

Abstract
Leishmaniases are an epidemic in various countries, and the parasite is developing resistance against available drugs. Thus, development of new drugs against Leishmania is an open area of investigation for synthetic organic chemists. To meet this challenge, a series of chromene-2-thione derivatives have been synthesized and docked into the active site of trypanothione reductase (TryR) enzyme required for redox balance of the parasite. These were screened on promastigote, axenic amastigote, and intracellular amastigote stages of Leishmania donovani and found to show high levels of antileishmanial activity together with minimal toxicity to human peripheral blood mononuclear cells. Compounds 3b and 3k were found to be the most active among the tested compounds. Although the compounds show moderate antileishmanial activity, they identify a chemical space to design and develop drugs based on these chromene-2-thione derivatives against the Leishmania parasite.
Keywords: chromene-2-thione, visceral leishmaniasis, molecular docking, trypanothione reductase, Lipinsky rule
The Leishmaniases are a spectrum of neglected parasitic diseases caused by different species of the genus Leishmania protozoan, which is transmitted to humans by the bite of female phlebotomine sand fly. In the human body, the parasites survive and multiply within phagolysosomes of macrophages as intracellular amastigotes. Leishmaniasis is endemic in 88 countries, and there are 500 000 new cases of its visceral form each year, leading to 50 000 deaths.1
It is mainly manifested in three forms viz. visceral leishmaniasis (VL), also known as kala-azar caused by Leishmania donovani; cutaneous leishmaniasis (CL) caused by Leishmania major, L. donovani, Leishmania tropica, and Leishmania aethiopica; and muco-cutaneous leishmaniasis (MCL) caused by Leishmania braziliensis. Among all leishmaniases, the visceral form is the most severe.2 In the absence of proper medication, it has a mortality rate of almost 100% independent of the immunological status of the patient.3−5
At the beginning of the VL epidemic in India in early 1970s, sodium stibogluconate (SSG) was the most effective antileishmanial drug; however, over the years, its efficacy declined, and currently, more than two-thirds of VL patients in the Bihar state of India do not respond to SSG.6 Furthermore, the long course of treatment of pentavalent antimony (SbV) often causes side effects such as myalgia, pancreatitis, cardiac arrhythmia, and hepatitis, leading to the reduction or cessation of treatment. Presently, amphotericin B and the oral anticancer drug miltefosine are considered the best therapeutic solutions for the treatment of VL.7 Considering the high toxicity and incomplete efficacy of the current clinical drugs, the search for new targets showing antileishmanial activity is very much desirable and would be of great relevance to both synthetic and medicinal chemists.
Coumarins (2H-chromene-2-ones) are among the best known oxygenated heterocycles and are present as a structural motif in numerous natural products. Interest in their chemistry continues unabated because of the broad range of biological activities displayed by this class of compounds.8−8d There are several examples of coumarin derivatives isolated from various families of plants, which have been tested and found to be effective against the promastigote form of Leishmania parasite within a range of 17–50 μg/mL for IC50 determination.9−9d Along with these naturally occurring coumarins, some synthetic coumarins such as 4-(3, 4-dimethoxyphenyl)-6,7-dimethoxy-2H-chromene-2-one have been shown to exhibit potent activity against L. donovani amastigotes (IC50, 1.1 μM).9d
We have selected our title compounds 2H-chromene-2-thione and benzo[f]chromene-2-thione for development and assessment against L. donovani. The compounds have already been reported to exhibit profound antioxidant activities10 but have not previously been tested against any Leishmania species.
Initially, the reaction of S,S-dimethyl trithiocarbonate with substituted acetophenone 1 in the presence of NaH afforded the β-oxodithioesters 2 in good yields (Scheme 1). Typical synthetic strategies to obtain the title compounds in excellent yields have been depicted in Scheme 1.
Scheme 1. Typical Synthetic Strategy for the Synthesis of 2H-Chromene-2-thione and Benzo[f]chromene-2-thione.
