

Abstract
The acute effect of the potent cyclin-dependent kinase (cdk) inhibitor (R)-roscovitine on Ca2+ channels inspired the development of structural analogues as a potential treatment for motor nerve terminal dysfunction. On the basis of a versatile chlorinated purine scaffold, we have synthesized ca. 20 derivatives and characterized their N-type Ca2+ channel agonist action. Agents that showed strong agonist effects were also characterized in a kinase panel for their off-target effects. Among several novel compounds with diminished cdk activity, we identified a new lead structure with a 4-fold improved N-type Ca2+ channel agonist effect and a 22-fold decreased cdk2 activity as compared to (R)-roscovitine. This compound was selective for agonist activity on N- and P/Q-type over L-type calcium channels.
Keywords: N/P/Q-type calcium channels, roscovitine, cdk2, selective agonist, Lambert−Eaton myasthenic syndrome, LEMS, neurological autoimmune disorder
Chemical communication in the nervous system is tightly regulated by the flux of calcium ions through certain subtypes of voltage-gated channels. A decrease in calcium flux at synapses can cause neurological diseases. Lambert–Eaton myasthenic syndrome (LEMS) is a neurological autoimmune disorder characterized by an autoimmune reduction of presynaptic P/Q-type (Cav2.1) calcium channels at the neuromuscular junction and a partial compensatory up-regulation of N-type (Cav2.2), L-type (Cav1), and R-type (Cav2.3) channels.1,2 N-type and P/Q-type channels appear to be the most relevant for the control of transmitter release as they selectively bind directly to and colocalize with transmitter release sites.3 The result is an overall calcium channel reduction at transmitter release sites that results in muscle weakness and is associated with compromised motor function. This disease is estimated to affect 1:100000 individuals in the United States;4 however, the true incidence of LEMS remains unknown as it is often undiagnosed in patients.5 Current treatment strategies are very limited, and those available are indirect and sometimes associated with undesirable side effects.
Selective calcium channel agonists that increase the ion flux through N- and P/Q-type calcium channels represent attractive potential therapeutics for LEMS and other neuromuscular diseases; however, to date, no such agonists have been identified. While (R)-roscovitine was originally developed as a cyclin-dependent kinase (cdk) inhibitor,6,7 several laboratories have subsequently demonstrated that it is also a potent agonist for N- and P/Q-type calcium channels,8−11 showing a concentration required for 50% efficacy (EC50) = 28–41 μM.12 N- and P/Q-type channels are the two major subtypes that regulate chemical transmitter release in the nervous system.13 The development of a potent agonist for N- and P/Q-type calcium channels could have a significant impact on the treatment of LEMS as well as other neurological diseases. Furthermore, the availability of a more potent N- and P/Q-selective agonist would be useful as an experimental tool to facilitate the study of calcium channel gating and regulation of cellular events. Accordingly, we embarked on developing analogues of (R)-roscovitine with decreased cdk activity and increased selectivity for these subsets of voltage-gated calcium channels.
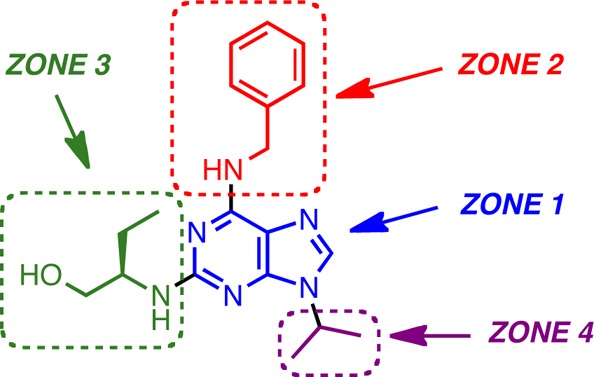
At the onset of our work, very little was known about structural modifications of (R)-roscovitine pertaining to its Ca2+ channel modulating properties.14 As summarized in Figure 1, the available literature activity data on roscovitine analogues disclosed only three similarly potent N-type calcium channel agonists among 12 structural analogues. We used a four-zone approach to analyze the literature results and to guide our initial round of medicinal chemistry structure–activity relationship (SAR) studies. In particular, zone 1, the core region, had only been superficially probed in the inactive analogues 1 and 2, which, however, differ too greatly from the purine scaffold of roscovitine to allow for any conclusions regarding the influence of core scaffold variations.12 Interestingly, translocation of the nitrogen atom in pyrazolo[1,5-a]-1,3,5-triazine 3,15−17 however, retained N-type Ca2+ channel agonist activity and confirmed that at least conservative modifications were tolerable.18 The literature data on zone 2 analogues were similarly very limited, as shown for compounds 4, 5, and 6 [e.g., (R)-olomoucine II].8,12 The former two trisubstituted N-6 amine analogues were devoid of any activity, suggesting that a hydrogen atom was required at N-6. Because the phenol group in 6 was able to substitute for roscovitine's benzene ring in zone 2 with minimal loss of activity, we concluded that zone 2 was amenable to further modifications. In contrast, analogues 7,128 (e.g., bohemine),12 and 9 [e.g., (S)-roscovitine]8,12 demonstrated that zone 3 was critical for the desired N-type Ca2+ channel agonist activity. It was also noteworthy and encouraging for the robustness of the SAR that upon the seemingly minor modification of an inversion of configuration in zone 3 in 9, the activity dropped ca. 14-fold.
