

Abstract
Hypoxia inducible factors (HIFs) are transcription factors that activate expression of multiple gene products and promote tumor adaptation to a hypoxic environment. To become transcriptionally active, HIFs associate with cofactors p300 or CBP. Previously, we found that arylsulfonamides can antagonize HIF transcription in a bioassay, block the p300/HIF-1α interaction, and exert potent anticancer activity in several animal models. In the present work, KCN1-bead affinity pull down, 14C-labeled KCN1 binding, and KCN1-surface plasmon resonance measurements provide initial support for a mechanism in which KCN1 can bind to the CH1 domain of p300 and likely prevent the p300/HIF-1α assembly. Using a previously reported NMR structure of the p300/HIF-1α complex, we have identified potential binding sites in the p300-CH1 domain. A two-site binding model coupled with IC50 values has allowed establishment of a modest ROC-based enrichment and creation of a guide for future analogue synthesis.
Keywords: hypoxia, solid tumors, p300, HIF arylsulfonamide inhibitors, binding model, QSAR, KCN1
Hypoxia is a condition in which insufficient oxygen is supplied to tissues. It is prevalent in fast-growing solid tumor tissues, due to inadequate development of vascular supply.1 The resulting perturbation in physiology renders the tumor more resistant to chemo- and radiotherapies.2 Tumors develop adaptive mechanisms to grow in a hypoxic microenvironment, including a switch to glycolytic metabolism and the activation of signals for the recruitment of new vasculature.3−5 The hypoxia inducible factor (HIF) pathway plays a crucial role for tumor cell growth under oxygen-deprived conditions.5 HIFs are a family of heterodimeric transcription factors composed of α- and β-subunits. The α-subunits are regulated at the post-translational level by a family of prolylhydroxylases (PHDs), which under normoxic conditions tag them with a hydroxyl group causing recognition by the Von Hippel Lindau protein, a component of an E3 ubiquitin ligase complex, and initiates their degradation by the proteasome. The PHDs require oxygen to function; hence, under hypoxia, HIFα subunits are stabilized and bind to the constitutively expressed β-subunits. The HIF heterodimer translocates to the nucleus and associates through its C-terminal activation domain (CAD) with the CH1 domains of cofactors p300 or CREB binding protein (CBP) to form a functional transcription factor. Activation of the HIF transcription pathway induces the expression of over a hundred genes, which encode proteins involved in the regulation of glucose metabolism, cell migration, and neovascularization.5,6
HIF-1α overexpression is a common occurrence in most human cancers and has been associated with drug resistance mechanisms.2,7−10 Thus, because of the central role of HIF in cancer, it has become an important target for cancer therapy using different approaches.9,11−15 In previous screening studies for the identification of small molecule inhibitors of the HIF pathway, we identified arylsulfonamides as a new scaffold that can interfere with HIF transcription using a hypoxia responsive element (HRE)-driven gene luciferase-based reporter assay.16−18 KCN1, one of the lead compounds (Figure 1), was further investigated and demonstrated strong antitumor activity against malignant gliomas, pancreatic cancer, and metastatic uveal melanoma in animal models.19−21 These studies further suggested that KCN1 might inactivate HIF transcriptional activity by disrupting the interaction of the p300/CBP cofactors with HIF-1α.21 In the present work, we provide initial evidence for a mechanism of action where KCN1 can bind to the CH1 domain of p300. Finally, we established a molecular model for the possible binding of KCN1 and its analogues to p300 through computational modeling of putative binding sites.
Figure 1.
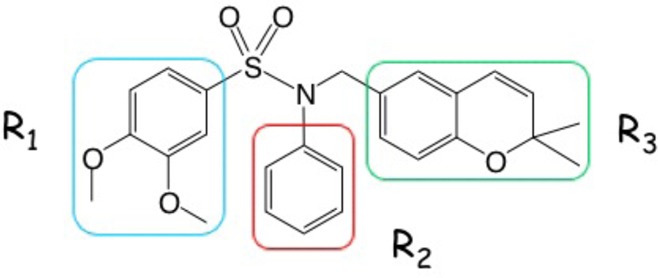
Structure of KCN1 highlighted by three key substituent groups.
