

Abstract
Two single-tryptophan variants were generated in a thermophilic alcohol dehydrogenase with the goal of correlating temperature-dependent changes in local fluorescence with the previously demonstrated catalytic break at ca. 30 °C (Kohen et al., Nature1999, 399, 496). One tryptophan variant, W87in, resides at the active site within van der Waals contact of bound alcohol substrate; the other variant, W167in, is a remote-site surface reporter located >25 Å from the active site. Picosecond-resolved fluorescence measurements were used to analyze fluorescence lifetimes, time-dependent Stokes shifts, and the extent of collisional quenching at Trp87 and Trp167 as a function of temperature. A subnanosecond fluorescence decay rate constant has been detected for W87in that is ascribed to the proximity of the active site Zn2+ and shows a break in behavior at 30 °C. For the remainder of the reported lifetime measurements, there is no detectable break between 10 and 50 °C, in contrast with previously reported hydrogen/deuterium exchange experiments that revealed a temperature-dependent break analogous to catalysis (Liang et al., Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9556). We conclude that the motions that lead to the rigidification of ht-ADH below 30 °C are likely to be dominated by global processes slower than the picosecond to nanosecond motions measured herein. In the case of collisional quenching of fluorescence by acrylamide, W87in and W167in behave in a similar manner that resembles free tryptophan in water. Stokes shift measurements, by contrast, show distinctive behaviors in which the active-site tryptophan relaxation is highly temperature-dependent, whereas the solvent-exposed tryptophan’s dynamics are temperature-independent. These data are concluded to reflect a significantly constrained environment surrounding the active site Trp87 that both increases the magnitude of the Stokes shift and its temperature-dependence. The results are discussed in the context of spatially distinct differences in enthalpic barriers for protein conformational sampling that may be related to catalysis.
Introduction
Understanding the role that protein dynamics play in enzyme catalysis is an area of intense research efforts and vigorous debate.1−6 Because timescales for protein motions range from femtoseconds (fs)/picoseconds (ps) to seconds,7 multiple kinetic and spectroscopic approaches are needed to interrogate the roles of slow versus fast motions in reaction rate acceleration. Historically, the analysis of kinetic isotope effects (KIEs) has contributed significantly to our understanding of the contribution of proximal and distal motions during C–H bond activation.8−13 Such analyses have included secondary KIEs,14,15 while being more generally associated with the properties of primary hydrogen KIEs and their temperature dependencies.13 Secondary KIEs have also been extended to implicate protein dynamics during heavy atom group transfer reactions.16,17 As spectroscopic techniques have been developed for macromolecular systems such as enzymes, there has been an explosion in the diversity of techniques used to probe protein dynamics. Namely, advances in techniques involving nuclear magnetic resonance (NMR),7,18−20 hydrogen/deuterium (H/D) exchange mass spectrometry,21−23 X-ray crystallography,24,25 single molecule methods,26−28 temperature-jump methods,29,30 and vibrational spectroscopy,31−33 are but a few of the experimental approaches available to characterize virtually any timescale of protein motion. However, obtaining evidence that relates a specific protein motion to the variables that control function is a much more elusive goal.
In the latter context, many ideas have emerged, with the goal of linking a protein’s catalytic efficiency to the extensive degrees of freedom accessible within these large systems. Proposals put forth include the involvement of networks of protein motions,2 the creation of near-attack conformers,34 rate-promoting protein modes,35 and protein conformational selection.7,36 The temperature-dependence of local probes of protein dynamics and electrostatics can be particularly informative and has been implemented via measurements of the Stark shift,37,38 Stokes shift,39−41 and T-jump fluorescence methods.42 Most of the completed studies have been implemented in the context of very low temperatures, characterizing features of the glass transition within proteins,43 or the impact of varying solvents.44 Herein, we provide a rigorous study that analyzes dynamical behavior at distinct functional locales within ht-ADH in the context of two functionally disparate temperature regimes.
Time-resolved fluorescence techniques, focusing either on the intrinsic fluorescence of tryptophan side chains or site specifically incorporated unnatural amino acids, are increasingly employed to measure fluorescence lifetimes, the time dependence of Stokes shifts, and the time constants for fluorescence quenching in the presence of exogenous ligands. Of particular interest is the time dependence of the reorganization of the surrounding medium in response to excitation at a single fluorescent side chain through the construction of time-resolved emission spectra (TRES), and the quantification of the corresponding time-dependent red shifts. Early investigations of time-dependent Stokes shifts measured the responses of homogeneous solvents of varying polarity to excited-state dipoles created in small molecules.45−48 In light of the recent increased interest in protein dynamics, time-dependent Stokes shifts have also been pursued in proteins to better understand structure–function relationships,41,49−51 rotameric interconversions,52,53 the role of biological water layers,54−57 and enzyme reaction coordinates.58 Moreover, Stern–Volmer analyses have been beneficial in understanding degrees of amino acid solvent exposure in proteins,51,59,60 interactions of quenchers with fluorophores,61,62 and characterization of protein conformational changes.63,64
Decades of kinetic and mass spectrometric data collected on a thermophilic alcohol dehydrogenase (ht-ADH) isolated from Bacillus stearothermophilus (B. stearothermophilus) have led to an integrated model that relates the properties of hydride transfer to the inherent properties of the conformational ensemble accessible to the enzyme. Arrhenius plots of the temperature-dependent hydride transfer within ht-ADH exhibit a discontinuous “break” at 30 °C in which the enthalpy of activation decreases by ca. 7 kcal/mol at higher temperatures (i.e., 22 kcal/mol < 30 °C; 15 kcal/mol > 30 °C).8 Moreover, the KIE data reveal that hydride transfer is temperature-dependent below 30 °C and temperature-independent above 30 °C. The aggregate kinetic data imply that the same enzyme can achieve two drastically different conformational ensembles with distinctive tunneling properties. Structural insight into the two temperature regimes of ht-ADH has come from the time and temperature-dependence of H/D exchange within the apoenzyme.22 Among twenty-one peptides analyzed in ht-ADH, representing almost full coverage of the protein sequence, five peptides were found to exhibit increased amide backbone solvent exchange with D2O with a transition temperature of 40–55 °C. Another five peptides located exclusively within the substrate-binding domain showed increased protein backbone deuteron incorporation, transitioning between 20–40 °C. These H/D exchange experiments in apoprotein have yielded a spatial resolution of the temperature-dependent change in protein flexibility that correlates with the catalytic behavior of ht-ADH. More recently, the impact of mutation of two cofactor binding domain residues (Leu176 and Val260) to side chains of reduced mass has led to a model in which the relative population of rigid, catalytically incompetent microstates and more flexible, tunneling-ready microstates govern not only the tunneling properties but also the magnitude of the experimental enthalpies and entropies of activation.65
The present work utilizes ps-resolved fluorescence spectroscopy to investigate fluorescence lifetimes, the temperature-dependence of time-dependent Stokes shifts, and collisional quenching of ht-ADH by acrylamide. In order to observe the enzyme dynamics with minimal perturbation, the red-edge excitation effect of tryptophan was exploited to minimize contributions from fluorescent tyrosines and phenylalanines.66 The native wild-type (WT) ht-ADH monomeric structure contains three tryptophans. Because lifetime measurements of WT protein cannot distinguish the origin of fluorescence without a priori knowledge about each locale, single-tryptophan variants were created such that two of the three tryptophans were replaced with combinations of either tyrosine or phenylalanine. Of the three single-Trp possibilities, two of them were found to have kinetic properties that closely mirror WT behavior. These constructs are W49F:W167Y (W87in), which fortuitously resides at the enzyme active site within van der Waals contact of the substrate-binding pocket, and W49F:W87F (W167in), a solvent-exposed, remote-site tryptophan found over 25 Å away from the catalytic center (Figure 1). Expanded representations of the side chains surrounding each tryptophan are shown in Figure 2. As will be presented herein, fluorescence lifetimes and time-dependent Stokes shifts reveal a greater sensitivity to environmental differences at each single tryptophan site than their collisional quenching behaviors. The data are discussed in the context of the unique properties of the W87in as they relate to catalysis.
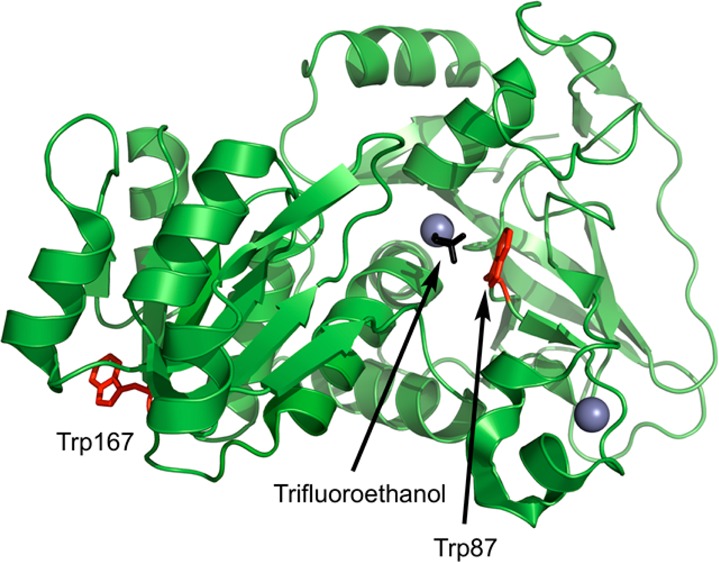
Figure 1.

Structure of monomeric ht-ADH (PDB: 1RJW). The relative locations of the two tryptophans (red) and substrate analogue trifluoroethanol (black) are represented with sticks. The catalytic zinc inside the active site (gray) is represented as a sphere.
Figure 2.
