

Abstract
Purpose
To identify genetic defects in a Chinese family with ectopia lentis (EL) and varicose great saphenous vein (GSV) and to analyze the correlations between phenotype and genotype.
Methods
Twenty-two (12 affected subjects and ten unaffected subjects) among 53 members of a Chinese family underwent complete physical, ophthalmic, and cardiovascular examinations. Genomic DNA was extracted from the leukocytes in the subjects’ peripheral blood. A minimum interval was achieved with linkage study and haplotype analysis. All 65 exons and the flanking intronic regions of fibrillin-1 (FBN1) were amplified with PCR and screened for mutations with direct Sanger sequencing. Molecular modeling was analyzed in an in silico study.
Results
The linkage study showed a strong cosegregation signal on chromosome 15. The non-parametric linkage analysis yielded a maximum score of 29.1(p<0.00001), and the parametric logarithm of the odds (LOD) score was 3.6. The minimum interval of the shared haplotype was rs1565863-rs877228. The best candidate gene in this region was FBN1. A novel mutation, c.3928G>A, p.1310G>S in exon 31, was identified in FBN1 and cosegregated well in the family. We applied molecular modeling to show the effect of this mutation on the fibrillin-1 structure. The mutation significantly distorts the calcium coordination, decreases the binding of the calcium ion in that motif, and affects the local calcium-binding epidermal growth factor (cbEGF) interface that depends on Ca binding.
Conclusions
FBN1-associated fibrillinopathies are a group of diseases with dynamic phenotype changes. Novel mutation p.1310G>S was first reported to cause Marfan syndrome (MFS). Our results expand the mutation spectrum in FBN1 and enhance our knowledge of genotype–phenotype correlations underlying FBN1 mutations.
Introduction
Mutations in the FBN1 gene (ID 2200; OMIM 134797) have been reported in patients with a series of fibrillinopathies, such as Marfan syndrome (MFS), isolated ectopia lentis (EL) [1], Weill-Marchesani syndrome, stiff skin syndrome, geleophysic andacrophysic dysplasia [2], and familial thoracic aortic aneurysms [3]. The estimated prevalence of MFS is approximately 1/5,000 worldwide [2]. Ectopia lentis is observed in approximately 80% of patients with MFS. MFS and EL are the two most common fibrillinopathies, and they share many clinical and genetic features. Mutations in TGFBR2 (ID 7048; OMIM 190182) and FBN1 are known to cause MFS, while FBN1, LTBP2 (ID 4053; OMIM 602091), and ADAMTSL4 (ID 54507; OMIM 610113) have been associated with EL.
MFS is diagnosed according to the Ghent criteria, which describe pleiotropic manifestations cardinally including lens dislocation, proximal aortic aneurysm, and long-bone overgrowth [4]. Isolated EL or predominant EL with some skeletal features does not satisfy the Ghent criteria because of the absence of aortic dilatation or dissection. Several studies recently aimed at comparing the old Ghent criteria to the revised criteria by studying patients with a suspected MFS phenotype and a known FBN1 mutation. Up to 15% of cases received a different clinical diagnosis, such as EL according to the revised criteria [5]. Nevertheless, these patients may still develop classic MFS later in life. Moreover, information about the genotype is essential for diagnosing or excluding MFS [6], which thus emphasizes the importance of molecular genetic analysis in patients with a suspected MFS phenotype.
We collected data from a large family (Figure 1) in southern China whose members have a history of lens dislocation and varicose great saphenous vein (GSV). Significant phenotypic variations were observed between the affected members in the family. Recently, one member (III:8) of the family showed aortic dissection and underwent surgery. In addition, one novel mutation, c.3928G>A, p.1310G>S in exon 31 of FBN1 was identified as cosegregating well in the family. Thus, we rediagnosed the family with MFS. Our findings demonstrated that FBN1-associated fibrillinopathies are a group of diseases with dynamic changes in phenotype. Although these patients did not initially meet the Ghent criteria, the patients may develop classic MFS later in life. Our results expanded the mutation spectrum of FBN1 and enhanced our knowledge of the phenotypic variability caused by different FBN1 mutations.
