations | Hematopoietic stem cells from bone marrow, peripheral blood, cord blood, etc. |
| Fetal liver stem cells | Oval cell populations | Mesenchymal stem cells |
| Induced pluripotent stem cells | Other cell types | Amniotic or placental stem cells |
Requires further knowledge in differentiation mechanisms and fate of transplanted cells.
Mature hepatocytes exhibit stem-like organ-repopulation capacity. Oval cells are morphologically identified with hepatobiliary and other markers. Whether nonparenchymal liver cell populations may generate hepatocytes is not resolved
More studies are needed to determine whether these cells could generate mature hepatocytes with cell therapy potential.
In WD, and also other genetic diseases, activation of endogenous stem cells could possibly be helpful for repairing liver injury, but these cells cannot by themselves be useful for correcting defective Cu excretion, unless healthy copies of ATP7B were to be simultaneously expressed. This will represent a combined cell/gene therapy approach, which requires ways to target specific cell types and needs to be validated with preclinical studies in various animal models.
The ability to generate highly differentiated hepatocytes from pluripotent cells with or without additional genetic modification to express ATP7B in such cells is a relatively distant goal at present, because many gaps remain in our understanding of differentiation and gene expression mechanisms to allow successful transplantation and disease correction with such cells. Nonetheless, progress in the field of stem cell biology and transplantation of stem cell–derived cells is proceeding at a rapid pace, and further breakthroughs may unexpectedly emerge.
Transplantation of other types of stem cells (e.g., hematopoietic stem cells, mesenchymal stem cells, amniotic stem cells) has also been proposed for various disorders, including chronic liver disease.42–45 Whether these types of cells could be of benefit in WD is uncertain. In this context, the fundamental considerations must include (1) whether these types of stem cells will actually engraft in the liver and differentiate into hepatocyte-like cells with the capability for hepatobiliary excretion of Cu, and (2) whether such stem cell–derived hepatocyte-like cells will also be capable of proliferating and replacing the required amounts of diseased liver in WD. Current evidence derived from cell transplantation studies in various organ systems does not support suggestions that hematopoietic, mesenchymal, or other types of stem cells could meet these requirements for replacing diseased hepatocytes in people, notwithstanding rare instances of hepatic transdifferentiation in selected animal models.45 Therefore, transplantation of such extrahepatic stem cells is not likely to be helpful, at least until robust mechanistic evidence has been gathered of their potential utility for WD in preclinical models.
Translating cell therapy for WD in people
To begin considering whether and when cell therapy will be appropriate for clinical trials, a number of key requirements must be met (Table 2). For example, cohorts of patients with well-defined and documented clinical courses of WD will be vital. In view of the rarity of WD in the general population, this could likely best be achieved by creating regional or national registries, under the supervison of dedicated experts. Potential candidates for such interventions in WD will likely include newly diagnosed people with relatively early disease, people with disease complications (e.g., liver failure or neurological presentations where rapid mobilization of Cu could be beneficial), and those with intolerance for drug therapy, Of course, the combination of existing drugs and cell or gene therapy approaches to mobilize Cu will likely be the way forward for early clinical trials.
Table 2.
Relevant criteria for clinical cell therapy in WD
| Criterion | Comments |
|---|---|
| Availability of subjects with well-defined and appropriately documented clinical disease | Needs establishment of patient registries and infrastructure for undertaking cell therapy |
| Well-characterized donor cells, including isolation and banking under cGMP conditions | Academic–private partnerships and greater funding for clinical trials will be helpful |
| Defined protocols for cell delivery, dosing, and hepatic targeting | Optimization required of specific protocols for clinical studies. Unified protocols for multicenter studies will be most effective |
| Persistence of transplanted hepatocytes should be indefinite | Allograft rejection and inflammation requires better controls and less toxic drugs |
| Liver replacement and repair should be quantifiable | Novel noninvasive assays for evaluating efficacy of cell therapy are desirable |
More work is certainly needed to provide understanding of relevant aspects of cell transplantation for WD (e.g., the effective routes, doses, single versus repeated cell transplantation). Similarly, it will be helpful to develop additional mechanisms to achieve liver repopulation with healthy cells (i.e., superior engraftment and proliferation in transplanted cells). Fortunately, animal studies have illuminated many of these areas. For instance, engraftment of transplanted cells has begun to be improved by the pharmacologial approach, where drugs in clinical use with vascular, anti-inflammatory, or trophic signaling properties have been found to be beneficial for liver repopulation,35,36 although their efficacy for WD needs to be tested. Cell therapy efforts for WD will be faciliated by methods of demonstrating whether transplanted cells have engrafted and begun to proliferate with replacement of liver. Similarly, effective endpoints to demonstrate therapeutic efficacy need to be established. The pool of donor organs will also have to be enlarged to procure the necessary supply of cells for transplantation. This process will benefit from superior methods for cryopresrvation and the banking of cells for transplantation. Control of allograft rejection will have to be better understood to achieve transplanted cell survival over a lifetime for correcting WD permanently.