Conceptually, KCN1 could inhibit HIF transcriptional function by disrupting the p300/HIF-1α interaction by binding separately to p300 or HIF-1α or by associating with the complex such that function is attenuated. Our previous studies have shown that KCN1 can disrupt the interaction between p300 or CBP with HIF-1α using coimmunoprecipitation studies, excluding the latter possibility.21 While p300 is able to maintain its 3D architecture even in the absence of HIF-1α, the C-terminal transactivation domain (CAD) of the latter is disordered when uncomplexed with p300.22,27 Therefore, a reasonable hypothesis is that KCN1 binds to p300 and thereby blocks induced fit by HIF-1α. We have utilized affinity pull down analysis with KCN1 coupled beads, 14C-KCN1 binding to p300, and surface plasmon resonance (SPR) measurements of the KCN1/p300-CH1 interaction to test this hypothesis.
Using nuclear extracts from hypoxic human glioma cells, binding of p300 and HIF-1α was probed in affinity pull down experiments with KCN1 immobilized on agarose beads through chemical cross-linking with an aliphatic linker (see the Supporting Information for the synthesis). Retained proteins were subjected to Western blot analysis. As Figure 2 illustrates, KCN1-coupled beads pulled down a fraction of the cellular p300 (+ lane), while little nonspecific binding was observed with beads alone used as a control (− lane). No detectable interaction between KCN1 and HIF-1α was observed. This experiment supports the hypothesis that KCN1 binds to p300, but the exact binding site is not revealed.
Figure 2.
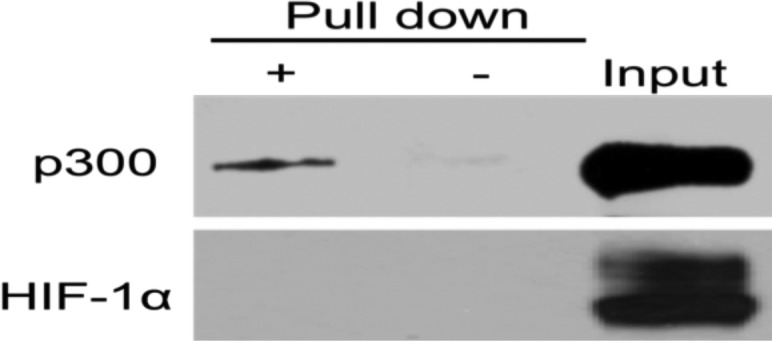
Western blot analysis pull down of p300 (upper panel) and HIF-1α (lower panel) proteins using KCN1-coupled agarose beads (+ lanes). A fraction (10%) of the cell extract before pull down was used as a control to verify protein expression in the cell extract (Input). Uncoupled beads were used as a control for nonspecific binding (− lanes).
Recombinant fusion peptides containing glutathione S-transferase (GST) and the CH1 domain of p300 (GST-p300-CH1) were produced in bacteria, purified, and then incubated with 14C-KCN1 (see the Supporting Information for the synthesis). After washing, the bound activity was counted in a scintillation counter and shown to be significantly greater than that obtained with GST-only peptides used as a control (Figure 3, left). This experiment supports the hypothesis that KCN1 can bind to the CH1 domain of p300.
Figure 3.

14C-KCN1 binds to the p300 CH1 domain. The recombinant proteins size, quantities, and integrity were verified by Coomassie-stained gel electrophoresis (right).
SPR methodology has been widely used for defining the kinetics and affinities for the interactions between a wide variety of macromolecular entities and high- and low-affinity small molecules.23−26 In the present instance, KCN1 was covalently tethered to a gold surface as shown in Figure 4.
Figure 4.

KCN1 attached to a gold surface.
Recombinant p300-CH1 peptides (after cleavage of the GST moiety) were streamed over the KCN1-enriched surface in a range of concentrations as illustrated by Figure 5. It shows that SPR signals respond to the different protein concentrations, which clearly indicates the existence of binding between KCN1 and p300-CH1. Analysis of the curve shapes and concentration dependence permits an estimate that KCN1 binds to p300-CH1 with a Kd value of ∼345 nM. Using a 1:1 kinetic binding model (A + B = AB), the association and dissociation processes were fitted to obtain the on (ka) and off (kd) rates separately. Consequently, the Kd value was calculated as kd/ka ∼ 345 nM. These results and the biochemical assays encouraged us to build a model of the p300-CH1/KCN1 complex and employ it for prospective generation of new analogs for bioactivity testing.
Figure 5.