Figure 1.

Pedigree of the extended family. Squares and circles indicate men and women, respectively. The solid symbols represent affected members. The slashed symbols denote deceased subjects. The patient above the arrow is the proband. Twenty-two DNA samples were collected from this family, including 12 affected subjects and ten unaffected subjects.
Methods
Clinical evaluations and sample collection
The family has lived in Chongming County, Shanghai, China, for several generations. Written informed consent was obtained. A statement indicating that the study was approved by the institutional Review Board (Peking Union Medical College Hospital, Peking Union Medical College, F-210) and that the study adhered to the tenets of the Declaration of Helsinki. All 53 members across five generations belong to the Han ethnic group (Figure 1). There were 16 affected members; seven were male, and nine were female. 16 patients were recruited from Chongming, Shanghai, China, including 7 males and 9 females. Evaluations of the intelligence of the family showed normal intelligence quotients. The family had no signs of consanguineous marriage. Evaluations of the intelligence of the family showed normal intelligence quotients. An affected adult has ocular signs and systemic signs. Ocular signs include ectopia lentis or myopia plus retinal detachment. Systemic signs include varicose great saphenous vein or arachnodactyly, with hyperextensible skin. The inheritance mode is autosomal dominant, based on the pedigree. Twenty-two members (12 affected subjects and ten unaffected subjects) in the pedigree consented, and were therefore included in the clinical evaluations (Table 1, Figure 2, and Figure 3) and molecular tests. A statement indicating that the study was approved by the Institutional Review Board (Peking Union Medical College Hospital, Peking Union Medical College, F-210).
Table1. Summary of clinical information of subjects with FBN1 mutation.
| Patient ID | III:2 | III:7 | III:8 | III:9 | III:12 | III:14 | IV:6 | IV:7 | IV:12 | IV:13 | IV:14 | V:4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age/Gender | 61/F | 59/M | 55/M | 42/F | 59/F | 67/F | 44/F | 41/M | 31/F | 28/F | 44/M | 8/F |
| Ocular system | ||||||||||||
| Ectopia lentis | + | + | + | + | + | + | + | + | - | - | + | - |
| Myopia | + | + | + | - | + | + | + | + | + | + | - | - |
| Strabismus | - | - | - | - | - | - | - | - | - | - | - | - |
| Glaucoma | - | - | - | + | - | - | - | - | - | + | - | - |
| Retinal detachment | - | + | - | - | - | - | - | - | + | + | - | - |
| Cardiovascular system | ||||||||||||
| Aortic root dimentsion (cm) | 2.2 | 2.4 | 2.3 | NA | NA | 2.5 | 2.1 | NA | 2.5 | 2.2 | 2.2 | NA |
| Mirral valve prolapse | - | - | - | - | - | - | - | - | - | - | - | - |
| Aortic aneurysm | - | - | + | - | - | - | - | - | - | - | - | - |
| Skeletal system | - | - | - | - | - | - | - | - | - | - | - | - |
| Height (cm) | 160 | 170 | 180 | 162 | 158 | 180 | 168 | 176 | 168 | 166 | 180 | 120 |
| Scoliosis | - | - | - | - | - | - | - | - | - | - | - | - |
| Arachnodactyly | - | - | + | - | - | - | - | + | + | - | - | - |
| Joint hypermobility | - | - | - | - | - | - | - | - | - | - | - | - |
| Pectus excavatum/ carinatum | - | - | - | - | - | - | - | - | - | - | - | - |
| Great Saphenous Vein | ||||||||||||
| Symptoms and sign | + | + | + | + | + | + | + | + | - | + | + | - |
| GSV surgery | - | + | - | - | - | + | - | - | - | - | - | - |
| Other manifestations | ||||||||||||
| Hyperextensible skin | + | + | + | - | + | + | + | + | + | + | + | - |
NA: not available
Figure 2.