Figure 1.

Summary of SAR model and literature data on roscovitine analogues evaluated for N-type Ca2+ channel agonist effects.
Because the literature data did not present any analogues of roscovitine with exclusive zone 4 modifications, the conclusions that could be drawn regarding the combined zone 3/4 alterations shown in Figure 1 were ambiguous. The N-9 methylated compound 10 (e.g., olomoucine) and (dimethylamino)olomoucine 11 were devoid of any activity,12 but this deficiency could also be attributed to the lack of appropriate functionality in the zone 3 side chain. Similarly, iso-olomoucine 12(12) was inactive either due to the shift in substitution in the imidazole ring, the lack of a 2-propyl group in zone 4, or, most likely, once again the absence of the (R)-2-amino-1-butanol side-chain at C-2 in zone 3.
In summary, the available literature information on the roscovitine activity profile as it pertained to N-type Ca2+ channel agonism/antagonism was very limited, and the combination of all previously observed effects did not yet allow for a rational second generation lead design and development. On the basis of the lack of activity resulting from minor modifications in zone 3 and the absence of significant SAR data from zone 1 scaffold alterations, we focused our studies on interrogating the purine scaffold further by variations of substituents in zones 2 and 4. Accordingly, for this first round of systematic SAR studies, we decided to retain the purine backbone as well as the (R)-2-amino-1-butanol side chain at C-2.
The synthetic strategy used for SAR investigations toward selective N-type Ca2+ channel agonists based on purines 13 is summarized in Scheme 1. Starting with the commercially available 2,6-dichloropurine 14,16N-9 alkylation of the purine with primary alkyl halides would lead to intermediate 15. Preferential nucleophilic aromatic substitution (SNAr) reaction at the more reactive C-6 position followed by displacement of the C-2 chloride in 16 with primary amines would provide target analogues 13.25
Scheme 1. Synthetic Strategy for Purine Side Chain Variations.