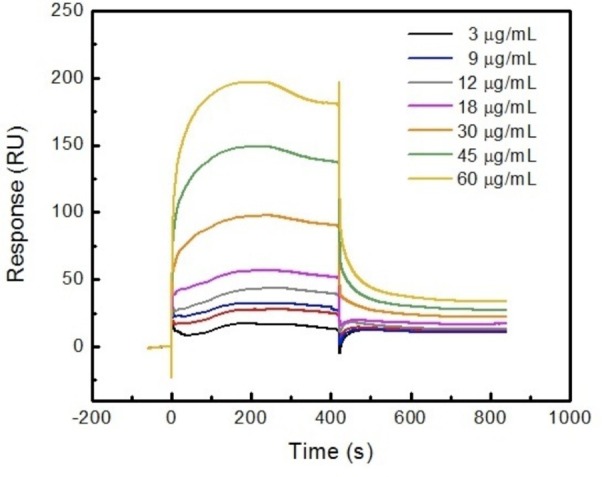
SPR sensorgrams after subtraction of control illustrate KCN1 binding to p300; protein concentrations from 3 to 60 μg/mL.
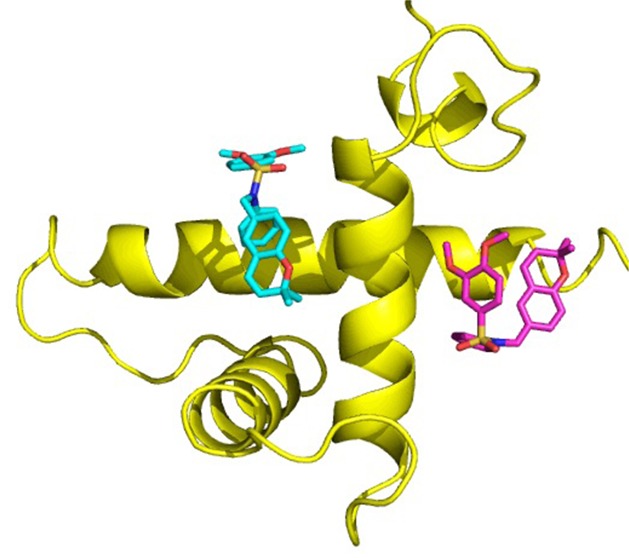
Neither the affinity pull down, radiolabeling binding assay, nor the SPR experiment provides information on the precise location of the presumed binding site for KCN1 on the CH1 domain of p300. In addition, no crystal structure of a small molecule complexed with p300 is available. Therefore, we sought to obtain binding site information from the structure of p300, previously reported mutants, and molecular docking. Figure 6 illustrates the structure of the p300(CH1 domain)/HIF-1α(CAD) complex derived by NMR spectroscopy (PDB code: 1L3E(27)). p300-CH1 consists of three major helices nearly perpendicular to each other. HIF-1α -CAD primarily presents loops that lay on the surface of p300, but two small helices (A and B) bind to orthogonal intersections of the helices of p300-CH1. The helix–helix interactions are characterized by a significant degree of hydrophobicity, which undoubtedly contributes to the mutual binding.
Figure 6.
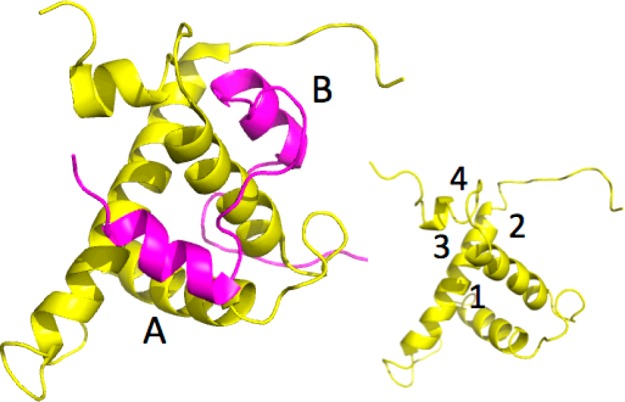
p300 (yellow)/HIF-1α (purple) complex; the insert reveals four potential binding sites for KCN1.