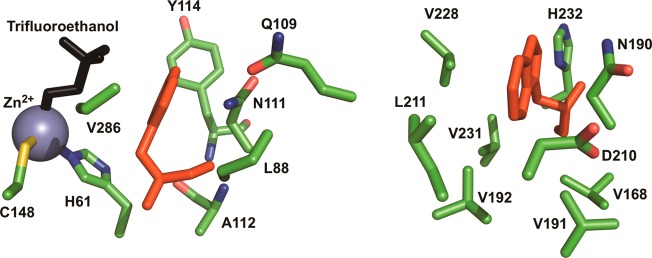
Closeups of residues surrounding Trp87 (left) and Trp167 (right). Tryptophans are shown in red. Residues with side-chain interactions within 6 Å of the tryptophan are shown for each locale. Amide backbone atoms were omitted for clarity.
Experimental Section
Site-Directed Mutagenesis and Protein Purification
W87in (W49F:W167Y) and W167in (W49F:W87F) were generated by following protocols in the QuikChange site-directed mutagenesis kit (Agilent Technologies). Mutant plasmids were generated and amplified using a PTC-2000 Peltier Thermal Cycler (MJ Research). Appropriate combinations of the following primers (Operon) and their respective reverse complements (not shown) were introduced to the PCR mixtures to generate the single tryptophan variants: W49F: 5′-CCGCTCACGGCGATTTCCCGGTAAAACCAAAAC-3′; W87F: 5′-CCGCGTTGGAATTCCTTTCTTATATTCTGCATGC-3′; W167Y: 5′-GTAACAGGGGCAAAACCAGGAGAATATGTAGCAATTTACGGTAT-3′. Changes to the pET-24b(+) Escherichia coli expression vector containing the B. stearothermophilus ht-ADH gene are underlined. Gene sequencing was performed by the UC Berkeley DNA Sequencing Facility. Plasmid purification for gene sequencing was executed using the materials and protocols contained within the QIAprep spin miniprep kit (QIAGEN). Tyrosine was chosen for position 167 due to significant activity loss (ca. 70%) after 30 min of incubation at 4 °C for W49F:W167F. Subsequent plasmid transformation, cell growth, and lysis are described elsewhere.22
The single-tryptophan variants were purified as previously described with some minor modifications.67 Because bound NADH quenches Trp87 fluorescence via FRET (Figure S1 in the Supporting Information), 70 mL of 1 mM adenosine 5′-monophosphate (Sigma-Aldrich) was used to elute the single tryptophan variants from the 5′-AMP Agarose affinity column. This was used in lieu of NAD+ to avoid any potential distortion of fluorescence measurements. After affinity column elution, the protein was spin concentrated to <500 μL using a 15 mL regenerated cellulose centrifugal spin filter (Millipore) then buffer exchanged three times to a 30× dilution with 25 mM KPi, 0.1 mM DTT, pH 7.0 (Fisher Scientific). The possibility of bound 5′-AMP to the enzyme was ruled out due to a high inhibition constant (KI = 2.3 mM) and the absence of shoulder absorbance at 260 nm in the purified product. Purified apoprotein was stored in 20–50 μL aliquots having a final concentration of at least 160 μM. Protein concentration was determined by Bradford assays68 and purity was determined by 10% SDS–PAGE.
Steady-State Kinetics
Enzyme turnover for W87in and W167in was quantified by monitoring the reduction of NADH (λmax = 340 nm) on a Cary50 spectrophotometer (Agilent Technologies) under varying concentrations of NAD+ (Sigma-Aldrich) and α,α-d2-benzyl alcohol (CDN Isotopes). NAD+ was freshly prepared before all kinetics assays by titrating a 200 mM stock solution to pH 7.0 with 0.5 M NaOH then diluting to a final concentration of 100 mM. The concentrations were simultaneously varied from 1–12 mM for NAD+ and from 2–16 mM for α,α-d2-benzyl alcohol (d-BnOH). All assays were dissolved in 50 mM potassium phosphate buffer to a final volume of 990 μL; reactions were then initiated using 10 μL of appropriately diluted enzyme. Initial rates were determined by measuring the slopes of the linear traces acquired for at least 3 min. Such conditions for turnover were assayed over nine temperatures ranging from 7–51.5 °C. Assays were incubated in a water bath before placement into the spectrophotometer’s Peltier-controlled cuvette cell. Kinetic parameters were obtained from a nonlinear fit to the 2D-Michaelis–Menten equation using DataFit (Oakdale Engineering); the corresponding activation parameters were obtained from nonlinear fits to the Arrhenius equation using DataFit.
Time-Dependent Activity Loss
The activity loss of W87in and W167in were tested under conditions that would reflect the stability of the protein spanning the duration of time-resolved fluorescence measurements (typically 2–3 h for measurements of 7 wavelengths at each temperature). Each respective variant was diluted to a concentration of 3–4 μM and incubated for 4 h at 10 °C, 30 °C, and 50 °C. The activity was measured under saturating conditions (10 mM NAD+, 15 mM d-BnOH) at ten time points ranging from 2 min to 4 h after enzyme incubation in triplicate.
Excessive activity loss is defined as leading to less than 75% of initial activity after a 2 min incubation. For W87in, there was little difference in protein stability profiles at 10 and 30 °C, while enzyme inactivation is somewhat faster at 50 °C (Figure S2 in the Supporting Information). On the basis of the observations, W87in samples were changed after 150 min of measurement between 10 and 30 °C and every 90 min at temperatures above 30 °C.
For W167in, the protein stability profiles indicated a smaller temperature-dependence at low temperatures. W167in does not show excessive loss of activity at 10 or 30 °C over a 4 h incubation period, although it is less stable than W87 in a high temperature (Figure S2 in the Supporting Information). For W167in, samples were not changed for lifetime measurements collected between 10–30 °C and replaced every 75 min at temperatures above 30 °C.
Other Controls
The oligomeric state and secondary structure of the single-Trp mutants were verified by size-exclusion chromatography and CD, respectively, using methods previously described.66 Circular dichroism (CD) spectra comparing W87in and W167in to the WT secondary structure are shown at 30 °C (Figure S3 in the Supporting Information). No major changes in secondary structure were observed across all variants relative to WT, verifying that the conservative substitutions contained within each variant do not cause a large change in overall protein structure. Gel filtration chromatograms are also shown at 4 °C (Figure S4 in the Supporting Information). Both variants elute predominantly as a tetramer, and all oligomeric distributions elute at retention times correlated to the appropriate molecular weight.
Steady-State Fluorescence
Fluorescence emission spectra of the single-Trp mutants were collected on a custom built Fluorolog-3 spectrofluorometer (Horiba Jobin-Yvon). Excitation was achieved with a 450W xenon lamp. The light was focused using a double Czerny Turner excitation monochromator (1 nm bandpass) with 1200 grooves/mm blazed at 330 nm. Photons from sample emission were focused using a single Czerny−Turner monochromator (10 nm bandpass) with 1200 grooves/mm blazed at 500 nm. The excitation and emission optics were calibrated using the lamp spectral maximum at 467 nm and the water Raman scattering band at 397 nm, respectively, using HPLC-grade water in a quartz cuvette.
For ht-ADH emission spectra, the excitation wavelength was set at 291 nm, and the emission spectra were collected from wavelengths spanning 305–395 at 0.5 nm increments. Spectra were collected over temperatures ranging from 10 to 50 °C in 5 °C intervals with a water bath controlling the cell temperature. Samples were equilibrated for 10 min at each respective temperature in a quartz cuvette (Starna Cells) before collecting emission spectra. All protein emission spectra reported were corrected for background fluorescence and Raman scattering. Peak emission wavelengths were determined by fitting the corrected spectra to a Gaussian using DataFit.
Picosecond-Resolved Fluorescence Spectroscopy
The time-correlated single photon counting technique (TCSPC) was used to obtain fluorescence decays. Selective excitation of the single tryptophan in each mutant was achieved using a Nano-LED 295 having a typical full-width at half-maximum pulse of <1.2 ns with a 1 MHz repetition rate. The Nano-LED was powered by a FluoroHub photon-counting controller. Single-photon signals were detected by a TBX-04 photomultiplier tube detection module. The photon-counting hub window contained 1024 channels with a time-to-amplitude conversion range of 56 ps/channel. Lifetimes were measured using reverse mode counting with a 75 ns coaxial delay and 0 ns sync delay. Magic angle (55°) conditions were employed to eliminate lifetime distortions resulting from rotational motion. Fluorescence decays of the single-Trp variants were obtained from 310–370 nm in 10 nm intervals with a 10 nm emission bandpass. Instrument response functions were collected once at each temperature using only 50 mM KPi, pH 7.0, with the emission monochromator set at 291 nm. All optical settings and delays were kept constant when acquiring the instrument response function. All decays were collected with a peak preset of 10000 counts. The time resolution of the instrument is calculated to be 120 ps. Procedures for sample incubation during data acquisition were performed in the same manner as listed under Steady-State Fluorescence (above) and executed at the same nine temperatures ranging from 10 to 50 °C in 5 °C intervals.