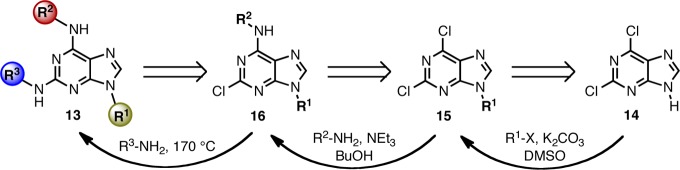
Zone 4 modifications R1 were readily achieved by subjecting dichloropurine 14 to deprotonation in DMSO in the presence of a mild base such as potassium carbonate, followed by the addition of alkyl bromides or iodides at 16–18 °C. Primary alkyl halides (R1 = Me, Pr) provided a similar yield (64–78%) to the secondary alkyl halide (R1 = i-Pr, 65%). Microwave irradiation at 120 °C for 20 min in n-BuOH and triethylamine in the presence of various aryl- and alkylamines converted intermediates 15 to the C-6 aminated purines 16 in moderate-to-good yields (52–95%). Finally, aminolysis at C-2 under forcing conditions (heating neat in a sealed flask at 170 °C in the presence of the R3-amine for 8–15 h) introduced the (R)-2-amino-1-butanol side chain to give target molecules 13 in variable yields (25–93%).26
Table 1 presents an overview of the synthetic analogues prepared using this pathway, as well as their N-type Ca2+ channel agonist and cdk2 kinase inhibitory properties. While most of the new derivatives were ineffective at enhancing N-type Ca2+ channel activity, an encouraging result was obtained with 13d, which showed a ca. 2-fold increased agonism and a 22-fold decreased cdk2 kinase activity versus the standard, (R)-roscovitine. We attributed the decreased kinase activity of 13d to the replacement of the i-propyl side chain at R1 with the more flexible n-propyl group, a hypothesis that was supported by the similarly decreased cdk2 activity of 13g. In addition, previous docking and experimental studies of roscovitine–cdk2 interactions also convey the significant effect of N-9 substituents on active site binding.14
Table 1. Chemical Structures and N-Type Ca2+ Channel Agonist Properties of Roscovitine Analogues 13 [R3 = (R)-2-Amino-1-butanol].
| structure |
yields (%) |
N-type Ca2+ channel activity | Cdk2 activity | ||||
|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | 14 → 15 | 15 → 16 | 16 → 13 | EC50 ± SEM (μM)a | EC50 ± SEM (μM)b |
| (R)-roscovitine | i-Pr | Bn | 27.58 ± 1.65 | 0.151 ± 0.004 | |||
| 13a | Pr | CH2(biphenyl) | 78 | 91 | 59 | >100 | ND |
| 13b | Pr | CH(Ph)2 | 78 | 65 | 67 | >100 | ND |
| 13c | Pr | (CH2)2Ph | 78 | 85 | 32 | >100 | ND |
| 13d | Pr | Bn | 78 | 81 | 66 | 14.23 ± 2.71 | 3.34 ± 0.05 |
| 13e | Pr | Ph | 78 | 59 | 49 | >100 | ND |
| 13f | Pr | CH2(3-Py) | 78 | 71 | 66 | >100 | ND |
| 13g | Pr | CH2CH(CH2)2 | 78 | 95 | 62 | >100 | 3.63 ± 0.42 |
| 13h | Me | CH2(biphenyl) | 64 | 86 | 62 | >100 | ND |
| 13i | Me | CH(Ph)2 | 64 | 71 | 63 | >100 | ND |
| 13j | Me | (CH2)2Ph | 64 | 83 | 64 | >100 | ND |
| 13k | Me | Bn | 64 | 68 | 61 | >70 | 1.44 ± 0.02 |
| 13l | Me | Ph | 64 | 65 | 54 | >100 | ND |
| 13m | Me | CH2CH(CH2)2 | 64 | 95 | 59 | >100 | ND |
| 13n | Me | CH2(3-Py) | 64 | 78 | 37 | >100 | ND |
| 13o | Pr | CH2[(p-trifluoromethyl)phenyl] | 78 | 61 | 67 | >100 | ND |
| 13p | i-Pr | CH2CH(CH2)2 | 65 | 52 | 85 | >100 | ND |
| 13q | i-Pr | (CH2)2Ph | 65 | 82 | 34 | >100 | ND |
| 13r | i-Pr | CH2(3-Py) | 65 | 86 | 61 | >100 | ND |
| 13s | i-Pr | CH2(biphenyl) | 65 | 76 | 71 | >100 | ND |
| 13t | i-Pr | CH(Ph)2 | 65 | 72 | 64 | >100 | ND |
| 13u | i-Pr | CH2[(2-methyl)5-thiophenyl] | 65 | 94 | 81 | 11.29 ± 1.48 | 0.262 ± 0.0002 |
| 13v | i-Pr | CH2[(m-trifluoromethyl)phenyl] | 65 | 60 | 25 | >100 | ND |
| 13w | Me | CH2[(2-methyl)5-thiophenyl] | 64 | 86 | 93 | 30.02 ± 1.87 | 3.04 ± 0.17 |
| 13x | Pr | CH2[(2-methyl)5-thiophenyl] | 78 | 89 | 91 | 7.21 ± 0.86 | 3.29 ± 0.43 |
Ca2+ channel agonist EC50 values were determined by whole cell perforated patch clamp recordings of deactivation currents from N-type channels as described in the Experimental Procedures. Cpd 13k was only soluble up to 200 μM in saline with 1% DMSO, thus limiting our ability to accurately determine agonist properties.
