

Abstract
Raloxifene is a 2nd-generation selective estrogen receptor modulator used for the prevention and treatment of osteoporosis and the prevention of breast cancer in postmenopausal women. Raloxifene is extensively metabolized by glucuronidation to form raloxifene-6-glucuronide (ral-6-Gluc) and raloxifene-4′-glucuronide (ral-4′-Gluc). The goal of the present study was to determine whether functional polymorphisms in active UGTs could play a role in altered raloxifene glucuronidation in vivo. Using homogenates from HEK293 UGT-overexpressing cell lines, raloxifene was shown to be glucuronidated primarily by the hepatic UGTs 1A1 and 1A9 and the extra-hepatic UGTs 1A8 and 1A10; no detectable raloxifene glucuronidation activity was found for UGT2B enzymes. Functional UGT1A1 transcriptional promoter genotypes were significantly (ptrend=0.005) associated with ral-6-Gluc formation in human liver microsomes, and, consistent with the decreased raloxifene glucuronidation activities observed in vitro with cell line over-expressing UGT1A8 variants, the UGT1A8*2 variant was significantly (p=0.023) correlated with total raloxifene glucuronide formation in human jejunum homogenates. While ral-4′-Gluc exhibited 1/100th the anti-estrogenic activity of raloxifene itself as measured by binding to the estrogen receptor, raloxifene glucuronides comprised ∼99% of the circulating raloxifene dose in raloxifene-treated subjects, with ral-4′-Gluc comprising ∼70% of raloxifene glucuronides. Plasma ral-6-Gluc (ptrend=0.0025), ral-4′-Gluc (ptrend=0.001), and total raloxifene glucuronides (ptrend=0.001) were increased in raloxifene-treated subjects who were predicted slow metabolizers [UGT1A8 (*1/*3)] vs intermediate metabolizers [UGT1A8 (*1/*1) or UGT1A8 (*1/*2)] vs fast metabolizers [UGT1A8 (*2/*2). These data suggest that raloxifene metabolism may be dependent on UGT1A8 genotype and that UGT1A8 genotype may play an important role in overall response to raloxifene.
Introduction
There was an estimated 226,870 subjects who developed breast cancer and 39,510 deaths arising from this disease in the United States in 2012 (1). Much of the treatment for estrogen-receptor positive (ER+) breast cancer in postmenopausal women has targeted the blocking of the ER-binding activity of estrogen or reducing estrogen synthesis. Tamoxifen, a first-generation selective estrogen receptor modulator (SERM), has been used for treatment and chemoprevention of breast cancer for over 25 years, but its long term use is associated with rare but serious adverse effects (2). Raloxifene is a second-generation SERM approved by the Food and Drug Administration (FDA) for the treatment and prevention of osteoporosis and the chemoprevention of invasive breast cancer in postmenopausal women (3). Raloxifene acts as an estrogen agonist in bone and liver to increase bone mineral density and decreases LDL-cholesterol (4) and exhibits strong anti-estrogen effects in breast and uterus (5). Recent clinical trials showed that raloxifene significantly reduced the incidence of breast cancer in high-risk women although not as effectively as tamoxifen (38% vs. 50%, respectively) (6). However, in contrast to tamoxifen, raloxifene does not cause endometrial proliferation (7). Though not as serious as those associated with tamoxifen, adverse effects associated with raloxifene include hot flashes, vaginal dryness and leg cramps, and thromboembolic events such as deep venous thrombosis, pulmonary emboli and retinal vein thrombosis (8).
Up to 60% of the raloxifene dose is absorbed rapidly after oral administration (8-11), but there is <2% bioavailability due mainly to extensive in vivo glucuronidation (8-11). Raloxifene is primarily excreted in feces, with less than 0.2% excreted as unchanged raloxifene and less than 6% eliminated as glucuronide conjugates in urine. In addition to the hepatic metabolism of raloxifene, several studies suggest that the intestine may play an important role in raloxifene metabolism (12-16). Previous studies have demonstrated the presence of two raloxifene glucuronides in the plasma of women taking raloxifene, raloxifene-6-β-glucuronide (ral-6-Gluc) and raloxifene-4′-β-glucuronide (ral-4′-Gluc), with a plasma ral-4′-Gluc:ral-6-Gluc ratio of ∼8:1. Unconjugated raloxifene comprises less than 1% in human plasma (9-11, 17).
