

Abstract
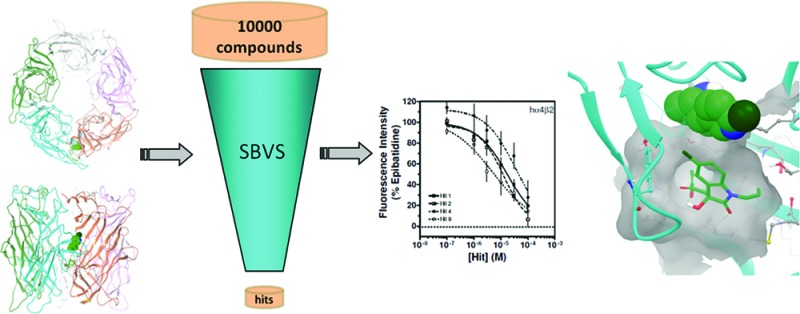
We performed a hierarchical structure-based virtual screening utilizing a comparative model of the human α4β2 neuronal nicotinic acetylcholine receptor (nAChR) extracellular domain. Compounds were selected for experimental testing based on structural diversity, binding pocket location, and standard error of the free energy scoring function used in the screening. Four of the eleven in silico hit compounds showed promising activity with low micromolar IC50 values in a calcium accumulation assay. Two of the antagonists were also proven to be selective for human α4β2 vs human α3β4 nAChRs. This is the first report of successful discovery of novel nAChR antagonists through the use of structure-based virtual screening with a human nAChR homology model. These compounds may serve as potential novel scaffolds for further development of selective nAChR antagonists.
Keywords: nAChR, nicotinic acetylcholine receptor, virtual screening, allosteric, noncompetitive antagonists, AChBP
Neuronal nicotinic receptors (nAChRs) play crucial roles in several physiological functions including memory and learning, attention, pain perception, and body temperature regulation. Dysfunction of this family of receptors has been implicated in a variety of diseases and disorders including attention deficit hyperactivity disorder (ADHD), Alzheimer's disease, Parkinson's disease, schizophrenia, and nicotine addiction.1−4 For several decades, active investigations have been undertaken by researchers across the globe to discover potent chemical modulators of nAChRs.5 Discovery of subtype selective chemical agents of nAChRs would enhance the pharmacological understanding of the specific roles played by the different subtypes of these receptors in various disease conditions. At present, a majority of the investigations focus on the development of modulators that target the nAChR orthosteric site.6 However, identification of subtype selective chemical agents by this approach is challenging due to the high degree of physicochemical similarity among the orthosteric sites of all nAChRs.6,7 Therefore, an alternate approach to modulate nAChR function is to target allosteric sites on the receptors.8,9
Our group previously identified a novel class of small molecules that (1) act as antagonists of nAChRs, 2) do not compete for binding at the orthosteric site, and 3) enhance the binding of agonist to the orthosteric site.9,10 Altogether, we classified these small molecules as negative allosteric modulators (NAMs). Through blind docking and molecular dynamics (MD) simulations with a human α4β2 (hα4β2) nAChR homology model, the binding site of these NAMs was identified.11 We hypothesized that these NAMs affect the dynamics of a long loop that is linked to channel gating. This loop, called the C loop, closes around agonists to initiate channel opening; however, these NAMs prevent C loop closure while not competing with agonist binding in the nearby orthosteric site.9 Using this previously identified allosteric binding site, we employed structure-based virtual screening (VS) to identify new small molecules that target this site.