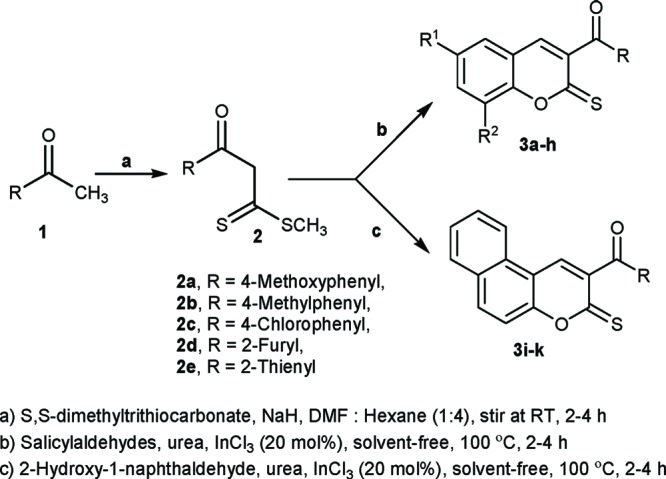
The β-oxodithioesters 2 were subjected to cyclocondensation with various 2-hydroxy aromatic aldehydes in the presence of InCl3 and urea to give chromene-2-thione in excellent yields (Scheme 1 and Figure 1).11
Figure 1.

Various synthesized potential conjugates against L. donovani.
Trypanothione reductase (TryR) is a key drug target enzyme involved in the redox metabolism of the parasite, and inhibition of TryR may disrupt the redox balance of the parasite leading to parasite death.12−12d Thus, organic molecules may be designed, which can bind the active site of TryR leading to its inhibition.
AutoDock 4.2 was used to perform molecular docking of small molecules on macromolecular protein by treating the ligand as conformationally flexible. Autodock 4.2 also has a free-energy scoring function, which uses an AMBER force field to estimate the free energy of binding of a ligand to its target. Our in silico analysis suggests TryR as a possible target of these compounds. These compounds show satisfactory docking with acceptable statistics near the active site of TryR.13 Detailed results of analysis (free energy of binding values for each ligand along with the description of interacting residues) are presented in Table 1.
Table 1. Docking Statistics of the Compounds with TryR and Their Interaction with the Protein.
| entry | binding energy | H-bond | possible hydrophobic interactions |
|---|---|---|---|
| 3a | –7.78 | 2JK6:Met-400(A):NH | Pro-398, Phe-396, Glu-467, Glu-466, Thr-463 from A chain; Leu-62, Lys-61 from B chain |
| 3b | –8.09 | Val-58, Lys-61, Leu-62, Thr-65 from B chain; Phe-396, Thr-397, Pro-398, Leu-399, Met-400, His-461 and Pro-462 from A chain | |
| 3c | –6.82 | Val-58, Lys-61, Leu-62, Thr-65 from B chain; Thr-463, Pro-398, Phe-396, Leu-399, Met-400 and Pro-462 from A chain | |
| 3d | –7.63 | 2JK6:Lys-61(B):HZ2 | Val-58, Lys-61, Leu-62 from B chain; Phe-396, Pro-398, Leu-399, Thr-463 and Ser-464 from A chain |
| 3e | –7.54 | Val-58, Lys-61, Leu-62, Thr-65 from B chain; Phe-396, Pro-398, Met-400, His-461 and Pro-462 from A chain | |
| 3f | –7.35 | Lys-61, Leu-62, Thr-65, Phe-396, Pro-398, Met-400 and Pro-462 | |
| 3g | –8.22 | 2JK6:Lys-61(B):HZ2 | Leu-62, Thr-65 from B chain; Phe-396, Thr-397, Pro-398, Met-400, Pro-462 and Thr-463 from A chain |
| 3h | –8.11 | Val-58, Lys-61, Leu-62, Thr-65 from B chain; Phe-396, Pro-398, Met-400, Pro-462 and Thr-463 from A chain | |
| 3i | –9.20 | Val-58, Lys-61, Leu-62, Thr-65 from B chain; Phe-396, Leu-399, Met-400, Thr-463, Glu-466 and Glu-467 from A chain | |
| 3j | –8.17 | 2JK6:Lys-61(B):HZ2 2JK6:Leu-399(A):HN | Phe-396, Pro-462, Thr-463 and Ser-464 from A chain |
| 3k | –8.61 | 2JK6:Ser-464(A):SH and OH | Val-58, Lys-61, Leu-62 from B chain; Phe-396, Pro-398, Leu-399 and Met-400 from A chain |
The enzyme (TryR) is a dimer consisting of two active sites. Active sites are formed by the interaction of both chains. The following is the description of residues around one of the active sites.