The battery of tests for evaluating the benefits of cell therapy in WD has been refined (Table 3). Moreover, noninvasive modalities based on principles of molecular imaging have been developed to both diagnose WD and to establish whether cell therapy has reconstituted ATP7B function. 40,46 Studies with radiocopper in animals estabished that suitable complexes can be developed, such as Cu–histidine, which has been used for treating Cu deficiency in Menkes disease, to image ATP7B function by positron emission tomography with or without cell transplantation in LEC rats.46 These types of assays will permit the identification of defective Cu excretion even before onset of hepatic damage and without the need for invasive liver biopsy, which should make it easier for most people. As further suitable complexes of copper can be developed to improve diagnostic potential of noninvasive imaging in WD, this area will be of considerable value for developing cell and gene therapy applications.
Table 3.
Approaches for evaluating cell therapy in WD
| Assay | Usefulness |
|---|---|
| Serum ceruloplasmin | Not useful for evaluating success of cell therapy. |
| Serum or urine Cu levels | Of unknown or limited value for cell therapy. |
| Liver Cu content | Highly useful and specific. Difficult to sample by liver biopsy on multiple occasions. Potential for sampling errors owing to variable liver copper content related to areas with higher or lower numbers of transplanted cells |
| Molecular assays (e.g., ATP7B mRNA or ATP7B protein expression levels) | Highly useful and specific. Requires multiple liver samples. Potential sampling issues as above. |
| Liver histology and markers of tissue injury | Highly useful. Sampling issues as above. |
| Bile copper assays | Highly useful and specific. Bile may be collected by duodenal intubation. |
| Noninvasive reporter assays | Molecular imaging of ATP7B function has excellent potential. Reporter constructs could be introduced in transplanted cells to demonstrate extent of liver repopulation, (e.g., by imaging modalities). |
Acknowledgments
This work was supported in part by NIH Grants R01 DK071111, R01 DK088561, and P30 DK41296. The contribution over years of many staff members and trainees in generating primary data is gratefully acknowledged.
References
- 1.Rosencrantz R, Schilsky M. Wilson disease: pathogenesis and clinical considerations in diagnosis and treatment. Semin Liver Dis. 2011;31:245–59. doi: 10.1055/s-0031-1286056. [DOI] [PubMed] [Google Scholar]
- 2.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–85. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 3.Coffey AJ, et al. A genetic study of Wilson’s disease in the United Kingdom. Brain. 2013;136:1476–87. doi: 10.1093/brain/awt035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loudianos G, et al. Wilson’s disease in two consecutive generations: the detection of three mutated alleles in the ATP7B gene in two Sardinian families. Dig Liver Dis. 2013;45:342–5. doi: 10.1016/j.dld.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 5.Braiterman LT, et al. Distinct phenotype of a Wilson disease mutation reveals a novel trafficking determinant in the copper transporter ATP7B. Proc Natl Acad Sci U S A. 2014 Mar 24; doi: 10.1073/pnas.1314161111. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zischka H, et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J Clin Invest. 2011;121:1508–18. doi: 10.1172/JCI45401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joseph B, et al. Isolated small intestinal segments support auxiliary livers with maintenance of hepatic functions. Nat Med. 2004;10:749–53. doi: 10.1038/nm1057. [DOI] [PubMed] [Google Scholar]
- 8.Takebe T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:481–4. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 9.Bandi S, et al. Perturbations in ataxia telangiectasia mutant signaling pathways after drug-induced acute liver failure and their reversal during rescue of animals by cell therapy. Am J Pathol. 2011;178:161–74. doi: 10.1016/j.ajpath.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore SD, Cox DW. Expression in mouse kidney of membrane copper transporters Atp7a and Atp7b. Nephron. 2002;92:629–34. doi: 10.1159/000064075. [DOI] [PubMed] [Google Scholar]