Construction of Time-Dependent Stokes Shift Spectra
Time-resolved emission spectra (TRES) were constructed from fluorescence decay data by methods previously described in more thorough detail.69−71 Briefly, the function describing the fluorescence decay was determined by deconvoluting the instrument response function (IRF) from a sum of exponential decays at each respective wavelength, λ (eq 1):
| 1 |
where I(t) is the IRF, n represents the number of exponential components needed for fitting the decay data, αi represents the weighted amplitudes such that ∑i αi(λ) = 1, and τi represents the fluorescence decay constant for the ith component. One exponential was always used to begin fitting the decays, and additional exponentials were iteratively added until a reduced X2 < 1.25 was achieved for the residuals. After obtaining satisfactory fitting parameters, an H(λ) that is linearly proportional to the steady-state emission spectrum is calculated utilizing the steady-state emission, F(λ) (eq 2):
| 2 |
Equation 3 represents the points Γ(λ,t) that comprise the TRES, which are calculated at arbitrary time points t by multiplying H(λ) to the deconvoluted decay parameters extracted from the raw data represented by eq 1:
| 3 |
Equations 4 and 5 are used for fitting the experimental time points at arbitrary times (t) to a log-normal line shape as a function of frequency (ν):62,72
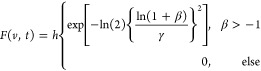 |
4 |
| 5 |
where νp is peak frequency that is time-dependent, h and Δ represent the respective peak heights and width, and γ is the asymmetry parameter. In accordance with previous treatments, emission timeslices were constructed until the νp reached the steady-state emission wavelength, νss measured in each respective single-Trp variant. The peak frequencies obtained from each time point were then plotted to construct the solvation correlation function (eq 6):
| 6 |
The red shift rate constant was obtained by fitting the solvation correlation function to a single or double exponential decay.
Stern–Volmer Analysis
The bimolecular quenching constant for acrylamide colliding with an excited state tryptophan was measured as a function of temperature in each single-Trp variant. All fluorescence lifetime decays were collected using the same instrumental setup and temperature ranges described in Picosecond-Resolved Fluorescence Spectroscopy and Steady-State Fluorescence. The emission wavelength was set to the peak wavelength for each respective single tryptophan variant. Fluorescence lifetimes were collected at concentrations ranging from 0–80 mM acrylamide in 20 mM increments. The reasons for the narrow range assayed versus other reports (typically up to 400–1000 mM acrylamide) were 2-fold: (1) the absorbance at 291 nm exceeded 0.1 at concentrations greater than 80 mM; (2) as a function of acrylamide concentration, [Q], contributions from static quenching (V) caused by ground-state complex formation are effectively minimized in the modified Stern–Volmer equation at the low concentration limit (eq 7):59,72−74
| 7 |
For this study the average lifetime, ⟨τ⟩, at each acrylamide concentration (eq 8) was calculated in order to construct the collisional Stern–Volmer plots (eq 9):
| 8 |
| 9 |
where kq is the second-order collisional quenching constant and ⟨τo⟩ is the average lifetime in the absence of acrylamide.
Results
Steady-State Kinetics
The use of single-Trp variants that can serve as appropriate spectroscopic analogues for WT requires that the kinetic features for each variant are similar to the WT counterpart. Arrhenius plots for both W87in and W167in (Figure S5 in the Supporting Information) exhibit the signature break first seen in WT.65 The enthalpies of activation for W87in and W167in at high and low temperatures, ΔH‡(hi) and ΔH‡(lo), are slightly lower than the barriers measured for WT. Regardless of these slightly depressed values, the low-temperature activation enthalpies of both variants are greater than three standard deviations from the WT high-temperature activation energy which is 14.5 ± 0.4 kcal/mol.8 Moreover, the ΔΔH‡ for both variants is within error of that observed in WT (Table 1).
Table 1. Kinetic and Activation Parameters for the ht-ADH Single-Trp Variantsa.
| mutant | kcat (s–1) | Km(NAD+) (mM) | Km(d-BnOH) (mM) | Dkcat/D(kcat/KM), NAD+ | Dkcat/D(kcat/KM), BnOH | KIEc | ΔH‡(lo)−ΔH‡(hi), (kcal/mol)d |
|---|---|---|---|---|---|---|---|
| WTb | 8.0 (1.0) | 1.1 (0.1) | 3.8 (0.5) | 1.0 (0.1) | 0.8 (0.2) | 3.1 (0.2) | 6.7 (0.9)e |
| W87in | 3.4 (0.2) | 1.8 (0.2) | 4.6 (0.6) | 1.0 (0.2) | 1.0 (0.2) | 2.5 (0.1) | 5.4 (0.4) |
| W167in | 3.0 (0.2) | 1.3 (0.3) | 6.7 (0.4) | 0.8 (0.1) | 0.9 (0.1) | 2.2 (0.1) | 5.7 (0.4) |
All kinetic parameters are reported at 30 °C.
From ref (65).
Defined as kcat(H-BnOH)/kcat(D-BnOH).
ΔH‡(lo) indicates ΔH‡ ≤ 30 °C; ΔH‡(hi) indicates ΔH‡ ≥ 30 °C.
ΔH‡(lo) = 22, 18.9, and 19 kcal/mol for WT, W87in, and W167in, respectively.
The conservative mutations made for both single-Trp variants also have only a minor impact on the limiting kinetic parameters in comparison to WT (Table 1). The kcat values, which report on rate-limiting hydride transfer, decrease by less than 3-fold at 30 °C and are within one standard deviation of each other. The KIE on kcat for both variants is decreased slightly, from 3.1 in WT to 2.5 for W87in and 2.2 for W167 in. While the depression in the KIE suggests that additional kinetic steps might be emerging that are isotopically insensitive, the ratio of the KIE on kcat and kcat/KM is close to unity for both W87in and W167in, supporting a kinetic mechanism in which hydride transfer largely limits catalysis under the conditions of both first-order (kcat) and second-order (kcat/KM) conditions. Finally, the KM values for substrate and cofactor are similar to one another and to the WT enzyme. These points indicate that steps associated with substrate/cofactor binding or product release are not contributing significantly as partially rate-determining steps in the single-Trp constructs.
Steady-State Fluorescence
The peak intensity-normalized steady-state emission spectra for W87in and W167in were determined at selected temperatures (Figure S6 in the Supporting Information). W87in peak emission occurs at 337.0 ± 0.5 nm for all temperatures; W167in peak emission occurs at 338.0 ± 0.5 nm. The temperature-dependence of the emission peak reveals two interesting features. The first is that the peak emission wavelength does not shift with increasing temperature. The second is that the emission intensity decreases linearly with increasing temperature (Figure S7 in the Supporting Information). In accordance with previously reported steady-state fluorescence behavior,65 these combined observations imply that there is no gross structural transition, disruption in solvation environment, or fundamental change in quenching behavior that could be the origin of the observed break in the Arrhenius plots for W87in and W167in.
Picosecond-Resolved Lifetime Decays for W87in and W167in
Figure 3 shows representative lifetime decays at the respective peak emission wavelength for each single-Trp variant at 30 °C. The residual errors and the associated χ2 goodness-of-fit value associated with fitting the decays to a given number of exponential components are listed below the lifetime traces. W87in was fit most appropriately with three exponential components having average lifetimes and amplitudes of 0.67 ns (8%), 2.17 ns (42%), and 4.64 ns (50%). W167in was fit with two exponential functions at 3.10 ns (8%) and 6.36 ns (92%). Though the fluorescence decay for W167in could be interpreted as predominantly single exponential in nature with more inherent error, the use of two lifetimes becomes increasingly important at the shorter emission wavelengths.
Figure 3.
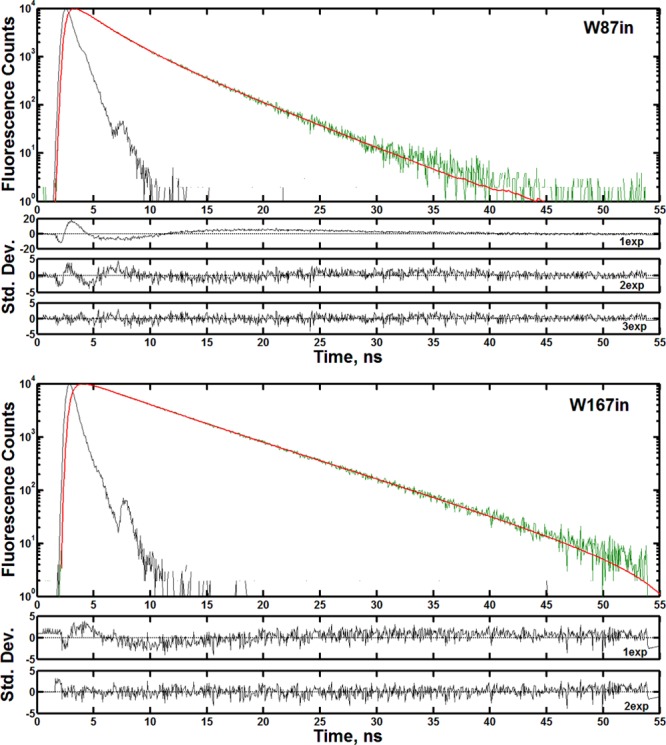
Representative lifetime decays and residual errors for W87in (top) and W167in (bottom) at 30 °C. Decays were collected at the peak emission wavelength determined from the steady-state emission spectra for each single-Trp variant. Each panel contains the instrument response function (black), the raw decay data (green), and the final fit from which lifetime data were derived (red). W87in was fit as a triexponential; W167in was fit as a biexponential. The panels showing the residuals underneath the fluorescence traces are listed in order of increasing number of exponentials. The associated χ2 values for W87in are 15.29 (top), 1.44 (middle), and 1.01 (bottom) for a one, two, and three-exponential fit, respectively. For W167in, the respective χ2 values were 1.90 (top) and 1.10 (bottom) for one and two exponentials.