Previous studies characterizing the family 1A UDP-glucuronosyltransferase (UGT) enzymes involved in the glucuronidation of raloxifene demonstrated that the hepatic UGTs 1A1 and 1A9 and the extra-hepatic UGTs 1A8 and 1A10 were active against raloxifene (14). A recent study suggested that the UGT1A1*28 allelic variant, which contains an A(TA)7TAA in the TATAA box of the UGT1A1 transcriptional promoter and is associated with decreased expression of the UGT1A1 gene (18), is associated with altered raloxifene pharmacokinetics (19). No studies have as yet been performed examining the role of genotypes in other active UGTs on raloxifene glucuronidation phenotype. The goal of the present study was to fully characterize the glucuronidating activity of individual UGT1A and UGT2B enzymes against raloxifene, and to compare the overall glucuronidating activity of variant active UGTs vs their wild type counterparts both in vitro and in vivo.
Materials and Methods
Chemicals and materials
Raloxifene, UDP-glucuronic acid (UDPGA), alamethicin, β-glucuronidase, β-actin and bovine serum albumin were purchased from Sigma-Aldrich (St. Louis, MO). Ral-6-Gluc, ral-4′-Gluc, raloxifene-d4, ral-6-Gluc-d4, and ral-4′-Gluc-d4 were purchased from Toronto Research Chemical (Toronto, ON, Canada). Dulbecco's modified Eagle's medium (DMEM), Dulbecco's phosphate-buffered saline (minus calcium chloride and magnesium chloride), fetal bovine serum, penicillin-streptomycin, and Geneticin (G-418) were purchased from Invitrogen (Carlsbad, CA). The BCA protein assay kit was purchased from Pierce Chemical (Rockford, IL).
Tissues and cell lines
A description of the normal human liver tissue specimens used for these studies and the methods used for liver microsomal preparation and protein quantification was provided previously (20). Normal jejunum tissues (n=46) were purchased from Sun Health Research Institute (Sun City, AZ) and were obtained from non-cancer subjects between 2.5 and 4 h post-mortem and flash-frozen at -70°C. Jejunum homogenates were prepared by tissue homogenization in Tris-buffered saline at 4°C and stored in 100 μL aliquot (10–20 mg protein/ml) at -80°C until use. Genomic DNA was extracted from liver and jejunum specimens using a Qiagen DNeasy Blood & Tissue extraction kit (Valencia, CA). All protocols involving the collection and analysis of tissue specimens were approved by the Institutional Review Board at Penn State University and were in accordance with assurances filed with and approved by the United States Department of Health and Human Services.
HEK293 cells stably transfected with and over-expressing individual wild-type and variant UGTs have been previously described (21-26). All of the UGT-over-expressing cell lines and homogenates used in these studies exhibited glucuronidation activity against known test substrates as previously described (21-24).
Plasma samples
Plasma samples were obtained from subjects entered into a clinical trial performed at Penn State University College of Medicine examining the combined effects of raloxifene on biomarkers of hormone independent breast cancer (27). All subjects were postmenopausal women with a breast density in excess of 25% and without a history of thromboembolic disorders and cardiovascular disease. Blood samples were collected from subjects within the two raloxifene-only treatment groups (30 or 60 mg daily doses) immediately prior to commencement of raloxifene treatment (time ‘0’) and at one or more time points (6, 12, 18 and 24 months) after the commencement of treatment. All subjects provided written consent and agreed to their tissues being used for genetic studies. Bloods were fractionated by centrifugation at 1200 g at 4°C for 5 min, and plasma and lymphocyte fractions were stored in 1 mL aliquots at -80°C until analysis or genomic DNA extraction as described above.
Glucuronidation assays
Glucuronidation activity assays were performed essentially as previously described (24) after an initial incubation of human liver microsomes (HLM; 15 μg), human jejunum homogenates (HJH; 10 μg), or homogenates from human UGT1A and UGT2B-over-expressing cells (2-100 μg). For glucuronidation activity rate assays, 2 μM raloxifene was utilized for HLM while 1 μM raloxifene was utilized for HJH, and assays were performed in duplicate for all specimens (n=105 HLM and n=46 HJH). For kinetic analysis, 0.0625-256 μM raloxifene were used for in vitro assays with UGT-over-expressing cell homogenates, three randomly-chosen HLM, and three randomly-chosen HJH, and was performed in triplicate in independent assays. All kinetic data for the analysis of UGT-overexpressing cell homogenates was analyzed after normalizing relative to UGT protein levels expressed in each of the overexpressing cell lines, performed by western blot analysis as described previously (24, 28).