- 11.Shafritz DA, Oertel M. Model systems and experimental conditions that lead to effective repopulation of the liver by transplanted cells. Int J Biochem Cell Biol. 2011;43:198–213. doi: 10.1016/j.biocel.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duncan AW, Dorrell C, Grompe M. Stem cells and liver regeneration. Gastroenterology. 2009;137:466–81. doi: 10.1053/j.gastro.2009.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Than NN, Newsome PN. Stem cells for liver regeneration. QJM. 2014 Jan 29; doi: 10.1093/qjmed/hcu013. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 14.Zhu S, et al. Mouse liver repopulation with hepatocytes generated from human fibroblasts. Nature. 2014;508:93–7. doi: 10.1038/nature13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yasui O, et al. Isolation of oval cells from Long-Evans Cinnamon rats and their transformation into hepatocytes in vivo in the rat liver. Hepatology. 1997;25:329–34. doi: 10.1053/jhep.1997.v25.pm0009021943. [DOI] [PubMed] [Google Scholar]
- 16.Irani AN, et al. Correction of liver disease following transplantation of normal hepatocytes in LEC rats modeling Wilson’s disease. Mol Ther. 2001;3:302–309. doi: 10.1006/mthe.2001.0271. [DOI] [PubMed] [Google Scholar]
- 17.Malhi H, et al. Early cell transplantation in LEC rats modeling Wilson’s disease eliminates hepatic copper with reversal of liver disease. Gastroenterology. 2002;122:438–447. doi: 10.1053/gast.2002.31086. [DOI] [PubMed] [Google Scholar]
- 18.Malhi H, et al. Mechanisms to repopulate liver with healthy donor cells for phenotypic correction in the LEC rat model of Wilson disease. Regen Med. 2008;2:165–173. doi: 10.2217/17460751.3.2.165. [DOI] [PubMed] [Google Scholar]
- 19.Joseph B, et al. Bile salt-induced pro-oxidant liver damage promotes transplanted cell proliferation for correcting Wilson disease in the LEC rat model. Hepatology. 2009;49:1616–24. doi: 10.1002/hep.22792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H, et al. Single liver lobe repopulation with wildtype hepatocytes using regional hepatic irradiation cures jaundice in Gunn rats. PLoS One. 2012;7:e46775. doi: 10.1371/journal.pone.0046775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okura H, et al. Transplantation of human adipose tissue-derived multilineage progenitor cells reduces serum cholesterol in hyperlipidemic Watanabe rabbits. Tissue Eng Part C Methods. 2011;17:145–54. doi: 10.1089/ten.tec.2010.0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ha-Hao D, et al. Chances and shortcomins of adenovirus-mediated ATP7B gene transfer in Wilson disease: proof of principle demonstrated in a pilot study with LEC rats. Z Gastroenterol. 2002;40:209–16. doi: 10.1055/s-2002-25151. [DOI] [PubMed] [Google Scholar]
- 23.Merle U, et al. Lentiviral gene transfer ameliorates disease progression in Long-Evans cinnamon rats: an animal model for Wilson disease. Scand J Gastroenterol. 2006;41:974–82. doi: 10.1080/00365520600554790. [DOI] [PubMed] [Google Scholar]
- 24.Gupta S, Rajvanshi P, Lee CD. Integration of transplanted hepatocytes in host liver plates demonstrated with dipeptidyl peptidase IV deficient rats. Proc Natl Acad Sci USA. 1995;92:5860–5864. doi: 10.1073/pnas.92.13.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Vree JM, et al. Correction of liver disease by hepatocyte transplantation in a mouse model of progressive familial intrahepatic cholestasis. Gastroenterology. 2000;119:1720–30. doi: 10.1053/gast.2000.20222. [DOI] [PubMed] [Google Scholar]
- 26.Rajvanshi P, et al. Human serum albumin microspheres approximate initial organ-specific biodistributions of transplanted hepatocytes and are effective cell surrogates for safety studies. Cell Transplant. 1998;7:275–283. doi: 10.1177/096368979800700306. [DOI] [PubMed] [Google Scholar]
- 27.Nagata H, Ito M, Shirota C, Edge A, McCowan TC, Fox IJ. Route of hepatocyte delivery affects hepatocyte engraftment in the spleen. Transplantation. 2003;76:732–4. doi: 10.1097/01.TP.0000081560.16039.67. [DOI] [PubMed] [Google Scholar]