Structure-based VS represents a rational method to identify novel scaffolds for a particular binding site when the experimental structure of the target is known or when it can be modeled.12 This method has been extensively used in the development of several pharmacological compounds, particularly anticancer agents.13 Recent efforts have been undertaken to improve the enrichment of structure-based VS results.14 Presently, structure-based rational drug design efforts targeting nAChRs are hindered due to the lack of high-resolution crystal structures. To date, structure-based VS for new nAChR ligands have been only applied to the homologous acetylcholine binding protein (AChBP).15−17 Although comparative protein modeling has been widely used for the purpose of drug design in the past several years, modeling membrane proteins, such as nAChRs, has been a challenging task. The large size of the pentameric nAChR and the highly dynamic loops known to be involved in the channel function represent hurdles in the development of reasonably accurate models for in silico screening. Therefore, developing a refined homology model in lieu of a high-resolution crystal structure and identifying a druggable allosteric site are key aspects in structure-based drug discovery of nAChR modulators. In this work, we report the methods employed to discover new chemotypes that may serve as new scaffolds for the development of subtype selective nAChR antagonists.
A previously described human α4β2 (hα4β2) extracellular domain comparative model was used as the starting point to identify a favorable receptor conformation for structure-based VS.10 The agonist epibatidine was docked to 25 receptor conformations that were extracted from a 5 ns MD simulation of the initial unbound model at regular 200 ps intervals (see the Supporting Information for methods details). The conformation to which epibatidine docked most similarly to the experimental binding mode revealed in the cocrystal structure with the homologous AChBP (PDB ID: 2BYQ) was selected as the VS target. The C loop in this selected conformation was in a partially “open” state. α-Cobratoxin, which binds in close proximity of the C loop, restricts its flexibility, thus acting as an antagonist (PDB ID: 1YI5),18 and similarly, compounds that can bind to this region may affect the C loop flexibility, which could eventually alter the channel gating. Hence, we concluded that the interface between the α4 and the β2 monomers, where C loop undergoes conformational movement to regulate the channel function, represents an attractive binding site for antagonist design (Figure S2 in the Supporting Information). The putative ligand binding subpockets in this region were probed using the SiteMap module of the Schrödinger molecular modeling suite 2008.19,20 The site probe and the previous docking studies10 allowed for a focused search space to be used during the VS. A Glide grid map of the epibatidine-bound α4β2 nAChR complex was generated over the SiteMap subpocket at the α4/β2 interface (46 Å × 46 Å × 46 Å box). The electronic ligand library, ChemBridge CNS-Set of 10000 compounds, was converted from 2D formatting to 3D structures using the LigPrep program in the Schrödinger Suite, which included enumeration of the protonation and tautomeric states. For compounds with chiral centers, stereoisomers were also generated up to a maximum of four configurations. Final coordinates were energy minimized using OPLS 2001 force field.
The structure-based VS was carried out using the Glide docking module of the Schrödinger Suite 2008.21,22 As outlined in Figure 1, the ligand library was first screened using Glide HTVS, which is specifically optimized to weed out the conspicuous nonbinders in an early stage with minimal computational expense. The top 50% of compounds on HTVS were passed to the subsequent more rigorous SP scoring. Finally, 50% of the top SP-ranked compounds were screened by Glide XP. Glide XP is the most resource intensive docking method of the Schrödinger Suite and is intended to minimize false positive hits.23 Although molecular docking has been established as a useful method in identifying bioactive compounds, scoring functions still represent a hurdle in the accuracy of VS.24 Hence, we retained 320 compounds with better than or equal to −6.0 kcal/mol Glide XP docking score for the final analysis. Because knowledge about the target under investigation has already been demonstrated as a key factor in enhancing the virtual screen enrichment, top compounds were visually inspected for key interactions with the receptor.25 From the docking results, we found two notable subpockets occupied by ligand clusters. The most prominent hot spot was observed toward the base of the C loop where most of the top-scored ligands displayed docking preference (see Figure 4). Another prominent subpocket was located in the same region as cobratoxin binding, close to the agonist binding site. We observed ligands with similar scaffolds in the top hits; therefore, compounds with diverse chemotypes were selected for in vitro assay in anticipation of obtaining structurally diverse compounds for further investigation. Eleven predicted active compounds and three negative control compounds (see Table S1 in the Supporting Information for ChemBridge ID numbers) were selected for bioassay based on structural diversity, binding pocket location, and standard error of the scoring results. Furthermore, the top compounds were redocked with AutoDock4 for cross-validation.26,27 In all cases, the Glide XP docking mode was reproducible with the AutoDock4 Lamarckian genetic algorithm, although to varying degrees of success. Each ligand was docked 100 independent times to the epibatidine-bound hα4β2 nAChR model with AutoDock4, and the results were clustered according to all-atom rmsd with a tolerance of 2 Å. The size of the docking clusters and the corresponding average docking scores for the poses that correspond with the Glide XP results are reported in Table 1.