p300-CH1 was extracted from the complex of Figure 6 and inspected graphically to reveal four possible clefts available for ligand binding at the junctions of the three helices (Figure 6 insert). Each was subjected to KCN1 docking with Glide followed by Prime MM-GBSA rescoring.28,29 Remarkably, the top two best-scoring sites (sites 1 and 2, Figures 6 and 7) are coincident with the binding locus of the two HIF-1α helices (A and B). Similar results were obtained with the structure of the CBP-CH1 cofactor determined by NMR.30 Examination of the detailed ligand-p300 interactions at site 1 reveals substantial hydrophobic contacts (Figure 8). The N-phenyl moiety is nestled in a deep pocket surrounded by Leu345, Leu346, Leu376, and Ile40031 complemented by π–π stacking with His349. The dimethoxyphenyl ring is located above Leu346, and one of the OMe units is stacked against Trp403, while the dimethylbenzopyran faces the side chains of Ile400 and Met379. Supplemental polar anchors are present for one of the aromatic methoxy groups (Lys350), the SO2 moiety (Lys404), and the benzopyran oxygen (Ser401 H-bond).
Figure 7.
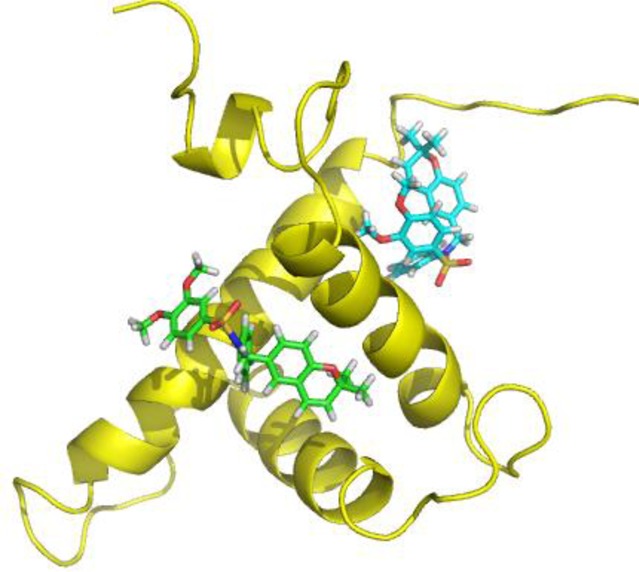
Two favored docking sites for KCN1 resulting from Glide/MM-GBSA scoring.
Figure 8.
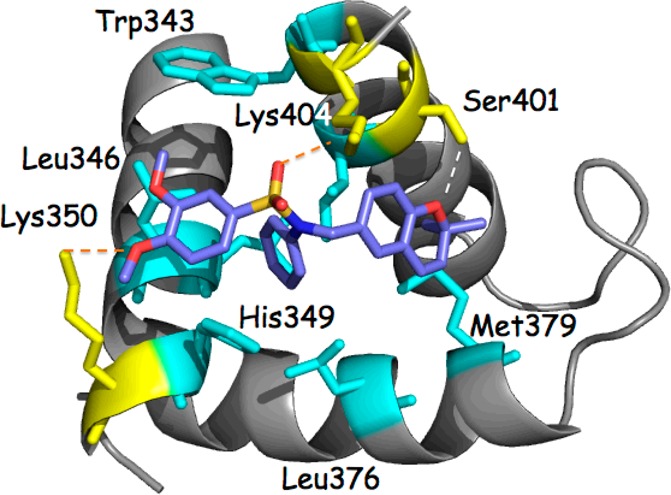
Detailed protein–ligand contacts for site 1 illustrating a substantial hydrophobic contact surface (cyan; Leu345 and Ile400 lie beneath the KCN1 ligand) and supplemental polar anchors (dotted lines; yellow).
These suggested outcomes are supported by a random mutation study for the p300/HIF-1α complex.32 Leu344, Leu345, Cys388, and Cys393 on p300 were determined to be crucial for interaction with HIF-1α (Figure 9). Cys388 and Cys393 coordinate to a zinc ion, explaining that disruption of the p300 zinc finger by random mutations at the two residues has interrupted the p300 structure and consequently its ability to interact with HIF-1α. As described above, KCN1 docking leads to intimate contacts between Leu344 and Leu345 and the ligand fragments R1 and R2 (Figure 1). In a similar vein, two residues critical for HIF-1α binding to p300, Leu818 and Leu822,32 form a hydrophobic cluster with p300 Leu344 and Leu345 side chains (Figure 9). Thus, it is reasonable to hypothesize that one or both of the helix–helix interaction sites in the p300/HIF-1α complex might serve as binding centers for KCN1 and its analogues.
Figure 9.