The residues involved in bonding and nonbonding interaction with ligands are as follows: Val-58, Lys-61, Leu-62, and Thr-65 from the B chain and Phe-396, Thr-397, Pro-398, Leu-399, Met-400, His-461, Pro-462, Thr-463, and Ser-464 from the A chain. The residues involved in hydrogen bonding with the ligands are as follows: Lys-61 from the B chain and Leu-399, Met-400, and Ser-464 from the A chain. These residues are found around the active sites Cys-52 and Cys-57 of the B chain. As most of the ligands are surrounded by the above residues, it can be concluded that these residues are highly conserved and play important functional roles. Thus, interaction of the ligands with these residues might be changing the active site structure, which leads to inhibition of the enzyme. Also, these ligands show low free energy of binding against TryR; thus, they bind with high affinity. All of the ligands mostly interact with residues of the A chain and very few residues of the B chain. The interaction of ligands with the enzyme is represented in Figure 2A. The figure shows that the lead molecules dock at a position near the active site of TryR, where the cofactor FAD (shown in red color) is bound. Ligand 3i shows the least free energy of binding followed by 3k, 3g, 3j, 3h, 3b, 3a, 3d, 3e, 3f, and 3c in that order.
Figure 2.
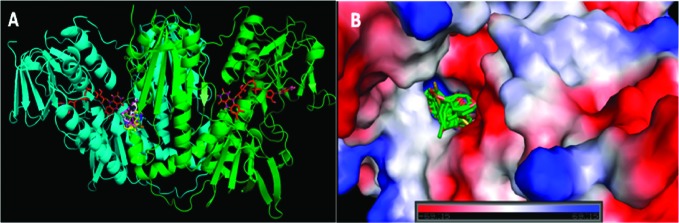
(A) Binding of the 11 ligands to the dimer TryR (PDB ID: 2JK6). (B) A vacuum electrostatics depiction of TryR (PDB ID: 2JK6) bound to all 11 ligands, showing protein contact potential.
The protein contact potential was generated for all of the docked conformations (Figure 2B). This depicted the whole surface of the protein. Red and blue colors show negative and positive charges, respectively. In this depiction, all of the ligands were shown to be bound inside a cavity formed at the interface of chain A and chain B, that is, the dimeric interface of the enzyme. Although the docked conformation was different for each ligand, the ligands are positioned at almost the same place, thus showing a consensus pattern of interactions with the residues near the active site. Figures showing interactions of each ligand with the protein are submitted as Supporting Information. It can be predicted that the inhibition modes of all of these ligands could be similar, because of their interactions with almost the same residues.
Our initial testing of these compounds against promastigote and amastigote stages of L. donovani showed that the compounds are active in IC50 range of 37–927 μM against axenic amastigotes, while IC50 was considerably higher (97–4634 μM) against promastigotes (Table 2). The mean of the IC50 values was found to be approximately five times lower against amastigotes than promastigotes, demonstrating the higher sensitivity of amastigotes to these compounds as compared to promastigotes.
Table 2. Antileishmanial Activity and Cytotoxicity of Chromene-2-thione Conjugates.
| IC50 (μM) |
cytotoxicity (%) |
||||
|---|---|---|---|---|---|
| entry | axenic amastigotes | promastigotes | intracellular amastigotes | at IC50 | at 2 × IC50 |
| 3a | 502 | 1398 | 16.63 | 14.82 | |
| 3b | 198 | 450 | 17 | 32.91 | 40.51 |
| 3c | 65 | 1224 | 26 | 12.31 | 25.96 |
| 3d | 524 | 1030 | 10.72 | 17.57 | |
| 3e | 83 | 4634 | 10.95 | 6.66 | |
| 3f | 927 | 4300 | 11.08 | 19.52 | |
| 3g | 446 | 744 | 9.40 | 33.14 | |
| 3h | 73 | 48 | 24 | 0.46 | 20.22 |
| 3i | 221 | 335 | 15.11 | 26.96 | |
| 3j | 37 | 2774 | 12.03 | 27.79 | |
| 3k | 36 | 97 | 22 | 28.84 | 24.63 |
| 3la | 0.061 | 0.465 | 16.02 | 24.40 | |
Compound 3l is amphotericin B.