Cdk1 cyclinB(h) inhibitory EC50 values were determined in duplicate at 0.2, 2, and 20 μM agent concentrations, with roscovitine as the positive control. ND = not determined.
The preference of the R1 group for the branched i-propyl with regard to cdk2 activity is quite pronounced, as shown for the methylated 13k, which also reduced the kinase activity ca. 8-fold. Unfortunately, all larger substituents introduced at this position ablated the agonist activity or decreased compound solubility to the extent that we were unable to measure activities (results not shown). We were hoping to use the R2 substituent in zone 2 to gain further channel selectivity, but unfortunately, all modifications of the benzyl group at this site, including the cyclo-propylmethyl group in 13g, led to complete loss of N-type Ca2+ channel activity. These negative results were hard to predict, given the dearth of information about zone 2 modifications prior to our work (Figure 1).
The first group at R2 that proved to be an effective mimic of the benzyl group in roscovitine was the methylthiophenyl-substituted 13u. This compound had almost 3-fold improved N-type Ca2+ channel agonism, even though it was still a low nanomolar cdk2 inhibitor. Because 13u was substituted with an i-propyl group at R1, we speculated that replacing this group with methyl or n-propyl while maintaining the methylthiophenyl group at R2 should give us reduced kinase inhibitory properties analogous to 13d, 13g, and 13k. Indeed, this turned out to be the case. Both 13w and 13x had an EC50 ≈ 3 μM against cdk2, but the R1 = methyl substitution in 13w also decreased the channel activity ca. 3-fold versus 13u. In contrast, 13x proved to be a considerably more potent N-type Ca2+ channel agonist with an EC50 = 7.2 μM for N-type channels. The effects of small changes in the R1 substitution on the N-channel affinity are quite remarkable. For 13x, we also examined agonist effects on P/Q-type (EC50 = 8.8 ± 1.1 μM) and L-type calcium channels (EC50 > 100 μM). Accordingly, 13x showed selectivity for N- and P/Q-type over L-type calcium channels. Furthermore, we profiled 13x against cdk1, cdk5, mitogen-activated protein kinase 1 (MAPK1), and myosin light chain kinase (MLCK) kinase targets and found inhibitory effects with EC50 = 20.56 ± 0.96, 3.03 ± 0.32, >20, and >20 μM, respectively, further illustrating the quite favorable low-activity kinase profile of this lead structure. In light of the large cellular ATP concentrations (in the 1–10 mM range),27 single-digit micromolar activities of 13x against some kinases are likely readily compensated for in vivo and are therefore not considered significant impediments from possible therapeutic applications of 13x as N/P/Q-type calcium channel agonist in LEMS.
In parallel to our synthetic studies, docking analyses to the cdk2/roscovitine complex were used to analyze structural parameters for interactions with (R)-roscovitine analogues and to lay the groundwork for future second generation SAR studies. The MVD algorithm for docking combines an algorithm for cavity detection in the 3D structure of the target protein with an optimization algorithm that evaluates different poses for the protein–ligand complex. Because the crystal structure of cdk2 (PDB ID: 3DDQ)23 contains (R)-roscovitine bound to the active site, it was possible to identify critical residues surrounding the binding pocket. The MolDock scoring function was in combination with the MolDock SE search algorithm and Tabu clustering.24 A search space volume of 25 Å radius encompassing the (R)-roscovitine binding domain was chosen for docking, and the ligands and catalytic pocket residues were allowed to be flexible during the simulation. Each ligand was docked iteratively into the chosen cavity in 10 independent runs, each of which consisted of 1500 steps. Poses generated from each run were subjected to Tabu clustering whereby the lowest energy pose below an energy threshold of 100 was generated as output. Thus, there were 10 poses per ligand ranked by energy. The lowest energy pose of the 10 poses per ligand was selected for visual inspection.