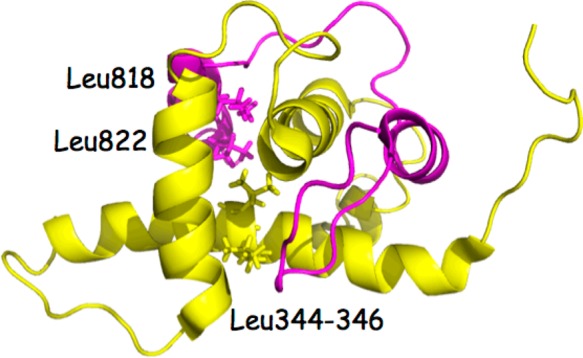
p300-HIF-1α complex illustrating the spatial relationship of residues critical for complex formation.
A series of recently reported KCN1 analogs were selected for quantitative structure–activity relationship (QSAR) investigation (Figure 1 and Tables S1 and S2 in the Supporting Information).16−18 The analogues were subjected to Glide docking at each of the two favored binding sites (Figure 6) followed by energy rescoring with Prime MM-GBSA to obtain estimates of binding free energy.28,29 The IC50 values for inhibition of HIF transcriptional activity in a glioblastoma cell reporter assay were obtained from single triplicate runs as previously described.16−18 To minimize possible biological variability from measurements made over time, an IC50 value for each analogue scaled to the corresponding KCN1 IC50 reference determined in the same experiment was employed for the correlations. Thus, the ratio of the IC50 value of the analogue and the corresponding IC50 value of KCN1 was used as a measurement of activity (ΔIC50 ∼ K) and applied to the QSAR analysis.
A variety of approaches employing full and split data sets, explcit ligand docking, and ligand-only pharmacophores failed to generate a quantitative SAR with r2 greater than 0.7. It would appear that significant scatter in the present data precludes development of a robjust QSAR. An alternative approach is the ROC (receiver operating characteristic) methodology,33 which uses a binary classifier to assess whether the predicted binding energy values are useful for enrichment of the active compounds. In the present context, we define true positives as compounds with an average IC50 value <650 nM. Five of 30 analogues fall in this category based on the cell-based reporter assay: 648, 306, 478, 378, and 280 nM activity (See Table 10, ref (17)).
Plotting selectivity versus sensitivity (i.e., the fraction of true positives vs the fraction of false positives at a given threshold setting), the AUCs (area under curve) for both sites 1 and 2 are ∼0.7, the ideal being 0.8–1. This indicates that the estimated energy values (MM-GBSA) can, to some extent, differentiate active from inactive analogues. Thus, we assume that compounds demonstrating favorable energies at both sites are likely to be active. In a test study carried out for the 30 available KCN1 analogues, only three survived: KCN1, KCN2, and KCN5, all of which are true positives. It is noteworthy that the latter two analogues are the most potent agents among the test molecules. This outcome indicates that the estimated energy values at both sites can enrich the active compounds. In the future, prior to a full-scale QSAR, structures with equal or better estimated potency values will be considered high priority for synthesis.
On the basis of affinity pull down analysis, 14C-KCN1 binding, KCN1/p300-CH1 SPR binding measurements, and evaluation of the NMR structure of the p300/HIF-1α complex, we provide a model illustrating how KCN1 and its analogues might antagonize the p300/HIF-1α interaction by occupying the two binding sites on p300-CH1 that are normally engaged by the two inducible helices of HIF-1α CAD when p300 and HIF-1α are associated. Because HIF-1α is disordered when uncomplexed with p300, it is reasonable to assume that occupancy of these two critical sites will prevent the induced fit with HIF-1α. On the basis of the large protein–protein interface and the energy needed to disrupt a pre-existing complex, we believe that KCN1 action most likely occurs when de novo-stabilized HIF-1α assembles into a 3D structure with p300. This also partly explains why an affinity approach with KCN1 beads on a cell extract with preformed HIF-1α-p300 complexes pulls down only a relatively small fraction of total p300. Another reason is that only a fraction of total p300 associates with HIF, as it is bound to other cellular proteins. The proposed two-site binding model explains many of the experimental results, provides a useful preliminary ROC analysis, and may prove applicable to the identification of novel KCN1 analogues with improved potency. Further validation of the model will be performed with an expanded set of analogues.