The temperature-dependence of the weighted-amplitude lifetimes is shown in Table 2. Interestingly, W167in reveals virtually no change in the distribution of the lifetimes at any given temperature, with the long lifetime component consistently carrying a weight of 92% to 93%. In W87in, however, there is a noticeable shift in the lifetime distributions between the two longest lifetime components. The longest lifetime’s amplitude systematically increases in weight from 46% to 55% going from 10 to 50 °C, while the middle lifetime’s amplitude decreases systematically from 46% to 40%. Arrhenius plots of the lifetime components were constructed for W87in (Figure 4) and for W167in (Figure S8 in Supporting Information). Only the shortest lifetime component in W87in has a detectable transition at 40 °C, with ΔH‡ = 12.7 ± 2.9 kcal/mol from 40 to 50 °C and ΔH‡ = 2.2 ± 0.5 kcal/mol from 10 to 40 °C. On the basis of the inherent errors of the lifetime measurements and the activation parameters, the differences in the activation energies are still greater than 3 standard deviations from one another, rendering it difficult to interpret the entire temperature regime as one linear model. Moreover, of the three individual measurements of τ1 at 50 °C (i.e., 0.25, 0.27, and 0.32 ns), even the longest lifetime of 0.32 ns cannot be statistically described by the linear regression from 10 to 40 °C. Hence, we propose that the molecular mechanism governing the fluorescence phenomena contained within τ1 is best characterized as two separate temperature regimes.
Table 2. Temperature-Dependent Fluorescence Decay Parameters for W87in and W167ina,b,c.
| temperature (°C) | α1d | τ1 (ns) | α2 | τ2 (ns) | α3 | τ3 (ns) |
|---|---|---|---|---|---|---|
| W87in | ||||||
| 10 | 0.07 | 0.79 | 0.46 | 2.57 | 0.47 | 5.39 |
| 15 | 0.07 | 0.77 | 0.45 | 2.44 | 0.48 | 5.23 |
| 20 | 0.07 | 0.72 | 0.45 | 2.35 | 0.48 | 5.05 |
| 25 | 0.06 | 0.58 | 0.45 | 2.19 | 0.49 | 4.81 |
| 30 | 0.08 | 0.67 | 0.42 | 2.17 | 0.50 | 4.64 |
| 35 | 0.04 | 0.51 | 0.43 | 2.01 | 0.53 | 4.47 |
| 40 | 0.04 | 0.52 | 0.42 | 1.91 | 0.54 | 4.27 |
| 45 | 0.06 | 0.47 | 0.41 | 1.81 | 0.53 | 4.08 |
| 50 | 0.02 | 0.28 | 0.42 | 1.64 | 0.56 | 3.80 |
| W167in | ||||||
| 10 | 0.07 | 3.43 | 0.93 | 6.94 | ||
| 15 | 0.07 | 3.42 | 0.93 | 6.82 | ||
| 20 | 0.07 | 3.43 | 0.93 | 6.66 | ||
| 25 | 0.07 | 3.26 | 0.93 | 6.46 | ||
| 30 | 0.08 | 3.10 | 0.92 | 6.36 | ||
| 35 | 0.07 | 3.03 | 0.93 | 6.11 | ||
| 40 | 0.08 | 2.76 | 0.92 | 5.90 | ||
| 45 | 0.08 | 2.83 | 0.92 | 5.70 | ||
Lifetimes were obtained at 337.0 nm for W87in and 338.0 nm for W167in.
The amplitudes are reported such that α1 + α2 + α3 = 1.
Lifetime errors are ±10% for τ1 and ±3% for τ2 and τ3.
No sub-nanosecond lifetimes were observed for W167in.
Figure 4.
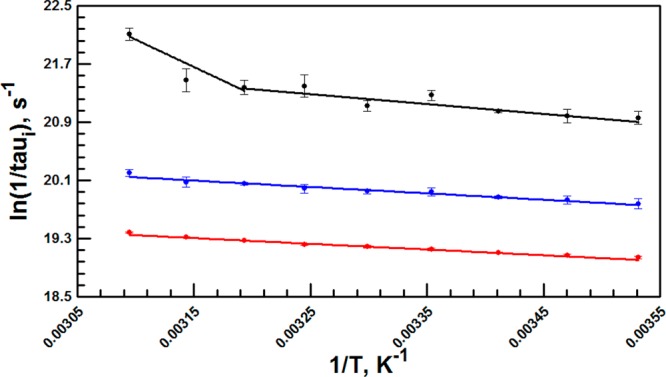
Arrhenius plots of fluorescence lifetimes in W87 in. A noticeable break is seen in the sub-nanosecond lifetime (black), but not for the two longer components (blue and red) (cf. Table 2).
The failure to detect this feature in the overall fluorescence can be ascribed to its relatively small amplitude. The presence of a third rapid decay process with W87in together with the overall distribution of lifetimes in relation to W167in is likely due to the proximity of W87in to the active site zinc ion. It is well-known that divalent metal ions can affect fluorescence lifetimes in the manner reported herein.75−78 The accelerated decrease in fluorescence lifetime above 40 °C suggests a transition to a conformational ensemble that places W87in in closer proximity to the active site metal ion.
Figure 5 illustrates the fluorescence lifetime decays as a function of temperature and emission wavelength. The traces shown in each panel were collected from wavelengths on the blue (310 nm), near the peak (340 nm), and on the red side (370 nm) of the emission spectrum. As expected, as the emission wavelength increases, the fluorescence lifetime increases in both single-Trp variants at all temperatures. For W167in, the traces are virtually identical at all temperatures going from 340 to 370 nm. On the other hand, W87in shows a reasonably significant increase in lifetime going from 340 to 370 nm at 10 °C, and these traces systematically merge closer together until they nearly overlap as the temperature is increased to 50 °C. Since the magnitude of the Stokes shift depends on the increase in fluorescence lifetimes in going from blue to red emission wavelengths, W87in is expected to exhibit a larger and more temperature-dependent Stokes shift than W167 in. These were determined, as shown below, from the magnitude of H(λ) and the subsequently calculated peak emission of the TRES (eqs 2 and 3).
Figure 5.

Representative lifetime decays for W87in (top) and W167in (bottom) shown as a function of temperature and wavelength. Within each mutant panel, the lifetime decays are shown at 10 °C (top), 30 °C (middle), and 50 °C (bottom). The decays correspond to wavelengths at 310 nm (blue), 340 nm (teal), and 370 nm (red) to demonstrate the fluorescence behavior on the blue, peak, and red areas of the emission spectrum for each single-Trp variant. The instrument response function is shown in black.
Temperature Dependence of the Time-Dependent Stokes Shifts in the Single-Trp Variants
Representative TRES are shown at 30 °C for each single-Trp variant in Figure 6 (left). The magnitude of the Stokes shift around Trp87 decreases with increasing temperature (Table 3), from a total shift of 486 cm–1 at 10 °C to a total shift of 100 cm–1 at 50 °C. This decrease in the shift’s magnitude is attributed to a systematically red-shifted time zero emission, υ(0), with increasing temperature. At all temperatures, the red shift for W167in is similar to that for W87in at the highest temperature only. This indicates that elevated temperature is necessary before the environment surrounding the initial excited state dipole of Trp87 resembles the more solvent-exposed surface Trp167.
Figure 6.

Selected Time-Resolved Emission Spectra for W87in (top left) and W167in (bottom left) at 30 °C. The spectral timeslices represent the percentage of the total red shift. Shown in both panels are 0% (black), one-third (blue), two-thirds (green), and 100% (red) completion. For W87in, the times needed to accomplish the respective shift percentage are at 0, 200, 500, and 1000 ps; for W167in, 0, 400, 1000, and 2000 ps. The time dependence of the red shift is derived from the solvation correlation function for W87in (top right) and W167in (bottom right). Decays are shown at 10 °C (black), 20 °C (blue), 30 °C (green), 40 °C (orange), and 50 °C (red). W87in was fit to a single exponential; W167in was fit to a biexponential. W167in was only analyzed to 45 °C; no 50 °C data are shown.
Table 3. Total Red Shift in Stokes Measurements for Each Single-Trp Variants as a Function of Temperature.
| Δυ (cm–1) |
||
|---|---|---|
| temperature (°C) | W87in | W167in |
| 10 | 483 | 100 |
| 15 | 490 | 120 |
| 20 | 402 | 114 |
| 25 | 365 | 99 |
| 30 | 255 | 111 |
| 35 | 234 | 109 |
| 40 | 146 | 96 |
| 45 | 187 | 122 |
| 50 | 100 | – |
The solvation correlation function, c(t), was constructed from the TRES peaks to quantify the time-dependent red shift for both single-Trp variants (Figure 6, right). The red shift for W87in was approximated as a single exponential at all temperatures. W167in was fit to a double exponential due to poor convergence using only a single exponential, but only the short lifetime calculated from the biexponential fit is used to analyze temperature-dependent red-shift behavior. The long lifetime component was typically measured at over 10 ns. This value is rendered unrealistic for excited-state reorganization within the protein when compared to the longest fluorescence lifetime of Trp167 at ca. 6 ns.
The protein-assisted environmental reorganization facilitating the red shift at Trp87 occurs in 1640 ps at 10 °C and accelerates by more than 10-fold to 150 ps as the temperature is increased to 50 °C (Table 4). The red shift times for W167in, on the other hand, show virtually no temperature-dependent trend with rates oscillating around the approximate value of W87in at 15 and 20 °C. There are clearly dynamical motions on the nanosecond timescale at position 167; however, these are controlled by TΔS‡ (11.2 ± 04 kcal/mol) rather than ΔH‡ (−0.2 ± 0.2 kcal/mol). The Arrhenius behavior for Trp87 by contrast (Figure 7) indicates a considerable enthalpic barrier (9.4 ± 0.3 kcal/mol) accompanied by a much more favorable TΔS‡ (34.2 ± 1.3 kcal/mol). The data are indicative of a unique structure at the active site that both stabilizes the excited state dipole (i.e., blue shifts the t = 0 emission wavelength) at low temperatures and is capable of much greater thermal excitation at elevated temperatures.