For validation purposes, docking was first applied to (R)-roscovitine, and the docked pose was computed to be very close to the position of the ligand in the X-ray structure (i.e., RMSD = 0.27 Å). The binding free energy, as estimated by the MolDock Score (arbitrary units) was −140, which indicated a strong interaction with cdk2 binding site. The same protocol was then applied to all (R)-roscovitine analogues. The docking studies demonstrated that analogues 13d, 13g, 13k, 13w, and 13x bound with a MolDock Score value between −127 and −143 and in a different orientation, less favorable than (R)-roscovitine. Compound 13u bound similarly to (R)-roscovitine with a MolDock Score of −142 (Figures 2 and 3). These results are consistent with the experimental data that show this compound to retain cdk2 activity, while 13d, 13g, 13k, 13w, and 13x displayed reduced cdk2 activities.
Figure 2.

Docking of 13d, 13g, 13k, 13w, 13x, and 13u to the cdk2/roscovitine complex. The electrostatic interaction surface at the binding site region is displayed and colored red for negative charge and blue for positive charge. Docking simulations were performed using Molegro Virtual Docker, taking into account side chain flexibility for all residues in the binding region.
Figure 3.
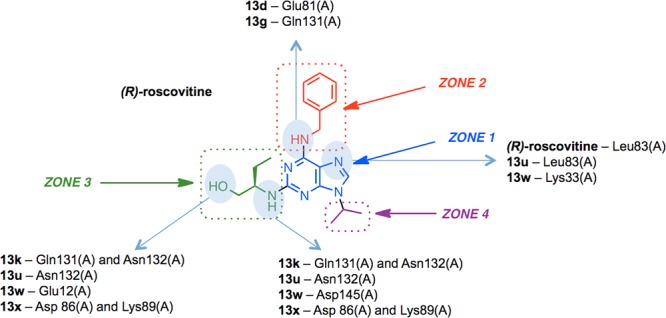
Hydrogen bond interactions between analogues 13d, 13g, 13k, 13u, 13w, 13x, and cdk2. Interacting chemical groups of the analogues are shown in blue ellipses on the (R)-roscovitine scaffold.
In summary, in ca. 20 synthetic analogues, we were able to generate a new lead structure 13x with a 4-fold improved N-type Ca2+ channel agonist efficacy and a 22-fold decreased cdk2 activity as compared to (R)-roscovitine. The SAR followed a logical trend, with small structural changes at R1 greatly influencing the cdk2 activity. Replacing the benzyl group at R2 with bioisosteric functions mostly led to compounds lacking N-type Ca2+ channel activity, with the notable exception of the 5-methylthiophene group, which considerably increased the desired agonism. Molecular docking studies were used to predict and analyze the structural trends leading to reduced cdk2 activity. We are now studying the in vivo effects of 13x as well as further improving its N- and P/Q-type Ca2+ channel activity profile. The long-term goal of our program is to develop selective voltage-gated N- and P/Q-type calcium channel agonists for the therapy of LEMS and other neurological diseases. The potential side effects of a use-dependent N- and P/Q-type calcium channel agonist that enhances calcium flux through calcium channels that are normally activated by action potential activity are expected to be few and manageable (assuming it does not cross the blood–brain barrier). This expectation is based on extensive clinical literature that uses an indirect method of achieving a similar outcome with potassium channel blockers such as 3,4-diaminopyridine (DAP).28,28b DAP prolongs the duration of the presynaptic action potential, and this indirectly increases the activation of all voltage-gated calcium channels in the peripheral nervous system. DAP is generally well tolerated by LEMS patients,28,28b with many of the side effects reported likely due to either broadening of action potentials in all neurons or the nonselective indirect effect of increasing calcium flux through all voltage-gated channels. On the basis of these previous reports, selective use-dependent N- and P/Q-type calcium channel agonists are predicted to be well tolerated.