Raloxifene glucuronidation was analyzed using a Waters ACQUITY ultra-pressure liquid chromatography-UV detector (UPLC/UV) system (Milford, MA) with a 1.7 μ ACQUITY UPLC BEH C18 analytical column (2.1 mm × 50 mm, Waters, Ireland) in series with a 0.2 μm Waters assay frit filter (2.1 mm, Waters, USA). The gradient elution conditions, using a flow rate of 0.5 ml/min, were as follows: starting with 5% acetonitrile and 95% buffer A (5 mM ammonium acetate, pH 5.0) for 1 min, a subsequent linear gradient to 100% acetonitrile over 5 min was performed and then maintained at 100% acetonitrile for 2 min. The wavelength for determination of raloxifene and its glucuronides was 274 nm. Raloxifene-glucuronides (ral-6-Gluc and ral-4′-Gluc) were confirmed by their stability in 1M NaOH and sensitivity to the treatment of β-glucuronidase. In addition, confirmation of raloxifene glucuronide formation was performed by loading up to 5 μL of incubation product onto an UPLC identical to that described above in tandem with a Waters TQD triple quadrupole MS system. By using a positive mode, the parent compound [M+H]+ peak and their corresponding glucuronide [M-Gluc.+H]+ peaks were characterized.
Determination of raloxifene metabolites in plasma
Stock solutions of raloxifene, ral-6-Gluc, ral-4′-Gluc and their deuterated internal standards were prepared in DMSO. Raloxifene, ral-6-Gluc and ral-4′-Gluc were combined into a standard stock solution and used to make a standard working solution from 25 ng/ml-25 μg/ml for raloxifene, and 100 ng/ml-100 μg/ml for ral-6-Gluc and ral-4′-Gluc. Deuterated internal standards were combined with final concentrations of 5 μg/ml for raloxifene-d4, 20 μg/ml for ral-6-Gluc-d4 and 20 μg/ml for ral-4′-Gluc-d4, and were kept at -20 °C before use.
Standard curves were constructed by plotting the ratio of analyte peak area to peak area of the corresponding internal standard versus analyte concentration for at least eight analyte concentrations. The standard working solution and deuterated internal standard were spiked into plasma from untreated women and mixed (125 μl), and 375 μl of the extraction solution (containing 49.9:49.9:0.2 methanol:acetonitrile:formic acid) was subsequently added to precipitate out proteins and extract raloxifene metabolite standards. After vortexing and subsequent centrifugation at 13,000 g for 20 min at 4°C, the supernatant was dried and the residue was reconstituted in 125 μl of reconstitution solution (50.0:49.9:0.1 acetonitrile:H2O:formic acid) to make a final concentration of 0.32-320 ng/ml for raloxifene, 1.28-1280 ng/ml for both ral-6-Gluc and ral-4′-Gluc, 60 ng/ml for raloxifene-d4, and 240 ng/ml for both ral-6-Gluc-d4 and ral-4′-Gluc-d4.
Due to the low concentration levels of raloxifene and its metabolites in the plasma from raloxifene-treated subjects, the plasma was preconcentrated 3.2-fold prior to loading onto the UPLC/MS/MS system. After spiking of deuterated internal standards into plasma (320 μl) from raloxifene-treated subjects and mixing, 960 μl of extraction solution was added to the plasma to precipitate out proteins and extract raloxifene metabolites. After centrifugation as described above, the supernantant was dried and reconstituted in 100 μl of reconstitution solution to keep analyte concentrations within the range of the calibration curve. The calculated concentrations from standard curves were divided by 3.2 to reflect the final raloxifene metabolites levels in plasma from raloxifene-treated subjects.
Calibration standards as well as the plasma sample extracts from each subject were analyzed by UPLC/MS/MS. Quantification of raloxifene, ral-6-Gluc and ral-4′-Gluc was performed using MRM of the transitions of m/z 474.2 → 112.2 for raloxifene, m/z 650.5 → 474.3 for ral-6-Gluc and ral-4′-Gluc, m/z 478.2 →116.2 for raloxifene-d4, and m/z 654.5 → 478.3 for ral-6-Gluc-d4 and ral-4′-Gluc-d4. The ral-6-Gluc and ral-4′-Gluc were distinguished by matching the retention time with the commercial standards in the chromatograms. The optimized MS conditions were: positive ionization mode, capillary voltage 3.0 kV, cone voltage 30 V, collision voltage 30 V, source temperature 150 °C and desolvation temperature 350 °C. Nitrogen was used as the desolvation and cone gas with the flow rate at 760 L/h and 50 L/h, respectively. Argon was used as the collision gas at flow rate of 0.1 L/h. The dwell time for each ion was 0.01 sec. All data were quantified by MassLynx™ NT 4.1 software with QuanLynx™ program (Waters Corp., Milford, MA, USA).