Depending upon the IC50 values of amastigotes and promastigotes, we selected some derivatives for intracellular macrophage susceptibility assay. The intracellular macrophage susceptibility assay of compounds 3b [6-bromo-3-(4-methoxybenzoyl)-2H-chromene-2-thione], 3c [3-(furan-2-oyl)-8-methoxy-2H-chromene-2-thione], 3h [3-(4-chlorobenzoyl)-8-methoxy-2H-chromene-2-thione], and 3k [3-(thien-2-oyl)-benzo[f]2H-chromene-2-thione] showed higher potency with IC50 values of 17, 26, 24, and 22 μM, respectively (Table 2).
The cytotoxicity of all of the synthesized compounds was determined against PBMC (peripheral blood mononuclear cells) at two concentrations (IC50 and twice that of IC50). None of the compounds showed more than 40% killing at a concentration twice that of the IC50 (Table 2). There is apparently only a weak or no correlation between log IC50 and binding energies. This may be due to differences in the bioavailability of these compounds.
Treatment options for VL are limited. A high level of parasite resistance has been developed for the available drug sodium antimony gluconate. The single oral drug miltefosine available for VL has limitations of a long half-life and toxicity issues. Paromomycin also shows adverse events during treatment. The antifungal drug amphotericin B deoxycholate requires long hospitalization and adverse effects such as renal toxicity.14 The lipid formulation of amphotericin B is the best of the available drug options for VL patients, but it is expensive. So, in the effort to develop an antileishmanial drug for the treatment of VL, we have tested these compounds against promastigote and amastigote stages of L. donovani, and the experimental observations showed that compounds 3b and 3k were found to be the most active. The increased potency of the compounds in the intracellular macrophage susceptibility assay leaves room for further exploration of the metabolism of the chemical compounds within the macrophage. Inhibition of TryR by compounds may lead to an imbalance in the parasite's redox potential, which weakens the parasite's defense against the oxidative pressure that they experience within the macrophage, and thus, an increased potency is obtained.
The ligands were also checked for compliance to the Lipinsky rule of five, and the results are summarized in Table 3. The rule states that a molecule likely to be developed as an orally active drug candidate should show no more than one violation of the following four criteria: It should not have more than five hydrogen bond donors, it should not have more than 10 hydrogen bond acceptors, it should not have molecular weight greater than 500 Da, and it should not have an octanol–water partition coefficient greater than 5. Molecular properties of all ligands were calculated by molinspiration, and it was found that no ligand showed any violation of the above criteria (Table 3). Therefore, these ligands have a good potential for eventual development as oral agents and can be potentially active drug candidates.
Table 3. Molinspiration Calculation of Properties for the Lipinsky Rulea.
| entry | nViol | natoms | miLog P | MW | nON | nOHNH | nrotb |
|---|---|---|---|---|---|---|---|
| acceptable range→ | <5 | <500 | <10 | <5 | |||
| 3a | 0 | 23 | 3.805 | 326.37 | 4 | 0 | 4 |
| 3b | 0 | 22 | 3.87 | 379.27 | 3 | 0 | 3 |
| 3c | 0 | 20 | 2.627 | 290.34 | 4 | 0 | 3 |
| 3d | 0 | 19 | 3.071 | 339.21 | 3 | 0 | 2 |
| 3e | 0 | 19 | 3.713 | 355.27 | 2 | 0 | 2 |
| 3f | 0 | 20 | 3.269 | 306.40 | 3 | 0 | 3 |
| 3g | 0 | 22 | 3.818 | 314.40 | 3 | 0 | 3 |
| 3h | 0 | 22 | 4.048 | 334.82 | 3 | 0 | 3 |
| 3i | 0 | 25 | 4.956 | 346.40 | 3 | 0 | 3 |
| 3j | 0 | 22 | 4.156 | 306.34 | 3 | 0 | 2 |
| 3k | 0 | 22 | 4.798 | 322.41 | 2 | 0 | 2 |
nViol, no. of violations; natoms, no. of atoms; miLog P, molinspiration predicted Log P; MW, molecular weight; nON, no. of hydrogen bond acceptors; nOHNH, no. of hydrogen bond donors; and nrotb, no. of rotatable bond.