Experimental Procedures
For the synthesis of 15, 16, and 13, see the Supporting Information. Biological evaluations of the effects of (R)-roscovitine derivatives on N-type calcium channels were initially performed using a tsA201 cell line19 that stably expresses all of the subunits of the N-type Ca2+ channel splice variant predominantly present in mammalian brain and spinal cord: Cav2.2 rnα1B-c (Cav 2.2 e[24a,Δ31a]), Cavβ3, and Cavα2δ1. For subsequent evaluation of effects on N-, P/Q-, or L-type channels, tsA-201 cells were transiently transfected with Cav2.2, Cav2.1, or Cav1.3, in combination with Cavβ3 and Cavα2δ1 (Addgene, Cambridge, MA) using FuGENE 6 (Promega, Madison, WI). All cells were maintained in DMEM supplemented with 10% fetal bovine serum. For the stable cell line expressing N-type channels, 25 μg/mL zeocin, 5 μg/mL blasticidin, and 25 μg/mL hygromycin were added as selection agents.
To assess the biological effects of (R)-roscovitine derivatives, whole-cell currents through Ca2+ channels were recorded using perforated patch methods as previously described.20−22 Briefly, the pipet solution consisted of 70 nM Cs2SO4, 60 mM CsCl, 1 mM MgCl2, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) at pH 7.4. Cultured cells were bathed in a saline composed of 130 mM choline chloride (ChCl), 10 mM tetraethylammonium chloride (TEA-Cl), 2 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES at pH 7.4. Patch pipettes were fabricated from borosilicate glass, and capacitive currents and passive membrane responses to voltage commands were subtracted from the data. Currents were amplified by an Axopatch 200B amplifier, filtered at 5 kHz, and digitized at 10 kHz for subsequent analysis using pClamp software (Axon Instruments/Molecular Devices, Sunnyvale, CA). A liquid junction potential of −11.3 mV was subtracted during recordings. To measure effects on calcium channel tail currents, the tail current integral was measured before and after application of a derivative, with the integral of each trace being normalized to its peak. All experiments were carried out at room temperature (22 °C). All (R)-roscovitine derivatives were dissolved in DMSO as a 100 mM stock and stored at −20 °C. For whole-cell recordings, (R)-roscovitine derivatives were diluted on the day of use into saline at a final concentration of 1–100 μM, and were bath-applied via a glass pipet in a ∼0.5 mL static bath chamber. Control recordings performed with 0.1–1% DMSO alone added to the drug delivery pipet solution revealed no significant effects on whole-cell Ca2+ currents. All other salts and chemicals were obtained from Sigma-Aldrich Chemical Company (St. Louis, MO).
Cdk1 cyclinB(h), cdk2 cyclinA(h), cdk5 p35(h), MAPK1(h), and MLCK(h) inhibitory activities were determined by Millipore UK Ltd. at 0.2, 2, and 20 μM agent concentration, with roscovitine as the positive control.
Flexible docking studies were performed using Molegro Virtual Docker (MVD, University of Aarhus, Denmark) to evaluate if the analogues bound to the (R)-roscovitine binding site of cdk2.23 The basis of these docking studies was a new hybrid search algorithm, that is, a guided differential evolution (DE), which combines DE optimization with a cavity prediction algorithm, which is dynamically used during the docking process. Briefly, all individual ligands were initialized, evaluated, and scored (Escore/MolDock Score) according to the fitness function, which is the sum of the intermolecular interaction energy between the ligand and the protein and the intramolecular interaction energy of the ligand: Escore = Einter + Eintra, with Einter being the ligand–protein interaction energy and Eintra being the internal energy of the ligand.24
EPLP is a piecewise linear potential using two different sets of parameters: one set for approximating the steric van der Waals term between atoms and the other stronger potential for hydrogen bonds. Eclash assigns a penalty of 1000 if the distance between two atoms (more than two bonds apart) is less than 2.0 Å. Thus, the Eclash term punishes infeasible ligand conformations.
Offspring were created using a weighted difference of the parent solutions, which were randomly selected from the population. If, and only if, the offspring was more fit, it would replace the parent. Otherwise, the parent survived and was passed on to the next generation, representing an iteration of the algorithm. The search process was terminated when the number of fitness evaluations exceeded the maximum number of evaluations permitted.
Acknowledgments
We thank Dr. Diane Lipscombe (Brown University) for kindly providing the stable N-type calcium channel tsA201 cell line and Michael Frasso (University of Pittsburgh) for the resynthesis of 13v.