Experimental Procedures
KCN1 linked to agarose beads (see the Supporting Information) was used to pull down KCN1-interacting proteins. Nuclear extracts prepared from hypoxic (1% O2) LN229 human glioma cells using the NE-PER kit (Pierce) were precleared with ethanolamine activated agarose beads to eliminate nonspecific protein binding to the beads. Bound proteins were separated by denaturing gel electrophoresis (SDS-PAGE), and Western blots were performed using anti-p300 and anti-HIF-1α antibodies as previously described.12,34
GST-only and GST-p300-CH1 proteins were expressed and purified as described previously.35 For the SPR experiments, p300-CH1 was cleaved from the purified GST fusion protein (10 mg) with 50 units of thrombin (GE Healthcare) in PBS at room temperature overnight, and its concentration was evaluated by the BCA Protein Assay (Thermo Scientific).
Binding and kinetics measurements were performed with a BIAcore T200 system and carboxylic acid coated sensor chips (CM-5 chips with five channels from BIAcore, two of which were used here). After activation of the surface with EDC/NHS, KCN1-amine (500 nM; see the Supporting Information for the synthesis of the free amine) in HBS-EP+ buffer was immobilized on the flow cell gold surface by covalent capture. One channel was used to immobilize KCN1-amine, and the other was left blank as a control. Recombinant p300-CH1 peptides in PBS were injected at a flow rate of 50 μL/min. A 1 M NaCl solution was used to dissociate the p300 from the KCN1 compound for surface regeneration. Injection of the protein (association) was followed by injection of running buffer (dissociation). To reduce the probability of nonspecific binding to the chip surface, 50 μL/L of surfactant P20 was added to the p300 buffers in the binding experiments.
Two-dimensional structures of KCN1 and its analogues were modeled by Chemdraw and then submitted to Ligprep in Maestro 9.0 to obtain 3D structures. The p300-CH1/HIF-1α CAD protein complex (PDB code 1L3E) was processed by the protein preparation wizard followed by removal of HIF-1α CAD. The p300 receptor was refined with the OPLS2005 force field by Impref minimization. Four docking sites, as shown in Figure 6, were selected for KCN1 Glide docking to determine the most favorable interaction pharmacophores. For other analogues, only sites 1 and 2 in Figure 6 were chosen for QSAR development. The receptor grid was generated at both presumed binding sites using the Glide receptor grid protocol without applying constraints. Ligand docking was accomplished with Glide SP precision (Maestro 9.0) for flexible docking of the ligands.28 The top 20 poses at each site were subsequently submitted to Prime MM-GBSA (all atoms of the receptor are frozen) for rescoring. The two best-scored KCN1 binding sites (1 and 2) were subsequently employed for linear regression and ROC evaluation.
For each ligand, the rescored top pose was selected, and the calculated MM-GBSA ΔG value was used as an estimate of binding energy. On the experimental side, certain compound IC50 values were measured at different times with KCN1 as a standard in each case. The ratio of the IC50 value of a given ligand to the corresponding IC50 value of KCN1 was used as a measurement of activity (ΔIC50 ∼ K) and applied to the attempted QSAR correlations. The relative binding energies (ΔE) and activities (K) were fitted by the equation ΔE = –RT ln K.
Glossary
Abbreviations
- HIF
hypoxia inducible factor
- CBP
CREB binding protein
- SPR
surface plasmon resonance
- PHDs
prolylhydroxylases
- QSAR
quantitative structure–activity relationship
- GST
glutathione S-transferase
Supporting Information Available