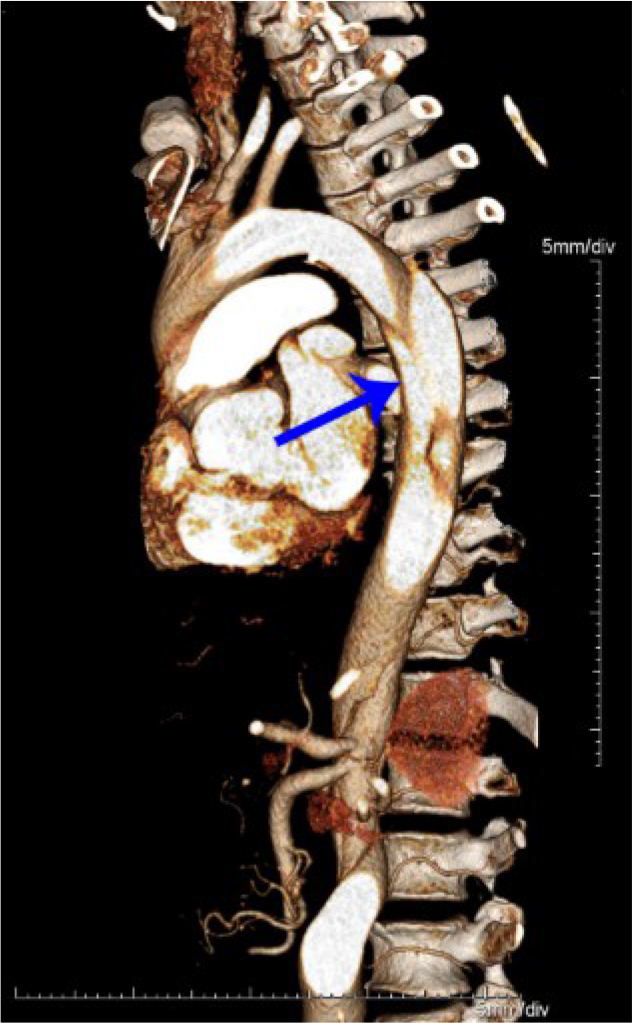
Computed tomography angiography of the aortic dissection of patient III:8. The descending aorta to the right common iliac artery with the main tear at the descending aorta highlighted by the arrow on computed tomography angiography.
Figure 3.
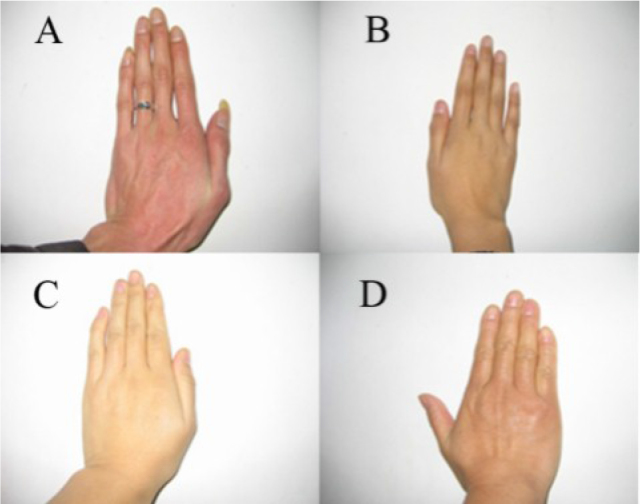
Photos of the patients’ hands. The long fingers of affected subjects are shown in A and B. A: Patient IV:7’s hand (adult) is 20.4 cm long. B: Patient V:4’s hand (12 years old) is 17.5 cm long. Family members’ hands that are normal length are shown in C and D. C: Patient IV:3’s hand (adult) is 17.5 cm long. D: Patient IV:2’s hand (adult) is 17 cm long.