Glossary
Abbreviations
- cdk
cyclin-dependent kinase
- LEMS
Lambert–Eaton myasthenic syndrome
- EC50
concentration required for 50% efficacy
- SAR
structure–activity relationship
- SNAr
nucleophilic aromatic substitution
- MAPK1
mitogen-activated protein kinase 1
- MLCK
myosin light chain kinase
Supporting Information Available
Experimental procedures, assay results, and spectral data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
This study was supported in part by the National Institutes of Health, the NIH P50 CMLD Program (GM067082 to P.W.), and a National Science Foundation Grant 0844604 (S.D.M.).
The authors declare no competing financial interest.
Dedication
† This paper is dedicated to the memory of Dr. Hiroyuki Nemoto.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Urbano F. J.; Pagani M. R.; Uchitel O. D. Calcium channels, neuromuscular synaptic transmission and neurological diseases. J. Neuroimmunol. 2008, 201–202, 136–144. [DOI] [PubMed] [Google Scholar]
- Motomura M.; Lang B.; Johnston I.; Palace J.; Vincent A.; Newsom-Davis J. Incidence of serum anti-P/Q-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J. Neurol. Sci. 1997, 147, 35–42. [DOI] [PubMed] [Google Scholar]
- Seagar M.; Lévêque C.; Charvin N.; Marquèze B.; Martin-Moutot N.; Boudier J. A.; Boudier J. L.; Shoji-Kasai Y.; Sato K.; Takahashi M. Interactions between proteins implicated in exocytosis and voltage-gated calcium channels. Philos. Trans. R. Soc. B, 1999, 354, 289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist S.; Stangel M. Update on treatment options for Lambert-Eaton myasthenic syndrome: Focus on use of amifampridine. Neuropsychiatr. Dis. Treat. 2011, 7, 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders D. B. Lambert-Eaton myasthenic syndrome: Diagnosis and treatment. Ann. N.Y. Acad. Sci. 2003, 998, 500–508. [DOI] [PubMed] [Google Scholar]
- Meijer L.; Raymond E. Roscovitine and other purines as kinase inhibitors. From starfish oocytes to clinical trials. Acc. Chem. Res. 2003, 36, 417–425. [DOI] [PubMed] [Google Scholar]
- Meijer L.; Bettayeb K.; Galons H.. (R)-Roscovitine (cyc202, seliciclib). In Inhibitors of Cyclin-Dependent Kinases as Anti-Tumor Agents; Smith P. J., Yue E. W., Eds.; CRC/Taylor & Francis: Boca Raton, 2007; pp 187–225. [Google Scholar]
- Cho S.; Meriney S. D. The effects of presynaptic calcium channel modulation by roscovitine on transmitter release at the adult frog neuromuscular junction. Eur. J. Neurosci. 2006, 23, 3200–3208. [DOI] [PubMed] [Google Scholar]
- De Stefino N. R.; Pilato A. A.; Dittrich M.; Cherry S. V.; Cho S.; Stiles J. R.; Meriney S. D. (R)-Roscovitine prolongs the mean open time of unitary N-type calcium channel currents. Neuroscience 2010, 167, 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z.; Chi P.; Bibb J. A.; Ryan T. A.; Greengard P. Roscovitine: A novel regulator of P/Q-type calcium channels and transmitter release in central neurons. J. Physiol. 2002, 540, 761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraei Z.; Anghelescu M.; Elmslie K. S. Slowed N-type calcium channel (CaV2.2) deactivation by the cyclin-dependent kinase inhibitor roscovitine. Biophys. J. 2005, 89, 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraei Z.; Elmslie K. S. The separation of antagonist from agonist effects of trisubstituted purines on Cav2.2 (N-type) channels. J. Neurochem. 2008, 105, 1450–1461. [DOI] [PubMed] [Google Scholar]
- Sheng Z.-H.; Westenbroek R. E.; Catterall W. A. Physical link and functional coupling of presynaptic calcium channels and the synaptic vesicle docking/fusion machinery. J. Bioenerg. Biomembr. 1998, 30, 335–345. [DOI] [PubMed] [Google Scholar]