- 28.Defresne F, et al. Biodistribution of adult derived human liver stem cells following intraportal infusion in a 17-year-old patient with glycogenosis type 1A. Nucl Med Biol. 2014;41:371–5. doi: 10.1016/j.nucmedbio.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Gupta S, et al. Studies on the safety of intrasplenic hepatocyte transplantation: Relevance to ex-vivo gene therapy and liver repopulation in acute hepatic failure. Human Gene Ther. 1993;4:249–257. doi: 10.1089/hum.1993.4.3-249. [DOI] [PubMed] [Google Scholar]
- 30.Gupta S, et al. Entry and integration of transplanted hepatocytes in liver plates occur by disruption of hepatic sinusoidal endothelium. Hepatology. 1999;29:509–519. doi: 10.1002/hep.510290213. [DOI] [PubMed] [Google Scholar]
- 31.Follenzi A, et al. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest. 2008;118:935–945. doi: 10.1172/JCI32748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joseph B, et al. Kupffer cells participate in early clearance of syngeneic hepatocytes transplanted in the rat liver. Gastroenterology. 2002;123:1677–1685. doi: 10.1053/gast.2002.36592. [DOI] [PubMed] [Google Scholar]
- 33.Krohn N, et al. Hepatocyte transplantation-induced liver inflammation is driven by cytokines-chemokines associated with neutrophils and Kupffer cells. Gastroenterology. 2009;136:1806–17. doi: 10.1053/j.gastro.2009.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joseph B, et al. Monocrotaline promotes transplanted cell engraftment and advances liver repopulation in rats via liver conditioning. Hepatology. 2006;44:1411–20. doi: 10.1002/hep.21416. [DOI] [PubMed] [Google Scholar]
- 35.Enami Y, et al. Hepatic stellate cells promote hepatocyte engraftment in rat liver after prostaglandin-endoperoxide synthase inhibition. Gastroenterology. 2009;136:2356–64. doi: 10.1053/j.gastro.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bahde R, et al. Endothelin-1 receptor A blocker darusentan decreases cell transplantation-induced hepatic perturbations and improves liver repopulation. Hepatology. 2014;59:1107–17. doi: 10.1002/hep.26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gagandeep S, et al. Transplanted hepatocytes engraft, survive and proliferate in the liver of rats with carbon tetrachloride-induced cirrhosis. J Pathol. 2000;191:78–85. doi: 10.1002/(SICI)1096-9896(200005)191:1<78::AID-PATH587>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 38.Sokhi RP, Rajvanshi P, Gupta S. Transplanted reporter cells help in defining onset of hepatocyte proliferation during the life of F344 rats. Am J Physiol Gastroint Liver Physiol. 2000;279:G631–G640. doi: 10.1152/ajpgi.2000.279.3.G631. [DOI] [PubMed] [Google Scholar]
- 39.Gupta S, et al. Hepatocytes exhibit superior transgene expression after transplantation into liver and spleen compared with peritoneal cavity or dorsal fat pad: Implications for hepatic gene therapy. Human Gene Ther. 1994;5:959–967. doi: 10.1089/hum.1994.5.8-959. [DOI] [PubMed] [Google Scholar]
- 40.Peng F, et al. Positron emission tomography of copper metabolism in the Atp7b (−/−) knock-out mouse model of Wilson’s disease. Mol Imaging Biol. 2012;14:70–8. doi: 10.1007/s11307-011-0476-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen KJ, et al. Liver cell transplantation leads to repopulation and functional correction in a mouse model of Wilson’s disease. J Gastroenterol Hepatol. 2004;19:1283–90. doi: 10.1111/j.1440-1746.2004.03451.x. [DOI] [PubMed] [Google Scholar]
- 42.Thomas JA, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. 2011;53:2003–2015. doi: 10.1002/hep.24315. [DOI] [PubMed] [Google Scholar]
- 43.Skvorak KJ, et al. Placental stem cell correction of murine intermediate maple syrup urine disease. Hepatology. 2013;57:1017–23. doi: 10.1002/hep.26150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terai S, et al. Status and prospects of liver cirrhosis treatment by using bone marrow-derived cells and mesenchymal cells. Tissue Eng Part B Rev. 2014 Jan 22; doi: 10.1089/ten.TEB.2013.0527. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 45.Eggenhofer E, et al. Allogeneic bone marrow transplantation restores liver function in Fah-knockout mice. Exp Hematol. 2008;36:1507–13. doi: 10.1016/j.exphem.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Bahde R, et al. Positron emission tomography with copper-64-histidine for nonivasive diagnosis of biliary copper excretion in LEC rat model of Wilson’s disease. J Nucl Med. 2012;53:961–8. doi: 10.2967/jnumed.111.092361. [DOI] [PMC free article] [PubMed] [Google Scholar]