Linkage analysis
Genomic DNA was extracted from peripheral blood leukocytes using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). A genome-wide scan for linkage was performed with an Illumina Infinium Human Linkage-12 panel containing 6090 single nucleotide polymorphism (SNP) markers (Illumina Inc., San Diego, CA). Four steps are used in the linkage method: 1) Establish a pedigree, 2) estimate recombination frequency, 3) calculate the LOD score, and 4) define a candidate region. Multipoint parametric and nonparametric linkage analysis was performed using MERLIN v 2.0.1. The parametric analysis was conducted assuming an autosomal dominant mode of inheritance with 0.99 penetrance. A disease allele frequency of 0.001 was used in the analysis. The genetic positions of the markers were provided by Illumina (City, CA). A LOD score greater than 3.0 was considered evidence of linkage.
Mutation screening
The best candidate gene in the linkage region is FBN1. PCR was performed to amplify the FBN1 exonic regions, as previously prescribed [7]. The final volume of 50 μl contained 40 ng genomic DNA, 10 pmol of each primer and 25 μl 2×Taq PCR master mix (Biomed Technologies, Beijing, China). PCRs were performed with denaturing at 94 °C for 5 min, followed by 30 cycles of a denaturing step at 94 °C, an annealing step at 56 °C-60 °C (depending on primer characteristics), each for 30 s and an extension step at 72 °C, for 60 s. A final extension step at 72 °C was performed for 7 min. Purified amplicons were sequenced by using an ABI 3730 Genetic Analyzer (ABI, Foster City, CA). Segregation tests were performed by directly sequencing other family members. One hundred normal Chinese controls were screened to verify the polymorphism. The FBN1 cDNA sequence (NC_000015.9) was used as the reference. TGFBR2 was also sequenced because of the venous changes.
Molecular modeling
The amino acid sequence of human fibrillin-1 was retrieved from the UniProtKB database. The domain structure of fibrillin-1 was determined using the Simple Modular Architecture Research Tool (SMART) [8,9]. The multiple sequence alignment of epidermal growth factor (EGF)-like Ca-binding motifs was performed using the algorithm for rapid automatic detection and alignment of repeats (RADAR). The initial mutant variant structures of the Ca-EGF motifs 20–22 (residues 1238–1362) were built with an automated protein-homology modeling server [10,11], using the protein structure of EGF domains 1,2,5 of human EMR2 as the structural template (PDB: 2bo2, subunit A). The mutant variant structure Gly1310Ser was generated and refined, and 3 ns was equilibrated in water at 37 °C, using the molecular visualization, modeling, and dynamics program YASARA [12,13].
Results
Clinical characteristics
The proband of the pedigree (III:8) was diagnosed by aortic dissection, and surgery was performed (Figure 1 and Figure 2). Other clinical features were considered, including EL and GSV, which led to our diagnosis of the family as having MFS [14]. However, the color sonography examination indicated that the diameter of the ascending aorta diameter was 2.1 cm, the diameter of the aortic root was 2.3 cm, and other measured values were within normal range. No abnormal findings were observed in the volumetric measurements of the atrium and the ventricle, which were conducted 3 years ago. The patients did not describe symptoms such as searing pain in the chest or back. Two years previously, IV:7 (41 years old) died suddenly from cerebral hemorrhage, which made us review his history. After doing so, we speculated that he may have had cerebral vascular disease. All other family members who harbor the mutation were found to have EL (over 28 years old) and GSV (since adolescence), but with no cardiovascular malformations. The family members’ height and weight fell within the normal adult range. Only three members (III:8, III:14, and IV:14) of the family have long bones; the others are within normal range. The heights of two affected subjects are within the lower limit of the normal range. The clinical information of the subjects with the FBN1 mutation is summarized in Table 1.