Table 4. Decay Times for the ht-ADH Single-Trp Variants’ Stokes Shiftsa.
| temperature (°C) | W87inc | W167inb,c |
|---|---|---|
| 10 | 1640 (80) | 1110 (30) |
| 15 | 1070 (60) | 1150 (90) |
| 20 | 1010 (40) | 920 (120) |
| 25 | 750 (20) | 1140 (180) |
| 30 | 460 (20) | 1010 (50) |
| 35 | 400 (20) | 1060 (140) |
| 40 | 220 (30) | 1110 (30) |
| 45 | 270 (30) | 770 (50) |
| 50 | 150 (30) | – |
Decay times are reported in picoseconds.
Only the fast component from the biexponential fit is reported.
Error reported in parentheses is the standard error.
Figure 7.
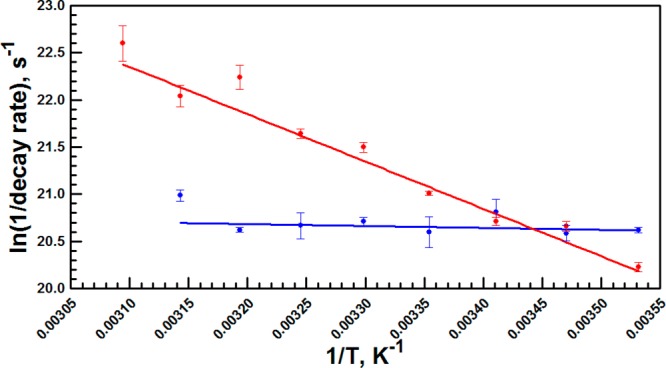
Arrhenius plots of the Stokes shift decay constants for W87in (red) and W167in (blue).
Collisional Stern–Volmer Quenching
Stern–Volmer plots are shown for select temperatures in Figure 8. At 40 °C and above, plots were only constructed out to 60 mM acrylamide for W87in due to apparent plateaus in the plot at 80 mM. The aggregate data indicate environments in both W87in and W167in that have relatively free and equal exposure to the solvent. The bimolecular rates for quenching at Trp167 are roughly only 2-fold higher than for W87in, and all rate constants measured are on the order of 108 M–1 s–1 (Table S1 in the Supporting Information). No Arrhenius break is observed at 30 °C for the temperature-dependent quenching behavior in either variant (Figure 9). Moreover, the enthalpy of activation for quenching in both variants is very close, 3.7 ± 0.1 and 4.7 ± 0.1 kcal/mol, respectively, and similar to the enthalpy of activation of 3.7 kcal/mol for acrylamide quenching of N-acetyl-tryptophanamide in water.72
Figure 8.
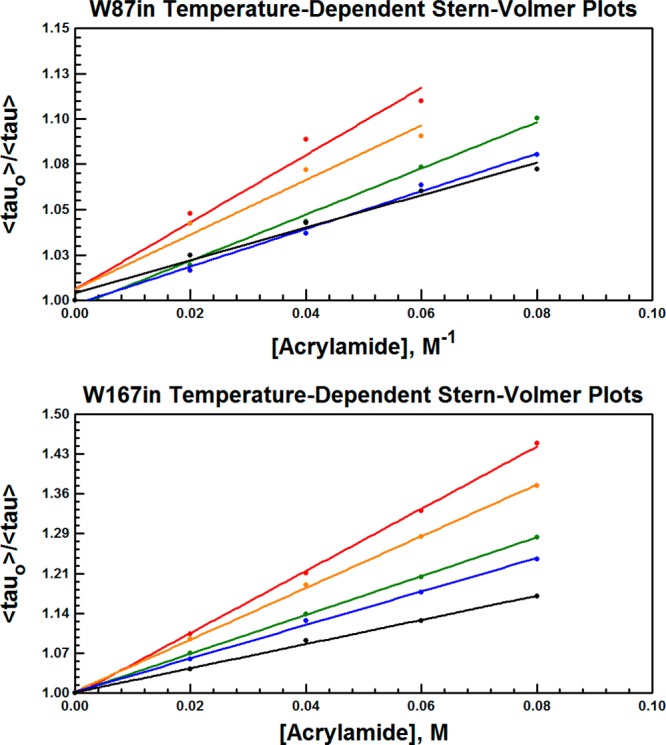
Collisional Stern–Volmer plots for W87in (top) and W167in (bottom). The representative temperatures are shown at 10 °C (black), 20 °C (blue), 30 °C (green), 40 °C (orange), and 50 °C (red).
Figure 9.
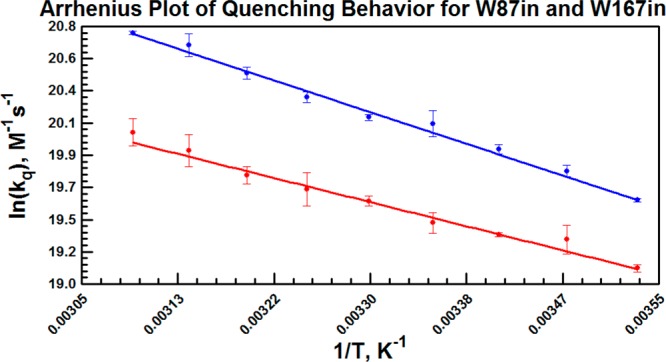
Arrhenius plots of the bimolecular quenching constants derived from the slopes plotted in Figure 9 for W87in (red) and W167in (blue).
Discussion
The role of protein motions is implicated in a wide range of enzymatic hydrogen tunneling processes. In particular, a class of nanosecond to picosecond motions has been invoked to explain changes in the temperature-dependence of the KIE that occurs for enzymes operating under nonoptimal conditions. Examples of the latter include a reduction of temperature into a nonphysiological range for thermophilic enzymes, the generation of site-specific mutants, and the study of reactions with slow substrate(s).13 Given the importance of an optimal hydrogen donor–acceptor distance in driving hydrogen tunneling, any perturbation away from this condition is expected to be accompanied, where possible, by the increasing participation of a donor–acceptor distance-sampling mode. Time-resolved fluorescence studies offer a means of interrogating motions within this timeframe, and for the present study, we chose the WT thermophilic alcohol dehydrogenase, ht-ADH, as an initial system to characterize. Importantly, ht-ADH undergoes a transition at 30 °C in both its tunneling and H/D exchange properties, raising the question of the extent to which of these previously characterized features will be linked to the nanosecond to picosecond motions accessible via fluorescence measurements. The ability to incorporate a single site-specific Trp into ht-ADH at two positions, the active site Trp87 together with Trp167 near the surface, allows spatial resolution of the phenomena under investigation. As shown in Figures S5 and S6 in the Supporting Information, both W87in and W167in demonstrate catalytic behavior and steady-state fluorescence properties similar to the native enzyme, making these constructs appropriate probes of the temperature transitions observed with native protein.
As described, fluorescence studies have been restricted to the apo-enzyme state. While it would have been valuable to interrogate the fluorescent properties of various binary or ternary complexes, the use of NADH was precluded due to FRET behavior with the active site Trp87 (cf., Figure 1 in the Supporting Information). The addition of NAD+ was also found unsuitable due to its excessive inner-filter effect of fluorescent photons. Finally, the high KD vales for alcohol substrate in binary complex formation led to concern regarding possible secondary effects on the protein structure and stability. The use of apo-enzyme for these studies is well-justified from the temperature-dependent transitions previously demonstrated in H/D exchange experiments applied to the apo-form of ht-ADH.22
Similarities in Solvent Accessibility at W87in and W167in
Collisional Stern–Volmer quenching by acrylamide was employed to report any differences between W87in and W167in with regard to solvent accessibility. The extent of quenching is governed by some combination of the inherent fluorescent lifetime (τo–1) and by kq[acrylamide], where kq is a second-order rate constant describing the collisional process between the steady-state concentrations of the excited state fluorophone (F*) and acrylamide. Invoking the steady-state assumption for the depopulation of F* leads to a linear expression shown in eq 9, where kq is extracted as a function of temperature (Figure 8). In principle, an acrylamide molecule can quench tryptophan fluorescence by accessing multiple pathways through the protein to the fluorescing residue. After entry, the acrylamide would then funnel its way to the fluorescent site in a manner that may be facilitated by protein fluctuations occurring on the nanosecond timescale.
As shown herein, the quenching efficiencies for either residue are roughly the same, regardless of their differences in solvent exposure, as indicated by the crystal structure. At 25 °C, both variants have collisional quenching constants between 2–5 × 108 M–1 s–1. Using the diffusion-controlled rate constant for acrylamide quenching of tryptophan in water (5.9 × 109 M–1 s–1), this results in a quenching efficiency range of ca. 5% for W87in and ca. 10% for W167in.73 Previously measured rate constants on the order of 107 M–1 s–1 have been observed for acrylamide quenching of buried tryptophans.72,79 This implies that both W87in and W167in contain a reasonably similar degree of solvent exposure, and that any impedance within the substrate-binding channel is imposing little or no constraint for the movement of acrylamide toward the Trp87.
Other studies have suggested that dynamical quenching can occur via a distance-dependent through-space interaction,61,62 although the quencher must still approach within 3 Å of the fluorophore.61,62 Trp87 resides >8 Å from the closest approach to bulk solvent, rendering it too far within the protein core to have random long-range interactions that distort the quenching behavior. Regardless of whether quenching is occurring as the result of direct collision or via some through-space interaction, we expected that fluctuations of the protein were likely to be involved in transport of the acrylamide to a distance close enough to facilitate fluorescence quenching. Nonetheless, the effect of temperature on the Stern–Volmer data reveals a very similar temperature-dependence at both positions. The two variants display a ΔH‡ close to 4 kcal/mol, consistent with the activation energy measured for acrylamide quenching of free N-acetyl-tryptophanamide in water.72 This further corroborates the notion that Trp87 near the catalytic center has a similar degree of exposure to solvent as Trp167.