Figure 1.
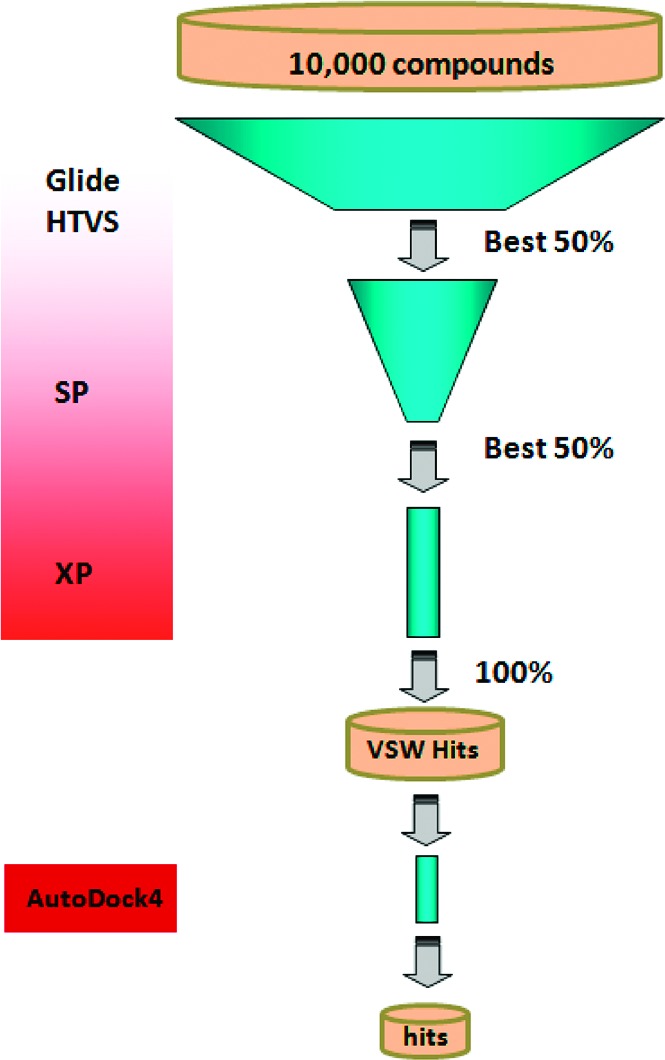
Hierarchical structure-based VS protocol scheme. A library of 10000 compounds from the ChemBridge CNS Set was evaluated in the fast high-throughput stage (HTVS) of GLIDE VSW. The top 50% of the compounds were further evaluated in the more CPU intensive SP stage. In the subsequent XP stage, 50% top compounds from SP docking were evaluated. Top compounds from the list were visually inspected for interactions. Finally, the selected compounds were cross-validated with AutoDock4.
Figure 4.

Molecular interactions of the compounds 1, 2, 4, and 9, represented by A, B, C, and D, respectively, predicted by Glide XP docking. The monomer chains (ribbon representation) and carbon atoms from each chain are differentially colored (α4 = cyan, and β2 = light orange). Epibatidine is shown as a stick representation with purple carbon, whereas the docked compounds are represented as stick green carbon. Relevant amino acid residues in the binding site are shown in ball and stick representation.