Within this manuscript, we present the efficient synthesis of a series of chromene-2-thiones, which show antileishmanial activity against L. donovani. Docking statistics and in vitro analysis against different forms of parasite suggested that compounds 3b and 3k were most active among the tested compounds. Although these compounds show a narrow therapeutic margin, there is chemical space to design and develop more selective and potent compounds. The viability of Leishmania in this work was determined by the MTT assay, which relies partially on mitochondrial functions. Recently, it was shown that chroman derivatives inhibit the mitochondrial bc1 complex, which influences the MTT reduction.15 The possibility that mitochondrial bc1 complex of the parasite also be an alternative target cannot be ruled out. Ligand docking studies could provide the first evidence for binding of compound in different areas of TryR. Thus, chromene-2-thiones can be a possible lead for the development of novel drug against leishmania.
Supporting Information Available
Information on compound synthesis and characterization data, methods for molecular docking and results, and protocols and results for various analyses on Leishmania parasite. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was carried out under financial support from the Council of Scientific and Industrial Research (Grant 01(2260)/08/EMR-II), the Department of Science and Technology (Grant SR/S1/OC-66/2009), New Delhi, and partly supported by NIAID, NIH Grant No. 1P50AI074321. R.K.V. is grateful to CSIR, New Delhi, for a senior research fellowship; G.K.V. is thankful to UGC, New Delhi, for a junior research fellowship; and V.K.P. is thankful to the Indian Council of Medical Research for a senior research fellowship.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chappuis F.; Sundar S.; Hailu A.; Ghalib H.; Rijal S.; Peeling R. W.; Alvar J.; Boelaert M. Visceral leishmaniasis: What are the needs for diagnosis, treatment and control ?. Nat. Rev. Microbiol. 2007, 5, 873–882. [DOI] [PubMed] [Google Scholar]
- Murray H. W.; Berman J. D.; Davies C. R.; Saravia N. G. Advances in leishmaniasis. Lancet 2005, 366, 1561–1577. [DOI] [PubMed] [Google Scholar]
- Grogl M.; Thomason T. N.; Franke E. D. Drug resistance in leishmaniasis: its implication in systemic chemotherapy of cutaneous and mucocutaneous disease. Am. J. Trop. Med. Hyg. 1992, 47, 117–126. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Kulshrestha A.; Singh R.; Salotra P. In vitro susceptibility of field isolates of Leishmania donovani to Miltefosine and amphotericin B: Correlation with sodium antimony gluconate susceptibility and implications for treatment in areas of endemicity. Antimicrob. Agents Chemother. 2009, 53, 835–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoli Z. M.; Rai M. E.; Gandapur A. S. Clinical presentation and management of visceral leishmaniasis. J. Ayub. Med. Coll. Abbottabad 2005, 17, 51–53. [PubMed] [Google Scholar]
- Croft S. L.; Sundar S.; Fairlamb A. H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S.; Jha T. K.; Thakur C. P.; Engel J.; Sindermann H.; Fischer C.; Junge K.; Bryceson A.; Berman J. Oral miltefosine for Indian visceral leishmaniasis. N. Engl. J. Med. 2002, 347, 1739–1746. [DOI] [PubMed] [Google Scholar]
- Fylaktakidou K. C.; Hadjipavlou-Litina D. J.; Litinas K. E.; Nicolaides D. N. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr. Pharm. Des. 2004, 10, 3813–3833. [DOI] [PubMed] [Google Scholar]