Molecular findings
A linkage peak was observed on chromosome 15; the highest parametric LOD score was 3.6, and the maximum non-parametric linkage (NPL) score was 29.1 (p<0.00001) between rs2004175 and rs877228 (Figure 4). The minimal shared haplotype was between rs1565863 and rs877228 (Figure 5). FBN1 was considered the candidate gene residing in the region. One novel missense mutation, c.3928G>A, p.1310G>S in exon 31 of FBN1 was identified (Figure 6). The missense mutation was present as a heterozygous allele in all affected members, but absent in normal members and 100 unrelated healthy controls. We applied molecular modeling to show the effect of this mutation on the fibrillin-1 structure. The mutation significantly distorts calcium coordination, decreases the binding of the calcium ion in that motif, and locally affects the calcium-binding epidermal growth factor (cbEGF) interface, which depends on Ca binding. No mutation was found in the TGFBR2 gene.
Figure 4.

Whole genome linkage analysis results for the family with ectopia lentis and varicose great saphenous vein. The x-axis shows each chromosome, and the y-axis shows the logarithm of the odds (LOD) score obtained. The linkage peak was observed on chromosome 15 with a parametric LOD score of 3.6 and a maximum non-parametric linkage (NPL) score of 29.1.
Figure 5.

The haplotype of this family. Genotypes of informative markers are shown for each individual. The chromosomal location (in Mbp) of each marker is listed along with the corresponding dbSNP reference number.
Figure 6.

Sequence analysis of the FBN1 gene. A: A novel heterozygous missense mutation c.3928G>A, p.1301Gly>Ser in exon 31 was identified in all affected family members. B: Unaffected subjects did not carry this change.
Molecular modeling
The protein structure of EGF motifs 20–22 was modeled using E-value = 1.7E-25, and the 31% sequence identity to EGF domains of human EMR2 was used as a structural template. In wild-type FBN1, the position of the Ca2+ ion is coordinated by three side-chain oxygens of Asn1341, Asp1322, and Glu1325. Mutation p.G1310S introduces a polar serine in a Ca-binding cavity (Figure 7).
Figure 7.

The atomic structure of the Ca-binding site and the functional consequences of the G1310S mutant variant are shown. Ca-binding site structures for the wild-type and the mutant variant are shown in beige and light blue, respectively. The Ca2+ ion is shown in green. The oxygen and hydrogen atoms are represented by red and white spheres, respectively.
Discussion
The phenotype of this family is unique, as we mentioned in a case report [14]. Law et al. reported disposition to aortic dilatation and varicose veins with affected members in a British family with a TGFBR2 mutation but without ocular lens dislocation [15]. We sequenced this gene and found no disease-causing variant. The results of the linkage study and mutation analysis confirmed the first association between FBN1 and this special MFS phenotype in EL and GSV of this Chinese family.
FBN1, which encodes a 350-kDa protein, is composed of three types of repeated modules. The EGF-like modules contain six highly conserved cysteine residues, which form disulphide bonds with each other and are critical in the stabilized folding of the domain [3]. FBN1 has 47 EGF-like modules, 43 of which contain a calcium-binding (cb), consensus sequence known as cbEGF-like modules [16]. FBN1 also contains the transforming growth factor β1-binding protein-like module (TGF; β1-BP-like module) [17]. This module has eight cysteine residues that form four disulfide bonds. The third type of module is a hybrid module [18]. Currently, more than 1,200 mutations in FBN1 have been identified. Among them, missense mutations account for the largest proportion (60%) [19].
Missense mutations in FBN1 may lead to diversiform clinical manifestation. Recently, a novel heterozygous missense mutation c.T2368A, p.C790S in FBN1 was observed in a large Pakistani family with autosomal dominant MFS [20]. All patients in the family, including four young children, developed ectopia lentis, myopia, and glaucoma, but with no defects in the cardiovascular system. A p.G214S mutation in exon 7 was reported in a Chinese family [21]. However, the prevalence of cardiovascular manifestations is not high in this family. In another study of a Taiwanese cohort, phenotype analysis revealed that only one third (47 of 157) of the families met the Ghent criteria [22]. Moreover, five cases with FBN1 mutations did not fulfill the Ghent criteria. Comparison with our family showed that only one member of the pedigree fulfilled the Ghent criteria. The predominant signs of this family are EL and GSV [14]. The EL sign appeared in the late 20s and GSV started in the second decade. Genetic analysis revealed that the novel missense mutation (c.3928G>A, p.1310G>S) in FBN1 is the underlying defect (Figure 4).