Table 1. In Silico Scoring and in Vitro Activities of Virtual Screen Hits against hα4β2 nAChRs.
| docking energy (kcal/mol) |
||||
|---|---|---|---|---|
| compd | Glide XP | AutoDock4 | AutoDock4 clustering (%) | percentage control (%) cont. ± SEMa |
| 1 | –8.57 | –9.17 | 82 | 47.7 ± 7.1 |
| 2 | –8.55 | –7.57 | 47 | 46.9 ± 1.7 |
| 3 | –8.43 | –7.91 | 63 | 83.2 ± 13.7 |
| 4 | –8.13 | –8.69 | 40 | 49.5 ± 11.7 |
| 5 | –7.92 | –8.03 | 60 | 72.5 ± 10.0 |
| 6 | –6.80 | –8.44 | 38 | 71.0 ± 20.1 |
| 7 | –6.72 | –7.94 | 61 | 102.8 ± 18.3 |
| 8 | –6.63 | –9.14 | 44 | 59.9 ± 2.6 |
| 9 | –6.61 | -7.73 | 70 | 51.1 ± 7.7 |
| 10 | –6.55 | –8.95 | 7 | 76.2 ± 14.6 |
| 11 | –6.42 | –8.30 | 28 | 59.8 ± 9.7 |
| N1 | –1.68 | –5.40 | 15 | 74.0 ± 4.8 |
| N2 | –1.61 | –6.24 | 10 | 83.3 ± 8.9 |
| N3 | –1.50 | –7.91 | 16 | 86.2 ± 16.9 |
Percentage inhibition of the agonist activity at 50 μM (n = 3).
The VS hits were experimentally tested with an in vitro calcium accumulation assay on HEK tsA201 cells stably expressing either hα4β2 nAChRs or hα3β4 nAChRs. An assay previously reported was used with slight modifications.9,28 Functional responses were quantified by first calculating the net fluorescence changes (the difference between control sham-treated and control agonist-treated groups). Net peak (maximum) fluorescence values during the third treatment period for both the control-agonist treatment group and the antagonist (with agonist) treatment group were determined. Results were expressed as a percentage of control-agonist groups. Out of the 11 in silico hits tested for the activity in a preliminary single concentration assay, four compounds demonstrated approximately 50% inhibition of hα4β2 nAChRs at concentrations of 50 μM (Figure 2). These four hits also showed little or no inhibitory activity on hα3β4 nAChRs at concentrations of 50 μM. These four hits, 1, 2, 4, and 9, as illustrated in Figure 2, were then tested in concentration–response studies (Figure 3 and Table 2).
Figure 2.

Preliminary in vitro validation of hit compounds on hα4β2 and hα3β4 nAChRs. The hit molecules, identified through structure-based VS, were validated using 50 μM concentrations on both hα4β2 and hα3β4 nAChRs, using the calcium accumulation assay. Values shown are meana ± SEMs of threeexperiments, performed in triplicate. Results are expressed as percentage of control, epibatidine-stimulated peak fluorescence levels.
Figure 3.

Concentration–response studies of hit molecules on hα4β2 and hα3β4 nAChRs. Hit compounds 1, 2, 4, and 9 were tested on both hα4β2 and hα3β4 nAChRs at the indicated concentrations, using the calcium accumulation assay. Values shown are means ± SEMs of 3–10 experiments, performed in triplicate. Results are expressed as percentage of control, epibatidine-stimulated peak fluorescence levels.
Table 2. Effects of Hit Compounds on hα4β2 and hα3β4 nAChRs.
| hα4β2 nAChRs |
hα3β4 nAChRs |
|||
|---|---|---|---|---|
| compd | IC50 (μM)a | nhb | IC50 (μM)a | nhb |
| 1 | 14.4 (7.5–27.8) | –1.0 | 46.3 (29.4–73.1) | –1.0 |
| 2 | 9.8 (6.5–14.7) | –0.9 | 19.6 (12.8–29.9) | –1.3 |
| 4 | 20.6 (7.6–55.4) | –0.8 | >100c | |
| 9 | 6.1 (3.1–11.6) | –0.6 | >100c | |
Geometric means, n = 3–10.
Hill coefficient.
No activity up to 100 μM.