Synthesis of KCN1-amine, immobilization of the latter on a gold surface and agarose beads, synthesis of 14C-KCN1, and a list of structures employed in the p300/KCN1 analogue analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by EmTechBio, the Southeastern Brain Tumor Foundation, the University Research Council, the NIH [R01 CA86335, CA116804 (E.G.V.M.), P30 CA138292 (Winship), GM084933 and GM86925 (B.W.), and P50 CA128301 (M.M.G.)], the V Foundation, the Max Cure Foundation, and the Samuel Waxman Cancer Research Foundation (E.G.V.M.). J.P.S. and Q.S. are likewise grateful to Prof. Dennis Liotta for generous support.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brat D. J.; Kaur B.; Van Meir E. G. Genetic modulation of hypoxia-induced gene expression and vascular proliferation: relevance to brain tumors. Front. Biosci. 2002, 8, d100–d116. [DOI] [PubMed] [Google Scholar]
- Lara P. C.; Lloret M; Clavo B.; Apolinario R. M.; Henríquez-Hernández L. A.; Bordón E.; Fontes F.; Rey A. Severe hypoxia induces chemo-resistance in clinical cervical tumors through MVP over-expression. Radiat. Oncol. 2009, 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belozerov V.; Van Meir E. G. Inhibitors of Hypoxia Inducible Factor-1 signaling. Curr. Opin. Invest. Drugs 2006, 7, 1067–1076. [PubMed] [Google Scholar]
- Kaur B.; Tan C.; Brat D. J.; Van Meir E. G. Genetic and hypoxic regulation of angiogenesis in gliomas. J. Neuro-Oncol 2004, 70, 229–243. [DOI] [PubMed] [Google Scholar]
- Semenza G. L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z.; Huang L. E.; Eckner R.; Bhattacharya S.; Jiang C.; Goldberg M. A.; Bunn H. F.; Livingston D. M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. 1996, 93, 12969–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos R.; van der Groep P.; Greijer A. E.; Shvarts A.; Meijer S.; Pinedo H. M.; Semenza G. L.; van Diest P. J.; Van der Wall E. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 2003, 97, 1573–1581. [DOI] [PubMed] [Google Scholar]
- Zhong H.; De Marzo A. M.; Laughner E.; Lim M.; Hilton D. A.; Zagzag D.; Buechler P.; Isaacs W. B.; Semenza G. L.; Simons J. W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [PubMed] [Google Scholar]
- Mun J.; Jabbar A. A.; Devi N. S.; Yin S.; Wang Y.; Tan C.; Culver D.; Snyder J. P.; Van Meir E. G.; Goodman M. M. Design and in vitro activities of N-alkyl-N-[(8-R-2,2-dimethyl-2H-chromen-6-yl)methyl]heteroarylsulfonamides, novel small molecule Hypoxia Inducible Factor-1 (HIF-1) pathway inhibitors and anti-cancer agents. J. Med. Chem. 2012, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur B.; Khwaja F. W.; Severson E. A.; Matheny S. L.; Brat D. J.; Van Meir E. G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro. Oncol. 2005, 7, 134–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belozerov V; Van Meir E. G. Hypoxia Inducible factor-1: A novel target for cancer therapy. Anti-Cancer Drugs 2005, 16, 901–909. [DOI] [PubMed] [Google Scholar]
- Narita T.; Yin S.; Gelin C. F.; Moreno C. S.; Yepes M.; Nicolaou K. C.; Van Meir E. G. Identification of a novel small molecule HIF-1a translation inhibitor. Clin. Cancer Res. 2009, 15, 6128–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-S.; Peng G.-Y.; Hicks J. M.; Weiss H. L.; Van Meir E. G.; Brenner M. K.; Yotnda P. Engineering human tumor-specific cytotoxic T cells to function in a hypoxic environment. Mol. Ther. 2008, 16, 599–606. [DOI] [PubMed] [Google Scholar]
- Post D. E.; Sandberg E. M.; Devi S. N.; Brat D. J.; Xu Z.; Tighiouart M.; Van Meir E. G. Targeted cancer-gene therapy using a HIF-dependent oncolytic adenovirus armed with interleukin-4. Cancer Res. 2007, 67, 6872–6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post D. E.; Devi N. S.; Li Z.; Brat D. J.; Kaur B.; Nicholson A.; Olson J. J.; Zhang Z.; Van Meir E. G. Cancer therapy with a replicating oncolytic adenovirus targeting the hypoxic microenvironment of tumors. Clin. Cancer Res. 2004, 10, 8603–8612. [DOI] [PubMed] [Google Scholar]