The most compelling observation from the quenching data at both sites is the lack of an enthalpic transition at 30 °C. Both Trp87 and Trp167 belong to peptides that exhibit increased solvent exchange above 30 °C as detected by H/D exchange under the EX2 condition.22 While this H/D exchange occurs over slow timescales, a hierarchy of motions may be expected to contribute to the local unfolding that allows transient access of the solvent D2O/OD– to the protein peptide backbone. The fact that collisional quenching of fluorescence is insensitive to the temperature transition that controls H/D exchange (and catalysis) could mean either that the timescale of motions controlling the fluorescence properties is not relevant or that the large degree of solvent exposure at both Trp87 and Trp167 results in an insensitivity of the quenching parameters to nanosecond motions. The Stokes shift data become especially relevant and informative in the latter context.
Temperature-Dependent Stokes Shifts Reveal Opposing Temperature-Dependent Effects at W87in and W167in
In general, the time-dependent Stokes shift reports on the evolution of the solvent’s electrostatic environment during the excited state of a fluorophore. Instantly after excitation, tryptophan is in its most electronically unfavorable environment because the surrounding residues have not yet rearranged to accommodate the newly generated electronic distortion, in accordance with the Franck–Condon (FC) principle. Hence, the TRES at τfc–1 is calculated to be the highest energy state for emission and is referred to as the FC state. During the excited state, the solvent reorganizes at a rate constant, ksol, producing more energetically favorable TRES with peaks shifting to red wavelengths until the steady-state emission wavelength at time τr–1 is reached.69−71
The temperature-dependence of the red shift decay rates reveals two opposing behaviors, featuring an active site environment at Trp87 that is highly temperature-dependent and a surface environment at Trp167 that is practically temperature-independent (Figure 7). This is accompanied by very different emission maxima at t = 0, with the W167in showing a much more red-shifted spectrum at the initial time point (Table 3). These two features are proposed to be intimately related to one another and suggest that significant amount of rearrangement at Trp167 is occurring on a much faster timescale than nanoseconds at all temperatures (cf. Table 4). Studies employing fluorescence upconversion techniques with femtosecond time resolution can report on distortions caused by water reorganization and water-coupled protein interactions having relaxation timescales on the order of 102–104 fs. Such solvation decays indicate much more drastic Stokes shifts of tryptophan fluorescence, relaxing nearly 800–1800 cm–1 to the red of their respective FC states.54−57 The fact that the overall shift for W87in is 486 cm–1 at 10 °C, in comparison to 100 cm–1 for W167in at the same temperature, is striking and shows that the environment at W87in is much more restricted, requiring an input of thermal energy before it can approach the behavior of W167in at 50 °C.
The origin of the factors controlling the respective enthalpic barriers can be rationalized according to the local structure around each tryptophan (Figure 2). Trp167 is slightly more accessible to solvent and resides in a homogeneous environment relative to Trp87. In fact, only two polar interactions exist near Trp167 (Figure 2). The side chain of Asn190 is a hydrogen-bonding partner with the indole nitrogen. His232 is the only other polar side chain interaction within 6 Å of Trp167 but is poorly aligned for any polar interactions because its imidazole moiety lies coplanar with the indole moiety. The sole Asn190 interaction with Trp167 is most likely responsible for stabilizing the excited state tryptophan, as the ca. 6 nanosecond lifetime is more than twice the value (2.7 ns) measured for free Trp in water.80 However, because of the scarce number of polar interactions at this site, the environment is unfavorable for accommodation of an excited-state fluorophore relative to the active site. This feature is proposed to result in a predominant relaxation at a faster rate than the time resolution of the present experiments. The presence of a very modest Stokes shift in the nanosecond regime for W167in may be due to a large number of van der Waals interactions (r-6 dependence) in the proximity of this side chain.
By contrast, W87in has a broad diversity of side chains interacting near the indole moiety. The catalytic Zn2+ with coordinated Cys148 and His61 are excellent candidates to quench inherent fluorescent lifetimes and are likely the origin of the sub-nanosecond component in the triexponential fluoresence lifetime analysis (Table 2). Asn111 and Gln109 are excellent hydrogen-bonding candidates that contain heavy atoms measured within 4 Å of the indole nitrogen. Gln109, Tyr114, and Ala112 also contribute amide backbone interactions to Trp87. This extremely heterogeneous array of interactions surrounding Trp87 can easily explain the large enthalpy of greater than 9 kcal/mol for the excited-state reorganization. The catalytic center is 8 Å from the bulk solvent, resulting in lower dielectric constants that may be expected to require a high-energy input to break the combination of ionic and dipole–dipole interactions (r-2 and r-3 dependence).
Relationship of the Fluorescence Data for ht-ADH to Kinetic and H/D Exchange Behavior
The spatial resolution of the Stokes shifts investigated for ht-ADH herein allows comparison to an earlier study of the temperature-dependence of H/D exchange within a thermophilic dihydrofolate reductase (DHFR).21 Despite the very different timescales of fluorescence and deuterium exchange studies, DHFR showed a similar pattern of a large local enthalpy (for unfolding) near the cofactor and substrate-binding site that contrasted with very small enthalpies (for local unfolding) at regions distant from the active site and near the protein surface. The studies of native DHFR led to the proposal of a largely entropy-driven local unfolding throughout the majority of the protein, which is the result of small enthalpic barriers for the interconversion of multiple conformers and reflects instead the probability of reaching a state amenable to solvent penetration and backbone exchange. The active site on the other hand, with its large number of specific interactions, was proposed to require substantial thermal activation to unfold sufficiently to expose backbone amides to the solvent.21 The common theme between the experiments with DHFR and the present study is the existence of extensive structure at the active site that is dominated by highly specific and concentrated dipolar and electrostatic interactions. These interactions are also likely to be the primary cause of the reorganization energy that is required to tune the electrostatics of the hydrogen donor and acceptor in C–H activation reactions.13 While the value of the enthalpy of activation for the Stokes shift at W87in for ht-ADH is approximately half of the barrier measured on catalysis above 30 °C, it is reasonable to assume that there is overlap among the environmental changes that affect both processes.
Among all of the fluorescent measurements on ht-ADH, the single one that shows any possible break with temperature analogous to catalysis and H/D exchange is a small amplitude sub-nanosecond fluorescence decay at Trp87 (Table 2 and Figure 4). This is completely absent from Trp167 and is ascribed to an interaction between Trp87 and the active site Zn2+. The arrangement of ligands to the active site metal catalyst may be expected to be very sensitive to temperature-dependent structural changes that likewise alter catalysis. The remainder of the fluorescent methods reflect motions than can be attributed to side chain rotations, backbone fluctuations, loop fluctuations, and translations of side chains.7,51,81 Motions of this type and timescale are functionally quite local and in the vicinity of Trp87 do not appear to correlate with the previously demonstrated transition in tunneling properties at 30 °C. We conclude that the nanosecond to picosecond motions detected from fluorescence measurements are likely spatially distinct from the similar time scale motions that control donor−acceptor distance sampling of the hydrogen tunneling coordinate under various conditions. Further, the latter motions may only be detectable in binary and tertiary complexes of enzymes. By contrast, the specific protein motions that generate a reversible trapping of ht-ADH into low enthalpy microstates below 30 °C are evident in τ1 for W87in (Table 2 and Figure 4), analogous to transitions seen earlier via H/D exchange. These properties point toward longer and more global motions controlling the transitions seen at 30 °C, warranting future studies of dynamics in ht-ADH on the microsecond timescale. Such motions may be directly linked to protein subunit interactions, as implicated from recent studies that show a connection between a Tyr25 side chain at the dimer interface of ht-ADH that dictates the presence of the Arrhenius break and the region of the active site where Trp87 resides.82 Extension of spectroscopic studies to mutant forms of ht-ADH known to profoundly alter the properties of the catalytically linked temperature transition are also likely to be quite informative.
Acknowledgments
This work was funded by a grant from the National Institutes of Health to J.P.K. (GM025765 and GM025765-32S1).