Compound 1 inhibited hα4β2 nAChRs with an IC50 value of 14.4 μM. Glide XP docked pose showed that this compound binds to the receptor with −8.57 kcal/mol binding energy, while AutoDock4 reconfirmed the pose with −9.17 kcal/mol of binding energy and 82% clustering. The compound binds in the pocket toward the base of the C loop with several interactions, the most apparent being an electrostatic interaction of the protonated piperidine nitrogen with β2Asp170 of the F loop (Figure 4A). This negatively charged residue likely plays a role in the closing movement of the C loop toward the β2 F-loop since α4Arg187 on the C loop is in close vicinity to form a salt bridge. The pyridinium methyl group points toward the hydrophobic “aromatic nest” formed by β2Trp56, α4Tyr92, and α4Trp148. The aromatic amine group of the ligand is in close proximity with the backbone carbonyl oxygen of β2Phe171 to form a hydrogen bond. Compound 2, despite being structurally distinct from compound 1, shows similar interactions with β2Asp170 and the interior hydrophobic residues (Figure 4B). The calcium accumulation assay shows that compound 2 inhibited hα4β2 nAChRs with an IC50 value of 9.8 μM. The AutoDock4 binding energy (−7.57 kcal/mol; 47% clustering) was comparable to Glide XP binding energy (−8.55 kcal/mol). The sulfone group is playing a role as a linker, keeping the aromatic ring in position to have favorable interaction with α4Tyr189. Compound 4 inhibited hα4β2 nAChRs with an IC50 value of 20.6 μM. The docking poses determined by both Glide XP (−8.13 kcal/mol) and AutoDock4 (−8.69 kcal/mol; 40% clustering) show the protonated morpholine ring nitrogen of compound 4 to form a favorable Coulombic interaction with the side chain of β2Asp170 (Figure 4C). Hydrogen bonds are formed by the amine linker with the β2Asn54 side chain and α4Tyr92 backbone carbonyl oxygen. The thiophene ring binds deep into the hydrophobic pocket where it shows good van der Waals interactions with hydrophobic residues α4Phe99, α4Val90, α4Trp148, β2Pro122, and β2Phe105. Interestingly, compound 4 extends deeper into the pocket as compared to the other two ligands. Compound 9 showed the most potent inhibition among the four hits with an IC50 value of 6.1 μM on hα4β2 nAChRs. Docking showed that the compound binds to a different pocket, adjacent to the docking site of the other three hits (Figure 4D). The furan ring of the compound buries into a small subpocket formed by several residues of β2 monomer and favorable van der Waals interactions with backbone atoms of the F loop residues β2Asp170, β2Asp169, β2Leu168, β2Ser167, and β2Ser37. The β2Asp169 backbone amide nitrogen acts as a hydrogen bond donor interacting with furan oxygen. The furan ring of compound 9 also forms π–π interaction with the β2Trp56 side chain. An aromatic ring would be ideal fragment in this pocket to interact with several of the nearby aromatic amino acids. An additional hydrogen-bonding interaction is observed between the heterocyclic carbonyl oxygen of compound 9 and the β2Ser167 hydroxyl group. The hydrocarbon tail attached to this ring shows hydrophobic contact with β2Met35, β2Phe118, and β2Leu120. The aromatic side chains of α4Tyr189 and α4Tyr196 of the C loop are within 5 Å of the ligand aromatic rings, showing beneficial contacts. Although the docking energy of 9 was inferior to other hits, the AutoDock4 docking conformations converged with strong clustering statistics (70%) to provide confidence in the binding mode.