- Hwu J. R.; Singha R.; Hong S. C.; Chang Y. H.; Das A. R.; Vliegen I.; De Clercq E.; Neyts J. Synthesis of new benzimidazole–coumarin conjugates as anti-hepatitis C virus agents. Antiviral Res. 2008, 77, 157–162. [DOI] [PubMed] [Google Scholar]
- Sardari S.; Mori Y.; Horita K.; Micetich R. G.; Nishibe S.; Daneshtalab M. Synthesis and antifungal activity of coumarins and angular furanocoumarins. Bioorg. Med. Chem. 1999, 7, 1933–1940. [DOI] [PubMed] [Google Scholar]
- Spino C.; Dodier M.; Sotheeswaran S. Anti-HIV coumarins from Calophyllum seed oil. Bioorg. Med. Chem. Lett. 1998, 8, 3475–3478. [DOI] [PubMed] [Google Scholar]
- Arango V.; Robledo S.; Séon-Méniel B.; Figadère B.; Cardona W.; Sáez J.; Otálvaro F. Coumarins from Galipea panamensis and their activity against Leishmania panamensis. J. Nat. Prod. 2010, 73, 1012–1014. [DOI] [PubMed] [Google Scholar]
- Ferreira M. E.; de Arias A. R.; Yaluff G.; de Bilbao N. V.; Nakayama H.; Torres S.; Schinini A.; Guy I.; Heinzen H.; Fournet A. Antileishmanial activity of furoquinolines and coumarins from Helietta apiculata. Phytomedicine 2010, 17, 375–378. [DOI] [PubMed] [Google Scholar]
- Iranshahi M.; Arfa P.; Ramezani M.; Jaafari M. R.; Sadeghian H.; Bassarello C.; Piacente S.; Pizza C. Sesquiterpene coumarins from Ferula szowitsiana and in vitro antileishmanial activity of 7-prenyloxycoumarins against promastigotes. Phytochemistry 2007, 68, 554–561. [DOI] [PubMed] [Google Scholar]
- Pierson J. T.; Dumetre A.; Hutter S.; Delmas F.; Laget M.; Finet J. P.; Azas N.; Combes S. Synthesis and antiprotozoal activity of 4-arylcoumarins. Eur. J. Med. Chem. 2010, 45, 864–869. [DOI] [PubMed] [Google Scholar]
- Singh O. M.; Devi N. S.; Thokchom D. S.; Sharma G. J. Novel 3-alkanoyl/aroyl/heteroaroyl-2H-chromene-2-thiones: Synthesis and evaluation of their antioxidant activities. Eur. J. Med. Chem. 2010, 45, 2250–2257. [DOI] [PubMed] [Google Scholar]
- Verma R. K.; Verma G. K.; Raghuvanshi K.; Singh M. S. An expedient route to highly functionalized 2H-chromene-2-thiones via ring annulation of β-oxodithioesters catalyzed by InCl3 under solvent-free conditions. Tetrahedron 2011, 67, 584–589. [Google Scholar]
- Fairlamb A. H.; Blackburn P.; Ulrich P.; Chait B. T.; Cerami A. Trypanothione: A novel bis(glutathionyl)spermidine cofactor for glutathione reductase in trypanosomatids. Science 1985, 227, 1485–1487. [DOI] [PubMed] [Google Scholar]
- Cinningham M. L.; Fairlamb A. H. Trypanothione Reductase from Leishmania Donovani: Purification, characterization and inhibition by trivalent Antimonials. Eur. J. Biochem. 1995, 230, 460–468. [DOI] [PubMed] [Google Scholar]
- Shukla A. K.; Patra S.; Dubey V. K. Evaluation of selected antitumor agents as subversive substrate and potential inhibitor of trypanothione reductase: An alternative approach for chemotherapy of Leishmaniasis. Mol. Cell. Biochem. 2011, 352, 261–270. [DOI] [PubMed] [Google Scholar]
- Venkatesan S. K.; Shukla A. K.; Dubey V. K. Molecular docking studies of selected tricyclic and quinone derivatives on trypanothione reductase of Leishmania infantum. J. Comput. Chem. 2010, 31, 2463–2475. [DOI] [PubMed] [Google Scholar]
- Baiocco P.; Colotti G.; Franceschini S.; Ilari A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [DOI] [PubMed] [Google Scholar]
- Chakravarty J.; Sundar S. Drug resistance in leishmaniasis. J. Global Infect. Dis. 2010, 2, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzote L.; Stamberg W; Patel A; Rosenau T.; Maes L; Cos P; Gille L. Synthetic chromanol derivatives and their interaction with complex III in Mitochondria from bovine, yeast, and leishmania. Chem. Res. Toxicol. 2011, 24, 1678–1685. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.