Glossary
Abbreviations:
- NAD+
nicotinamide adenine dinucleotide
- ht-ADH
thermophilic alcohol dehydrogenase from Bacillus stearothermophilus
- KIE
kinetic isotope effect
- WT
wild-type
- TRES
time-resolved emission spectra
- TCSPC
time-correlated single photon counting
- BnOH
benzyl alcohol
Supporting Information Available
Steady-state fluorescence spectra, Stern–Volmer rate constants, enzyme kinetics, CD spectra, and gel-filtration data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nagel Z. D.; Klinman J. P. Update 1 of: Tunneling and Dynamics in Enzymatic Hydride Transfer. Chem. Rev. 2010, 110, Pr41–Pr67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S.; Benkovic S. J. Relating Protein Motion to Catalysis. Annu. Rev. Biochem. 2006, 75, 519–41. [DOI] [PubMed] [Google Scholar]
- Schwartz S.; Schramm V. L. Enzymatic Transition States and Dynamic Motion in Barrier Crossing. Nat. Chem. Biol. 2009, 5, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrank T. P.; Bolen D. W.; Hilser V. J. Rational Modulation of Conformational Fluctuations in Adenylate Kinase Reveals a Local Unfolding Mechanism for Allostery and Functional Adaptation in Proteins. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16984–16989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Warshel A. At the Dawn of the 21st Century: Is Dynamics the Missing Link for Understanding Enzyme Catalysis?. Proteins: Struct., Funct., Bioinf. 2010, 78, 1339–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forconi M.; Porecha R. H.; Piccirilli J. A.; Herschlag D. Tightening of Active Site Interactions En Route to the Transition State Revealed by Single-Atom Substitution in the Guanosine-Binding Site of the Tetrahymena Group I Ribozyme. J. Am. Chem. Soc. 2011, 133, 7791–7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Lei M.; Thai V.; Kerns S. J.; Karplus M.; Kern D. A Hierarchy of Timescales in Protein Dynamics is Linked to Enzyme Catalysis. Nature 2007, 450, 913–916. [DOI] [PubMed] [Google Scholar]
- Kohen A.; Cannio R.; Bartolucci S.; Klinman J. P. Enzyme Dynamics and Hydrogen Tunneling in a Thermophilic Alcohol Dehydrogenase. Nature 1999, 399, 496–499. [DOI] [PubMed] [Google Scholar]
- Knapp M. J.; Rickert K.; Klinman J. P. Temperature-Dependent Isotope Effects in Soybean Lipoxygenase-1: Correlating Hydrogen Tunneling with Protein Dynamics. J. Am. Chem. Soc. 2002, 124, 3865–3874. [DOI] [PubMed] [Google Scholar]
- Hatcher E.; Soudackov A. V.; Hammes-Schiffer S. Proton-Coupled Electron Transfer in Soybean Lipoxygenase: Dynamical Behavior and Temperature Dependence of Kinetic Isotope Effects. J. Am. Chem. Soc. 2007, 129, 187–196. [DOI] [PubMed] [Google Scholar]
- Maglia G.; Allemann R. K. Evidence for Environmentally Coupled Hydrogen Tunneling During Dihydrofolate Reductase Catalysis. J. Am. Chem. Soc. 2003, 125, 13372–13373. [DOI] [PubMed] [Google Scholar]
- Pudney C. R.; Johannissen L. O.; Sutcliffe M. J.; Hay S.; Scrutton N. S. Direct Analysis of Donor-Acceptor Distance and Relationship to Isotope Effects and the Force Constant for Barrier Compression in Enzymatic H-Tunneling Reactions. J. Am. Chem. Soc. 2010, 132, 11329–11335. [DOI] [PubMed] [Google Scholar]
- Klinman J. P.; Kohen A. Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha Y.; Murray C. J.; Klinman J. P. Hydrogen Tunneling in Enzyme Reactions. Science 1989, 243, 1325–1330. [DOI] [PubMed] [Google Scholar]
- Roston D.; Kohen A. Elusive Transition State of Alcohol Dehydrogenase Unveiled. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 9572–9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Y.; Klinman J. P. Enzymatic Methyl Transfer: Role of an Active Site Residue in Generating Active Site Compaction That Correlates with Catalytic Efficiency. J. Am. Chem. Soc. 2011, 133, 17134–17137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P. A.; Vetticatt M. J.; Schramm V. L. Transition State Analysis of the Arsenolytic Depyrimidination of Thymidine by Human Thymidine Phosphorylase. Biochemistry 2011, 50, 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell J. R.; Dyson H. J.; Wright P. E. Effect of Cofactor Binding and Loop Conformation on Side Chain Methyl Dynamics in Dihydrofolate Reductase. Biochemistry 2004, 43, 374–383. [DOI] [PubMed] [Google Scholar]
- Baldwin A. J.; Kay L. E. NMR Spectroscopy Brings Invisible Protein States into Focus. Nat. Chem. Biol. 2009, 5, 808–814. [DOI] [PubMed] [Google Scholar]
- Osborne M. J.; Schnell J.; Benkovic S. J.; Dyson H. J.; Wright P. E. Backbone Dynamics in Dihydrofolate Reductase Complexes: Role of Loop Flexibility in the Catalytic Mechanism. Biochemistry 2001, 40, 9846–9859. [DOI] [PubMed] [Google Scholar]
- Oyeyemi O. A.; Sours K. M.; Lee T.; Resing K. A.; Ahn N. G.; Klinman J. P. Temperature Dependence of Protein Motions in a Thermophilic Dihydrofolate Reductase and its Relationship to Catalytic Efficiency. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10074–10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z.-X.; Lee T.; Resing K. A.; Ahn N. G.; Klinman J. P. Thermal-Activated Protein Mobility and its Correlation with Catalysis in Thermophilic Alcohol Dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9556–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M. M.; Rempel D. L.; Du Z. H.; Gross M. L. Quantification of Protein-Ligand Interactions by Mass Spectrometry, Titration, and H/D Exchange: PLIMSTEX. J. Am. Chem. Soc. 2003, 125, 5252–5253. [DOI] [PubMed] [Google Scholar]
- Fraser J. S.; Clarkson M. W.; Degnan S. C.; Erion R.; Kern D.; Alber T. Hidden Alternative Structures of Proline Isomerase Essential for Catalysis. Nature 2009, 462, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotte F.; Lim M. H.; Jackson T. A.; Smirnov A. V.; Soman J.; Olson J. S.; Phillips G. N.; Wulff M.; Anfinrud P. A. Watching a Protein as it Functions with 150-ps Time-Resolved X-ray Crystallography. Science 2003, 300, 1944–1947. [DOI] [PubMed] [Google Scholar]
- Flynn E. M.; Hanson J. A.; Alber T.; Yang H. Dynamic Active-Site Protection by the M. tuberculosis Protein Tyrosine Phosphatase PtpB Lid Domain. J. Am. Chem. Soc. 2010, 132, 4772–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W.; English B. P.; Luo G. B.; Cherayil B. J.; Kou S. C.; Xie X. S. Fluctuating Enzymes: Lessons from Single-Molecule Studies. Acc. Chem. Res. 2005, 38, 923–931. [DOI] [PubMed] [Google Scholar]
- Yang H.; Luo G.; Karnchanaphanurach P.; Louie T.-M.; Rech I.; Cova S.; Xun L.; Xie X. S. Protein Conformational Dynamics Probed by Single-Molecule Electron Transfer. Science 2003, 302, 262–266. [DOI] [PubMed] [Google Scholar]
- Phillips R. S.; Miles E. W.; McPhie P.; Marchal S.; Georges C.; Dupont Y.; Lange R. Pressure and Temperature Jump Relaxation Kinetics of the Conformational Change in Salmonella typhimurium Tryptophan Synthase L-Serine Complex: Large Activation Compressibility and Heat Capacity Changes Demonstrate the Contribution of Solvation. J. Am. Chem. Soc. 2008, 130, 13580–13588. [DOI] [PubMed] [Google Scholar]
- Ye M. P.; Zhang Q. L.; Li H.; Weng Y. X.; Wang W. C.; Qiu X. G. Infrared Spectroscopic Discrimination between the Loop and Alpha-Helices and Determination of the Loop Diffusion Kinetics by Temperature-Jump Time-Resolved Infrared Spectroscopy for Cytochrome c. Biophys. J. 2007, 93, 2756–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan G.; Hu Y.; Oyerinde O. F.; Su J.; Groves J. T.; Spiro T. G.; Conformational A. Switch to Beta-Sheet Structure in Cytochrome c Leads to Heme Exposure. Implications for Cardiolipin Peroxidation and Apoptosis. J. Am. Chem. Soc. 2007, 129, 504–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S. K.; Ji M. B. A.; Gaffney K. J.; Boxer S. G. Direct Measurement of the Protein Response to an Electrostatic Perturbation that Mimics the Catalytic Cycle in Ketosteroid Isomerase. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 16612–16617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaria J. N.; Dutta S.; Nydegger M. W.; Rock W.; Kohen A.; Cheatum C. M. Characterizing the Dynamics of Functionally Relevant Complexes of Formate Dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17974–17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur S.; Bruice T. C. The Near Attack Conformation Approach to the Study of the Chorismate to Prephenate Reaction. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12015–12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mincer J. S.; Schwartz S. D. Rate-Promoting Vibrations and Coupled Hydrogen-Electron Transfer Reactions in the Condensed Phase: A Model for Enzymatic Catalysis. J. Chem. Phys. 2004, 120, 7755–7760. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; Nussinov R.; Wright P. E. The Role of Dynamic Conformational Ensembles in Biomolecular Recognition. Nat. Chem. Biol. 2009, 5, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suydam I. T.; Snow C. D.; Pande V. S.; Boxer S. G. Electric Fields at the Active Site of an Enzyme: Direct Comparison of Experiment with Theory. Science 2006, 313, 200–204. [DOI] [PubMed] [Google Scholar]
- Fafarman A. T.; Sigala P. A.; Schwans J. P.; Fenn T. D.; Herschlag D.; Boxer S. G. Quantitative, Directional Measurement of Electric Field Heterogeneity in the Active Site of Ketosteroid Isomerase. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E299–E308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. W.; He T. F.; Guo L. J.; Stevens J. A.; Li T. P.; Wang L. J.; Zhong D. P. Mapping Solvation Dynamics at the Function Site of Flavodoxin in Three Redox States. J. Am. Chem. Soc. 2010, 132, 12741–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J. A.; Link J. J.; Kao Y. T.; Zang C.; Wang L. J.; Zhong D. P. Ultrafast Dynamics of Resonance Energy Transfer in Myoglobin: Probing Local Conformation Fluctuations. J. Phys. Chem. B 2010, 114, 1498–1505. [DOI] [PubMed] [Google Scholar]