- Tan C.; de Noronha R. G.; Devi N. S.; Jabbar A. A.; Kaluz S.; Liu Y.; Mooring S. R.; Nicolaou K. C.; Wang B.; Van Meir E. G. Sulfonamides as a new scaffold for hypoxia inducible factor pathway inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5528–5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid-Mooring S.; Jin H.; Devi N. S.; Jabbar A. A.; Kaluz S.; Liu Y.; Van Meir E. G.; Wang B. Design and synthesis of novel small-molecule inhibitors of the hypoxia inducible factor pathway. J. Med. Chem. 2011, 54, 8471–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun J.; Jabbar A. A.; Devi N. S; Liu Y.; Van Meir E. G.; Goodman M. M. Structure–activity relationship of 2,2-dimethyl-2H-chromene based arylsulfonamide analogs of 3,4-dimethoxy-N-[(2,2-dimethyl-2H-chromen-6-yl)methyl]-N-phenylbenzenesulfonamide, a novel small molecule hypoxia inducible factor-1 (HIF-1) pathway inhibitor and anti-cancer agent. Bioorg. Med. Chem. 2012, 20, 4590–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Ao L.; Rayburn E. R.; Xu H.; Zhang X.; Zhang X.; Wang M. H.; Wang H.; Van Meir E. G.; Zhang R.. KCN1, a novel synthetic sulfonamide anticancer agent: In vitro and in vivo anti-pancreatic cancer activities and preclinical pharmacology. Submitted for publication.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Kaluz S.; Yang H.; Van Meir E. G.; Grossniklaus H. E.. Efficacy of the novel small molecule HIF pathway inhibitor KCN1 in the control of micrometastases in experimental ocular melanoma. Manuscript in preparation. [Google Scholar]
- Yin S.; Kaluz S.; Devi S. N.; Jabbar A. A., ; de Norhona R.; Wang W.; Mun J.; Boreddy P. R.; Abbruscato T.; Zhang R.; Goodman M. M.; Nicolaou K. C., ; Van Meir E. G. Arylsulfonamide KCN1 inhibits in vivo glioma growth and blocks HIF signaling. :Submitted for publication.. doi: 10.1158/1078-0432.CCR-12-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Guzman R. N.; Wojciak J. M.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. CBP/p300 TAZ1 Domain Forms a Structured Scaffold for Ligand Binding. Biochemistry 2005, 44, 490–497. [DOI] [PubMed] [Google Scholar]
- Homola J.; Yeea S. S.; Gauglitzb G. Surface plasmon resonance sensors: Review. Sens. Actuators, B 1999, 54, 3–15. [Google Scholar]
- Pattnaik P. Surface plasmon resonance: applications in understanding receptor-ligand interaction. Appl. Biochem. Biotechnol. 2005, 126, 79–92. [DOI] [PubMed] [Google Scholar]
- Navratilova I.; Hopkins A. L. Emerging role of surface plasmon resonance in fragment-based drug discovery. Future Med. Chem. 2011, 3, 1809–1820. [DOI] [PubMed] [Google Scholar]
- de Mol N. J. Surface plasmon resonance for proteomics. Methods Mol. Biol. 2012, 800, 33–53. [DOI] [PubMed] [Google Scholar]
- Freedman S. J.; Sun Z. Y.; Poy F.; Kung A. L.; Livingston D. M.; Wagner G.; Eck M. J. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc. Natl. Acad. Sci. 2002, 99, 5367–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glide, version 5.5; Schrödinger, LLC: New York, NY, 2009. [Google Scholar]
- Prime, version 2.1; Schrödinger, LLC: New York, NY, 2009. [Google Scholar]
- Dames S. A.; Martinez-Yamout M.; De Guzman R. N.; Dyson H. J.; Wright P. E. Structural basis for HIF-1α/CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. 2002, 99, 5271–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This numbering used in the p-300/HIF-1α NMR structure (PDB code 1L3E) is offset by 222 for p300 by comparison with the full length p300 sequence (ref (27)). Thus, Leu123 and Leu124 are Leu345 and Leu346, respectively. Likewise, the HIF-1α numbering is offset by 784, leading to congruence between leucines 34/38 and 818/822, respectively.
- Gu J.; Milligan J.; Huang L. E. Molecular Mechanism of Hypoxia-inducible Factor 1α-p300 Interaction. J. Biol. Chem. 2001, 276, 3550–3554. [DOI] [PubMed] [Google Scholar]
- http://en.wikipedia.org/wiki/Receiver_operating_characteristic; accessed 5/25/2012.
- Tan C.; de Noronha R. G.; Roecker A. J.; Pyrzynska B.; Khwaja F.; Zhang Z.; Zhang H.; Teng Q.; Nicholson A. C.; Giannakakou P.; Zhou W.; Olson J. J.; Pereira M. M.; Nicolaou K. C.; Van Meir E. G. Identification of a novel small molecule inhibitor of the hypoxia-inducible factor-1 (HIF-1) pathway. Cancer Res. 2005, 65, 605–612. [PubMed] [Google Scholar]
- Kaluz S.; Kaluzová M.; Stanbridge E. J. Proteasomal inhibition attenuates transcriptional activity of hypoxia-inducible factor 1 (HIF-1) via specific effect on the HIF-1alpha C-terminal activation domain. Mol. Cell. Biol. 2006, 15, 5895–5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.