The majority of these missense mutations affects one of the cbEGF domains and often involves one of the six highly conserved cysteine residues within the cbEGF domains. Mutations leading to severe phenotypes are mostly found clustered in exons 24–32 [23–25], which encodes a central stretch of 12 cbEGF repeats. This stretch is important in the formation of a rigid rod-like structure, which might be involved in the formation of the microfibril assembly [26]. The novel missense mutation reported in this paper (c.3928G>A, p.1310G>S) affects a highly conserved residue of fibrillin-1. The SIFT score is 0.06, and the Polyphen2_HDIV_score is 0.986. The corresponding prediction is probably damaging. We applied molecular modeling to show the effect of this mutation at the fibrillin-1 structure (Figure 6). Two events occurred in our molecular dynamics simulations. First, a hydrogen bond was formed between the HG hydrogen of Ser1310 and the OE1 oxygen of Glu1325 (yellow, 2.75 Å), moving the side chain of Glu1325 and excluding it from the calcium ion coordination. Second, Asp1322 moved in a direction out of the Ca-binding cavity. These two events will significantly distort the calcium coordination, decrease the binding of the calcium ion in that motif, and affect the local cbEGF interface that depends on Ca-binding [27]. Therefore, we speculate that the mutation may change the local rigid rod-like structure of FBN1 and result in the phenotype observed in this Chinese family.
The phenotypes of fibrillinopathies are not only highly heterogeneous but also change with age. Follow-up revealed new manifestations: aortic dissection of the proband and cerebral hemorrhage in patient IV:7. Our findings indicated that clinical follow-up is critical in monitoring potentially life-threatening risks, regardless of whether cardiovascular abnormalities were found initially or not. Another important point raised by the results of this study is that molecular diagnosis should become an integral part of managing MFS. Our patients displayed clinical signs of MFS and GSV, which made an accurate diagnosis very challenging. Thus, a molecular diagnosis would allow us to perform a more accurate clinical diagnosis and genetic counseling. Adhering to the process “bedside-bench-bedside” would help clinicians provide better patient care.
In summary, a novel FBN1 mutation, p.1310G>S, was first reported to cause Marfan syndrome in a large family with lens dislocation and varicose great saphenous vein. Our results expanded the mutation spectrum in FBN1 and enhanced our knowledge of genotype-phenotype correlations underlying FBN1 mutations.
Acknowledgments
We are very grateful to all participating patients and their relatives. The research leading to these results received funding from the Ministry of Human Resource and Social Security of the People’s Republic of China (2009) and the Foundation Fighting Blindness USA (CD-CL-0808–0470-PUMCH) to R.S. Author Contributions The authors declare no conflict of interest. Ruifang Sui conceived the project, designed the experiments and prepared the manuscript. Fei Xu performed the genetic experiments. Qing Fu analyzed the data and wrote the paper. Peng Liu was involved in clinical evaluations of patients and prepared the Figures and Tables. Feng Wang and Hui Wang were involved in the linkage analysis. Wei Shen and Lin Liu were involved in ophthalmic examinations. Yuri V. Sergeev did the structural modeling.