- Abbyad P.; Shi X. H.; Childs W.; McAnaney T. B.; Cohen B. E.; Boxer S. G. Measurement of Solvation Responses at Multiple Sites in a Globular Protein. J. Phys. Chem. B 2007, 111, 8269–8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie B. N.; Deng H.; Desamero R.; Callender R. Large Scale Dynamics of the Michaelis Complex in Bacillus stearothermophilus Lactate Dehydrogenase Revealed by a Single-Tryptophan Mutant Study. Biochemistry 2013, 52, 1886–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbyad P.; Childs W.; Shi X. H.; Boxer S. G. Dynamic Stokes Shift in Green Fluorescent Protein Variants. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20189–20194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinenko K. L.; Webber N. M.; Meech S. R. Internal Conversion in the Chromophore of the Green Fluorescent Protein: Temperature Dependence and Isoviscosity Analysis. J. Phys. Chem. A 2003, 107, 2616–2623. [Google Scholar]
- Chakrabarti S. K.; Ware W. R. Nanosecond Time-Resolved Emission Spectroscopy of 1-Anilino-8-Naphthalene Sulfonate. J. Chem. Phys. 1971, 55, 5494–5498. [Google Scholar]
- Bagchi B.; Oxtoby D. W.; Fleming G. R. Theory of the Time Development of the Stokes Shift in Polar Media. Chem. Phys. 1984, 86, 257–267. [Google Scholar]
- Ruggiero A. J.; Todd D. C.; Fleming G. R. Subpicosecond Fluorescence Anisotropy Studies of Tryptophan in Water. J. Am. Chem. Soc. 1990, 112, 1003–1014. [Google Scholar]
- Jimenez R.; Fleming G. R.; Kumar P. V.; Maroncelli M. Femtosecond Solvation Dynamics of Water. Nature 1994, 369, 471–473. [Google Scholar]
- Chen J. J.; Toptygin D.; Brand L.; King J. Mechanism of the Efficient Tryptophan Fluorescence Quenching in Human Gamma D-Crystallin Studied by Time-Resolved Fluorescence. Biochemistry 2008, 47, 10705–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha S.; Sahu K.; Roy D.; Mondal S. K.; Roy S.; Bhattacharyya K. Slow Solvation Dynamics at the Active Site of an Enzyme: Implications for Catalysis. Biochemistry 2005, 44, 8940–8947. [DOI] [PubMed] [Google Scholar]
- Jha A.; Ishii K.; Udgaonkar J. B.; Tahara T.; Krishnamoorthy G. Exploration of the Correlation between Solvation Dynamics and Internal Dynamics of a Protein. Biochemistry 2011, 50, 397–408. [DOI] [PubMed] [Google Scholar]
- Ferreira S. T. Fluorescence Studies of the Conformational Dynamics of Parvalbumin in Solution Lifetime and Rotational Motions of the Single Tryptophan Residue. Biochemistry 1989, 28, 10066–10072. [DOI] [PubMed] [Google Scholar]
- Maglia G.; Jonckheer A.; De Maeyer M.; Frere J. M.; Engelborghs Y. An Unusual Red-Edge Excitation and Time-Dependent Stokes Shift in the Single Tryptophan Mutant Protein DD-Carboxypeptidase from Streptomyces: The Role of Dynamics and Tryptophan Rotamers. Protein Sci. 2008, 17, 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W. H.; Kao Y. T.; Zhang L. Y.; Yang Y.; Wang L. J.; Stites W. E.; Zhong D. P.; Zewail A. H. Protein Surface Hydration Mapped by Site-Specific Mutations. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13979–13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W. H.; Wang L. J.; Lu W. Y.; Boechler A.; Sanders D. A. R.; Zhong D. P. Dissection of Complex Protein Dynamics in Human Thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 5366–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Y.; Wang L. J.; Kao Y. T.; Qiu W. H.; Yang Y.; Okobiah O.; Zhong D. P. Mapping Hydration Dynamics Around a Protein Surface. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18461–18466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon O. H.; Yoo T. H.; Othona C. M.; Van Deventer J. A.; Tirrell D. A.; Zewail A. H. Hydration Dynamics at Fluorinated Protein Surfaces. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17101–17106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs W.; Boxer S. G. Solvation Response along the Reaction Coordinate in the Active Site of Ketosteroid Isomerase. J. Am. Chem. Soc. 2010, 132, 6474–6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftink M. R.; Jameson D. M. Acrylamide and Oxygen Fluorescence Quenching Studies with Liver Alcohol-Dehydrogenase Using Steady-State and Phase Fluorometry. Biochemistry 1982, 21, 4443–4449. [DOI] [PubMed] [Google Scholar]
- Kim S. J.; Chowdhury F. N.; Stryjewski W.; Younathan E. S.; Russo P. S.; Barkley M. D. Time-Resolved Fluorescence of the Single Tryptophan of Bacillus-Stearothermophilus Phosphofructokinase. Biophys. J. 1993, 65, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J. R.; Zelent B.; Gryczynski I.; Kusba J.; Johnson M. L. Distance-Dependent Fluorescence Quenching of Tryptophan by Acrylamide. Photochem. Photobiol. 1994, 60, 205–214. [DOI] [PubMed] [Google Scholar]
- Strambini G. B.; Gonnelli M. Fluorescence Quenching of Buried Trp Residues by Acrylamide Does Not Require Penetration of the Protein Fold. J. Phys. Chem. B 2010, 114, 1089–1093. [DOI] [PubMed] [Google Scholar]
- Weber J.; Senior A. E. Features of F-1-ATPase Catalytic and Noncatalytic Sites Revealed by Fluorescence Lifetimes and Acrylamide Quenching of Specifically Inserted Tryptophan Residues. Biochemistry 2000, 39, 5287–5294. [DOI] [PubMed] [Google Scholar]
- Secundo F.; Russo C.; Giordano A.; Carrea G.; Rossi M.; Raia C. A. Temperature-induced conformational change at the catalytic site of Sulfolobus solfataricus alcohol dehydrogenase highlighted by Asn249Tyr substitution. A hydrogen/deuterium exchange, kinetic, and fluorescence quenching study. Biochemistry 2005, 44, 11040–11048. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Dong M.; Bahnson B. J.; Klinman J. P. Impaired Protein Conformational Landscapes as Revealed in Anomalous Arrhenius Prefactors. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10520–10525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J. R.Fluorescence Spectroscopy of Biomolecules. In Encyclopedia of Molecular Biology and Molecular Medicine, Meyers R. A., Ed. VCH Publishers: New York, NY, 1995. [Google Scholar]
- Nagel Z. D.; Meadows C. W.; Dong M.; Bahnson B. J.; Klinman J. P. Active Site Hydrophobic Residues Impact Hydrogen Tunneling Differently in a Thermophilic Alcohol Dehydrogenase at Optimal Versus Nonoptimal Temperatures. Biochemistry 2012, 51, 4147–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. M.; Rapid A. and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–54. [DOI] [PubMed] [Google Scholar]
- Badea M. G.; Brand L. Time-Resolved Fluorescence Measurements. Methods Enzymol. 1979, 61, 378–425. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.; Cherek H.; Laczko G.; Gratton E. Time-Resolved Fluorescence Emission-Spectra of Labeled Phospholipid-Vesicles, as Observed Using Multi-Frequency Phase-Modulation Fluorometry. Biochim. Biophys. Acta 1984, 777, 183–193. [Google Scholar]
- Horng M. L.; Gardecki J. A.; Papazyan A.; Maroncelli M. Subpicosecond Measurements of Polar Solvation Dynamics: Coumarin-153 Revisited. J. Phys. Chem. 1995, 99, 17311–17337. [Google Scholar]
- Eftink M. R.; Ghiron C. A. Exposure of Tryptophanyl Residues and Protein Dynamics. Biochemistry 1977, 16, 5546–5551. [DOI] [PubMed] [Google Scholar]
- Komiyama T.; Miwa M. Fluorescence Quenching as an Indicator for the Exposure of Tryptophyl Residues in Streptomyces Subtilisin Inhibitor. J. Biochem. 1980, 87, 1029–1036. [PubMed] [Google Scholar]
- Kenoth R.; Swamy M. J. Steady-Sate and Time-Resolved Fluorescence Studies on Trichosanthes Cucumerina Seed Lectin. J. Photochem. Photobiol. B 2003, 69, 193–201. [DOI] [PubMed] [Google Scholar]
- Varnes A. W.; Wehry E. L.; Dodson R. B. Interactions of Transition-Metal Ions with Photoexcited States of Flavins: Fluorescence Quenching Studies. J. Am. Chem. Soc. 1972, 94, 946–950. [DOI] [PubMed] [Google Scholar]
- Cook R. L.; Langford C. H. Metal-Ion Quenching of Fulvic-Acid Fluorescence Intensities and Lifetimes: Nonlinearities and a Possible 3-Component Model. Anal. Chem. 1995, 67, 174–180. [Google Scholar]
- Tabak M.; Sartor G.; Neyroz P.; Spisni A.; Cavatorta P. Interaction of Copper and Nickel Ions with Tryptophan and Glycyltryptophan: Time Resolved Fluorescence Study. J. Lumin. 1990, 46, 291–299. [Google Scholar]
- Kim S. J.; Lewis M. S.; Knutson J. R.; Porter D. K.; Kumar A.; Wilson S. H. Characterization of the Tryptophan Fluorescence and Hydrodynamic Properties of Rat DNA-Polymerase-Beta. J. Mol. Biol. 1994, 244, 224–235. [DOI] [PubMed] [Google Scholar]
- Eftink M. R.; Hagaman K. A. Fluorescence Lifetime and Anisotropy Studies with Liver Alcohol-Dehydrogenase and Its Complexes. Biochemistry 1986, 25, 6631–6637. [DOI] [PubMed] [Google Scholar]
- Boens N.; Qin W. W.; Basaric N.; Hofkens J.; Ameloot M.; Pouget J.; Lefevre J. P.; Valeur B.; Gratton E.; Vandeven M.; et al. Fluorescence Lifetime Standards for Time and Frequency Domain Fluorescence Spectroscopy. Anal. Chem. 2007, 79, 2137–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; Dyson H. J.; Wright P. E.; An N. M. R. Perspective on Enzyme Dynamics. Chem. Rev. 2006, 106, 3055–3079. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Cun S. J.; Klinman J. P. Identification of a Long-range Protein Network That Modulates Active Site Dynamics in Extremophilic Alcohol Dehydrogenases. J. Biol. Chem. 2013, 288, 14087–14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.