- For a discussion of SAR effects pertaining to the cdk2 activity profile of roscovitine, seeOtyepka M.; Krystof V.; Havlicek L.; Siglerova V.; Strnad M.; Koca J. Docking-based development of purine-like inhibitors of cyclin-dependent kinase-2. J. Med. Chem. 2000, 43, 2506–2513. [DOI] [PubMed] [Google Scholar]
- Popowycz F.; Fournet G.; Schneider C.; Bettayeb K.; Ferandin Y.; Lamigeon C.; Tirado O. M.; Mateo-Lozano S.; Notario V.; Colas P.; Bernard P.; Meijer L.; Joseph B. Pyrazolo[1,5-a]-1,3,5-triazine as a purine bioisostere: Access to potent cyclin-dependent kinase inhibitor (R)-roscovitine analogue. J. Med. Chem. 2009, 52, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oumata N.; Bettayeb K.; Ferandin Y.; Demange L.; Lopez-Giral A.; Goddard M.-L.; Myrianthopoulos V.; Mikros E.; Flajolet M.; Greengard P.; Meijer L.; Galons H. Roscovitine-derived, dual-specificity inhibitors of cyclin-dependent kinases and casein kinases 1. J. Med. Chem. 2008, 51, 5229–5242. [DOI] [PubMed] [Google Scholar]
- Bettayeb K.; Sallam H.; Ferandin Y.; Popowycz F.; Fournet G.; Hassan M.; Echalier A.; Bernard P.; Endicott J.; Joseph B.; Meijer L. N-&-N, a new class of cell death-inducing kinase inhibitors derived from the purine roscovitine. Mol. Cancer Ther. 2008, 7, 2713–2724. [DOI] [PubMed] [Google Scholar]
- Meriney S. D. Unpublished results.
- Lin Y.; McDonough S. I.; Lipscombe D. Alternative splicing in the voltage-sensing region of N-type Cav2.2 channels modulates channel kinetics. J. Neurophys. 2004, 92, 2820–2830. [DOI] [PubMed] [Google Scholar]
- White M. G.; Crumling M. A.; Meriney S. D. Developmental changes in calcium current pharmacology and somatostatin inhibition of calcium current in chick parasympathetic neurons. J. Neurosci. 1997, 17, 6302–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazejian B.; DiGregorio D. A.; Vergara J. L.; Poage R. E.; Meriney S. D.; Grinnell A. D. Direct measurements of presynaptic calcium and calcium-activated potassium currents regulating neurotransmitter release at cultured Xenopus nerve-muscle synapses. J. Neurosci. 1997, 17, 2990–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.; Meriney S. D. The effects of presynaptic calcium channel modulation by roscovitine on transmitter release at the adult frog neuromuscular junction. Eur. J. Neurosci. 2006, 23, 3200–3208. [DOI] [PubMed] [Google Scholar]
- Bettayeb K.; Oumata N.; Echalier A.; Ferandin Y.; Endicott J. A.; Galons H.; Meijer L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [DOI] [PubMed] [Google Scholar]
- Thompson E. E.; Kornev A. P.; Kannan N.; Kim C.; Ten Eyck L. F.; Taylor S. S. Comparative surface geometry of the protein kinase family. Protein Sci. 2009, 18, 2016–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selective replacements of heterocyclic chloride substituents with carbon nucleophiles, see, for example,Wipf P.; George K. M. Regioselective palladium-catalyzed cross-coupling reactions of 2,4,7-trichloroquinazoline. Synlett 2010, 644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oumata N.; Ferandin Y.; Meijer L.; Galons H. Practical synthesis of roscovitine and CR8. Org. Process Res. Dev. 2009, 13, 641–644. [Google Scholar]
- Kemp G. J.; Meyerspeer M.; Moser E. Absolute quantification of phosphorus metabolite concentrations in human muscle in vivo by 31P MRS: A quantitative review. NMR Biomed. 2007, 20, 555–565. [DOI] [PubMed] [Google Scholar]
- Oh S. J.; Claussen G. G.; Hatanaka Y.; Morgan M. B. 3,4-Diaminopyridine is more effective than placebo in a randomized, double-blind, cross-over drug study in LEMS. Muscle Nerve 2009, 40, 795–800. [DOI] [PubMed] [Google Scholar]
- Titulaer M. J.; Lang B.; Verschuuren J. J. Lambert-Eaton myasthenic syndrome: From clinical characteristics to therapeutic strategies. Lancet Neurol. 2011, 10, 1098–1107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.