In addition to hα4β2 nAChR activity, all compounds were also tested against hα3β4 receptors. Of the four hit compounds, two showed selectivity for hα4β2 vs hα3β4 nAChRs. Compound 9 was the most potent and selective compound on hα4β2 nAChRs among the hits and showed no hα3β4 inhibition up to 100 μM. The comparison of the putative binding site residues of compound 9 reveals several differences between the β4 and β3 subunits. The most evident difference is the amino acid substitution β2Thr58Lys, which would cause an unfavorable steric clash with the hydrocarbon tail from the heterocyclic scaffold. This difference has been previously described for other molecules that also bind to this allosteric binding site.10 This pocket seems to harbor a few more differences, which include β2Met35Gln, β2Lys162Met, β2Phe118Leu, and β2Gln33Lys. Another notable difference between β2 and β4 subunits is on the F loop where a series of consecutive amino acids are unconserved between the subunits. These include β2Leu168Met, β2Val165Thr, β2Glu164Pro, β2Ser163Thr, and β2Lys162Met substitutions. These substitutions, especially the conformationally restricting β2Glu164Pro, could affect the dynamics of the hα3β4 F loop. This might be one of the reasons for the minor difference in the relative selectivity of compounds 1 and 2 with 3- and 2-fold difference in selectivity, respectively. Compound 4, which binds deeper into the pocket, making more interactions with the interface residues, also showed higher affinity for hα4β2 nAChRs as compared to hα3β4 nAChRs. Although most of the amino acid residues in the close proximity to the putative docked binding pocket of compound 4 are conserved, we observed α4Asp96Val and α4Arg187Ile substitution at the α/β interface. Residue α4Asp96 pairs with β2Lys126 to form a salt bridge toward the interior of the channel in the hα4β2 nAChR. Another potential salt bridge is formed between F loop β2Asp170 and C loop α4Arg187. These two salt bridges are absent in the hα3β4 nAChR due to the amino acid substitutions α4Arg187Ile and α4Asp96Val. This suggests a possible difference in the dynamics of these nAChR subtypes and thus a difference in the binding pockets.
In conclusion, the structure-based in silico screening was successful in discovering four novel negative allosteric nAChR scaffolds, two of which showed higher affinity for hα4β2 as compared to hα3β4 nAChR receptors. Optimization of these compounds could potentially yield potent subtype-selective antagonists. These compounds can also be used as chemical probes to further characterize the receptor pharmacology and possibly lead to the development of therapeutic agents for the treatment of neurological diseases.
Acknowledgments
We thank Professor Jon Lindstrom, University of Pennsylvania (Philadelphia, PA), for providing HEK ts201 cells.
This work was partially supported by the NIH/NIDA R21 Grant (5R21DA029433) to D.B.M. and C.L. and computing resource units provided by the Ohio Supercomputer Center. Financial support for B.J.H. is from the National Institutes of Health National Institute on Drug Abuse Diversity Supplement. Financial support for R.E.P. is from the American Foundation for Pharmaceutical Education.
Supporting Information Available
The overall workflow scheme, Chembridge IDs of the compounds, computational methods, and details of in vitro experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Barik J.; Wonnacott S. Molecular and cellular mechanisms of action of nicotine in the CNS. Handb. Exp. Pharmacol. 2009, 173–207. [DOI] [PubMed] [Google Scholar]
- Rahman S.; Lopez-Hernandez G. Y.; Corrigall W. A.; Papke R. L. Neuronal nicotinic receptors as brain targets for pharmacotherapy of drug addiction. CNS Neurol. Disord.: Drug Targets 2008, 7, 422–441. [DOI] [PubMed] [Google Scholar]
- Shimohama S. Nicotinic receptor-mediated neuroprotection in neurodegenerative disease models. Biol. Pharm. Bull. 2009, 32, 332–336. [DOI] [PubMed] [Google Scholar]
- Fowler C. D.; Arends M. A.; Kenny P. J. Subtypes of nicotinic acetylcholine receptors in nicotine reward, dependence, and withdrawal: Evidence from genetically modified mice. Behav. Pharmacol. 2008, 19, 461–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arneric S. P.; Holladay M.; Williams M. Neuronal nicotinic receptors: A perspective on two decades of drug discovery research. Biochem. Pharmacol. 2007, 74, 1092–1101. [DOI] [PubMed] [Google Scholar]