References
- 1.Lönnqvist L, Child A, Kainulainen K, Davidson R, Puhakka L, Peltonen L. A novel mutation of the fibrillin gene causing ectopia lentis. Genomics. 1994;19:573–6. doi: 10.1006/geno.1994.1110. [DOI] [PubMed] [Google Scholar]
- 2.Hoffjan S. Genetic dissection of marfan syndrome and related connective tissue disorders: an update 2012. Mol Syndromol. 2012;3:47–58. doi: 10.1159/000339441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Francke U, Berg MA, Tynan K, Brenn T, Liu W, Aoyama T, Gasner C, Miller DC, Furthmayr HA. Gly1127Ser mutation in an EGF-like domain of the fibrillin-1 gene is a risk factor for ascending aortic aneurysm and dissection. Am J Hum Genet. 1995;56:1287–96. [PMC free article] [PubMed] [Google Scholar]
- 4.De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–26. doi: 10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 5.Faivre L, Collod-Beroud G, Ades L, Arbustini E, Child A, Callewaert BL, Loeys B, Binquet C, Gautier E, Mayer K, Arslan-Kirchner M, Grasso M, Beroud C, Hamroun D, Bonithon-Kopp C, Plauchu H, Robinson PN, De Backer J, Coucke P, Francke U, Bouchot O, Wolf JE, Stheneur C, Hanna N, Detaint D, De Paepe A, Boileau C, Jondeau G. The new Ghent criteria for Marfan syndrome: what do they change. Clin Genet. 2012;81:433–42. doi: 10.1111/j.1399-0004.2011.01703.x. [DOI] [PubMed] [Google Scholar]
- 6.Sheikhzadeh S, Kade C, Keyser B, Stuhrmann M, Arslan-Kirchner M, Rybczynski M, Bernhardt AM, Habermann CR, Hillebrand M, Mir T, Robinson PN, Berger J, Detter C, Blankenberg S, Schmidtke J. von KY. Analysis of phenotype and genotype information for the diagnosis of Marfan syndrome. Clin Genet. 2012;82:240–7. doi: 10.1111/j.1399-0004.2011.01771.x. [DOI] [PubMed] [Google Scholar]
- 7.Sui RF, Wei HB, Zhao JL, Hu SY, Wang B, Huang SZ, Dong M. Novel mutation of fibrillin 1 gene cause ectopia lentis in a Chinese family. Zhonghua Yan Ke Za Zhi. 2004;40:828–31. [PubMed] [Google Scholar]
- 8.Ponting CP, Schultz J, Milpetz F, Bork P. SMART: identification and annotation of domains from signalling and extracellular protein sequences. Nucleic Acids Res. 1999;27:229–32. doi: 10.1093/nar/27.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–4. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnold K, Kiefer F, Kopp J, Battey JN, Podvinec M, Westbrook JD, Berman HM, Bordoli L, Schwede T. The Protein Model Portal. J Struct Funct Genomics. 2009;10:1–8. doi: 10.1007/s10969-008-9048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–92. doi: 10.1093/nar/gkn750. PMID:18931379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krieger E, Nabuurs SB, Vriend G. Homology modeling. Methods Biochem Anal. 2003;44:509–23. doi: 10.1002/0471721204.ch25. [DOI] [PubMed] [Google Scholar]
- 13.Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77(Suppl 9):114–22. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen W, Fu Q, Sui R, Wu J, Liu L. Unique phenotype in a Chinese family pedigree: ectopia lentis with varicose great saphenous vein. Eye (Lond) 2010;24:1614–7. doi: 10.1038/eye.2010.82. [DOI] [PubMed] [Google Scholar]
- 15.Law C, Bunyan D, Castle B, Day L, Simpson I, Westwood G, Keeton B. Clinical features in a family with an R460H mutation in transforming growth factor beta receptor 2 gene. J Med Genet. 2006;43:908–16. doi: 10.1136/jmg.2006.042176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khau Van Kien P, Baux D, Pallares-Ruiz N, Baudoin C, Plancke A, Chassaing N, Collignon P, Drouin-Garraud V, Hovnanian A, Martin-Coignard D, Collod-Beroud G, Beroud C, Roux AF, Claustres M. Missense mutations of conserved glycine residues in fibrillin-1 highlight a potential subtype of cb-EGF-like domains. Hum Mutat. 2010;31:E1021–42. doi: 10.1002/humu.21131. [DOI] [PubMed] [Google Scholar]
- 17.Meng B, Li H, Yang T, Huang S, Sun X, Yuan H. Identification of a novel FBN1 gene mutation in a Chinese family with Marfan syndrome. Mol Vis. 2011;17:2421–7. [PMC free article] [PubMed] [Google Scholar]
- 18.Collod-Béroud G, Boileau C. Marfan syndrome in the third Millennium. Eur J Hum Genet. 2002;10:673–81. doi: 10.1038/sj.ejhg.5200876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson PN, Booms P, Katzke S, Ladewig M, Neumann L, Palz M, Pregla R, Tiecke F, Rosenberg T. Mutations of FBN1 and genotype-phenotype correlations in Marfan syndrome and related fibrillinopathies. Hum Mutat. 2002;20:153–61. doi: 10.1002/humu.10113. [DOI] [PubMed] [Google Scholar]
- 20.Micheal S, Khan MI, Akhtar F, Weiss MM, Islam F, Ali M, Qamar R, Maugeri A, den Hollander AI. Identification of a novel FBN1 gene mutation in a large Pakistani family with Marfan syndrome. Mol Vis. 2012;18:1918–26. [PMC free article] [PubMed] [Google Scholar]
- 21.Dong J, Bu J, Du W, Li Y, Jia Y, Li J, Meng X, Yuan M, Peng X, Zhou A, Wang L. A new novel mutation in FBN1 causes autosomal dominant Marfan syndrome in a Chinese family. Mol Vis. 2012;18:81–6. [PMC free article] [PubMed] [Google Scholar]
- 22.Hung CC, Lin SY, Lee CN, Cheng HY, Lin SP, Chen MR, Chen CP, Chang CH, Lin CY, Yu CC, Chiu HH, Cheng WF, Ho HN, Niu DM, Su YN. Mutation spectrum of the fibrillin-1 (FBN1) gene in Taiwanese patients withMarfan syndrome. Ann Hum Genet. 2009;73:559–67. doi: 10.1111/j.1469-1809.2009.00545.x. [DOI] [PubMed] [Google Scholar]
- 23.Stheneur C, Faivre L, Collod-Beroud G, Gautier E, Binquet C, Bonithon-Kopp C, Claustres M, Child AH, Arbustini E, Ades LC, Francke U, Mayer K, Arslan-Kirchner M, De Paepe A, Chevallier B, Bonnet D, Jondeau G, Boileau C. Prognosis factors in probands with an FBN1 mutation diagnosed before the age of 1 year. Pediatr Res. 2011;69:265–70. doi: 10.1203/PDR.0b013e3182097219. [DOI] [PubMed] [Google Scholar]
- 24.Loeys B, Nuytinck L, Delvaux I, De Bie S, De Paepe A. Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med. 2001;161:2447–54. doi: 10.1001/archinte.161.20.2447. [DOI] [PubMed] [Google Scholar]
- 25.Kainulainen K, Karttunen L, Puhakka L, Sakai L, Peltonen L. Mutations in the fibrillin gene responsible for dominant ectopia lentis and neonatal Marfan syndrome. Nat Genet. 1994;6:64–9. doi: 10.1038/ng0194-64. [DOI] [PubMed] [Google Scholar]
- 26.Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Marziliano N, Dietz HC, Halliday D, Beroud C, Bonithon-Kopp C, Claustres M, Muti C, Plauchu H, Robinson PN, Ades LC, Biggin A, Benetts B, Brett M, Holman KJ, De Backer J, Coucke P, Francke U, De Paepe A, Jondeau G, Boileau C. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–66. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen SA, Iqbal S, Lowe ED, Redfield C, Handford PA. Structure and interdomain interactions of a hybrid domain: a disulphide-rich module of the fibrillin/LTBP superfamily of matrix proteins. Structure. 2009;17:759–68. doi: 10.1016/j.str.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]