- Jensen A. A.; Frolund B.; Liljefors T.; Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem. 2005, 48, 4705–4745. [DOI] [PubMed] [Google Scholar]
- Albuquerque E. X.; Pereira E. F.; Alkondon M.; Rogers S. W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura R. F.; Hogenkamp D. J.; Li W. Y.; Tran M. B.; Belluzzi J. D.; Whittemore E. R.; Leslie F. M.; Gee K. W. Negative allosteric modulation of nicotinic acetylcholine receptors blocks nicotine self-administration in rats. J. Pharmacol. Exp. Ther. 2007, 323, 907–915. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cestari T. F.; Henderson B. J.; Pavlovicz R. E.; McKay S. B.; El-Hajj R. A.; Pulipaka A. B.; Orac C. M.; Reed D. D.; Boyd R. T.; Zhu M. X.; Li C.; Bergmeier S. C.; McKay D. B. Effect of novel negative allosteric modulators of neuronal nicotinic receptors on cells expressing native and recombinant nicotinic receptors: implications for drug discovery. J. Pharmacol. Exp. Ther. 2009, 328, 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson B. J.; Pavlovicz R. E.; Allen J. D.; Gonzalez-Cestari T. F.; Orac C. M.; Bonnell A. B.; Zhu M. X.; Boyd R. T.; Li C.; Bergmeier S. C.; McKay D. B. Negative allosteric modulators that target human alpha4beta2 neuronal nicotinic receptors. J. Pharmacol. Exp. Ther. 2010, 334, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovicz R. E.; Henderson B. J.; Bonnell A. B.; Boyd R. T.; McKay D. B.; Li C. Identification of a negative allosteric site on human α4β2 and α3β4 neuronal nicotinic acetylcholine receptors. PLoS ONE 2011, 6, e24949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andricopulo A. D.; Salum L. B.; Abraham D. J. Structure-based drug design strategies in medicinal chemistry. Curr. Top. Med. Chem. 2009, 9, 771–790. [DOI] [PubMed] [Google Scholar]
- Geromichalos G. D. Importance of molecular computer modeling in anticancer drug development. J. BUON 2007, 12Suppl. 1S101–S118. [PubMed] [Google Scholar]
- Waszkowycz B. Towards improving compound selection in structure-based virtual screening. Drug Discovery Today 2008, 13, 219–226. [DOI] [PubMed] [Google Scholar]
- Babakhani A.; Talley T. T.; Taylor P.; McCammon J. A. A virtual screening study of the acetylcholine binding protein using a relaxed-complex approach. Comput. Biol. Chem. 2009, 33, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utsintong M.; Talley T. T.; Taylor P. W.; Olson A. J.; Vajragupta O. Virtual screening against alpha-cobratoxin. J. Biomol. Screen. 2009, 14, 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulens C.; Akdemir A.; Jongejan A.; van Elk R.; Bertrand S.; Perrakis A.; Leurs R.; Smit A. B.; Sixma T. K.; Bertrand D.; de Esch I. J. Use of acetylcholine binding protein in the search for novel alpha7 nicotinic receptor ligands. In silico docking, pharmacological screening, and X-ray analysis. J. Med. Chem. 2009, 52, 2372–2383. [DOI] [PubMed] [Google Scholar]
- Bourne Y.; Talley T. T.; Hansen S. B.; Taylor P.; Marchot P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake alpha-neurotoxins and nicotinic receptors. EMBO J. 2005, 24, 1512–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Moitessier N.; Englebienne P.; Lee D.; Lawandi J.; Corbeil C. R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153Suppl. 1S7–S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert M. H. Targeted scoring functions for virtual screening. Drug Discovery Today 2009, 14, 562–569. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- Huey R.; Morris G. M.; Olson A. J.; Goodsell D. S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [DOI] [PubMed] [Google Scholar]
- McKay D. B.; Chang C.; Gonzalez-Cestari T. F.; McKay S. B.; El-Hajj R. A.; Bryant D. L.; Zhu M. X.; Swaan P. W.; Arason K. M.; Pulipaka A. B.; Orac C. M.; Bergmeier S. C. Analogs of methyllycaconitine as novel noncompetitive inhibitors of nicotinic receptors: Pharmacological characterization, computational modeling, and pharmacophore development. Mol. Pharmacol. 2007, 71, 1288–1297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.