UGT genotyping
Genomic DNA from human liver, human jejunum and human lymphocytes was used to genotype the UGT1A1*28 allele, and the UGT1A8 codons 173 (Ala>Gly; rs1042597) and 277 (Cys>Tyr; rs17863762) SNPs. For the UGT1A1 TATAA box polymorphism genotype, DNA was PCR amplified as previously described (29) using sense and antisense primers: 5′-GAGTATGAAATTCCAGCCAGTTCAAC-3′ and 5′-TCCACTGGGATCAACAGTATCTT-3′ (corresponding to -224 to -198 and +107 to +85 relative to the UGT1A1 ATG translation start site, respectively), resulting in an amplicon of 331 bp with the TATAA box polymorphism near the middle of the amplicon. After running on a 1.0% agarose gel and extraction using a Qiagen gel extraction kit (Valencia, CA), purified PCR products were sequenced using an ABI Hitachi 3730XL DNA Analyzer, with sequencing confirmed using both the forward and reverse amplification primers described above. Sequencing results were confirmed by visual inspection of the TATAA box chromatogram peaks. The UGT1A1*28 allele was in Hardy-Weinberg equilibrium and indicated an allelic frequency of 35% in subjects from whom HLM samples were obtained and 34% in subjects taking raloxifene from whom plasma raloxifene metabolites were analyzed.
The UGT1A8 coding SNPs were genotyped by real-time PCR using ABI Taqman Drug Metabolism Genotyping Assays (C__11742072_10 for rs1042597 and C__34418788_20 for rs17863762) according to manufacturer's protocols. The three UGT1A8 alleles were in Hardy-Weinberg equilibrium with allelic frequencies of 76.2% for UGT1A8*1, 20.2% for UGT1A8*2, and 3.6% for UGT1A8*3 in subjects from whom HJH samples were obtained and 71.3%, 26.4% and 2.3%, respectively, in subjects taking raloxifene from whom plasma raloxifene metabolites were analyzed.
UGT mRNA expression in human jejunum
RNA was extracted from all 46 jejunum specimens using the Qiagen RNeasy Mini Kit (Valencia, CA) according to the manufacturer's protocol. After digestion with DNAse I digestion, RNA concentrations were determined using a Nanodrop ND-1000 spectrophotometer. RNA purity was assessed by absorbance ratios A260/A280 (>1.9) and A260/A230 (>1.8). RNA integrity was determined using an Agilent 2100 Bioanalyzer with Agilent RNA 6000 Nano chips, and all 46 jejunum samples used in this study had an RIN>4.0 with clearly visible 28S and 18S rRNA bands. Reverse transcription (RT) and real-time PCR was performed for five randomly-selected jejunum RNA specimens as previously described (30) to assess the relative expression levels of UGTs 1A1, 1A8, 1A9 and 1A10. Real-time PCR was carried out using a 25 ng RNA equivalent of cDNA, and expression levels were normalized to the expression of the GAPDH gene. Quadruplicate real-time PCRs were performed for each cDNA sample analyzed using a 10 μL final reaction volume according to manufacturer's protocols (assay IDs: UGT1A1, Hs02511055_s1; UGT1A8, Hs01592482_m1; UGT1A10, Hs02516990_s1; UGT1A9, Hs02516855_sH; GAPDH, Hs99999905_m1). Reactions were performed in a 384-well plate using the ABI 7900 HT Sequence Detection System under the following conditions: 1 cycle at 50°C for 2 min, 1 cycle at 95°C for 10 min, and 40 cycles of 95°C for 15 sec and 60°C for 1 min. Relative quantification (RQ) of UGT1A expression was calculated using the ΔΔCt method as previously described (30).
Estrogen receptor (ER)-binding assay
Competitive binding assays of raloxifene, ral-6-Gluc and ral-4′-Gluc with the estrogen receptor was performed essentially as previously described (31) by incubating the cytosolic fraction of MCF-7 cells (500 μg total protein) with 10−9 M 3H-labeled-estradiol (E2) and between 10−11 to 10−6 M of competitor (raloxifene, ral-6-Gluc, or ral-4′-Gluc). Data were expressed as the percentage of specific binding of 3H-E2 for the ER when competitor was not present. The relative binding affinity (RBA) for each test compound was calculated as IC50 which was normalized to that of E2.
Statistical analysis
The Student's t-test (2-sided) was used for comparing kinetic values of glucuronidation formation for UGT wild-type versus variant overexpressing cell lines, and for comparing raloxifene Gluc formation rates in HLM and HJH between two different genotypes. The one-way ANOVA trend test was used to examine the overall effect of UGT genotypes on raloxifene glucuronide formation in HLM and HJH while the Jonckheere-Terpstra trend test was used to examine the overall effect of UGT genotypes on raloxifene glucuronide levels in human plasma. Kinetic constants were determined using the Michaelis-Menten Model in Graphpad Prism 5 software (La Jolla, CA).
Results
Previous studies have demonstrated that HLM catalyze the formation of two glucuronides of raloxifene, raloxifene-6-Gluc and raloxifene-4′-Gluc (14, 32). Similar to that observed in previous studies (14, 32), two major peaks with retention times of 2.70 and 2.97 min were observed by UPLC-MS/MS in in vitro raloxifene glucuronidation assays with HLM (Figure 1A) and HJH (Figure 1B). The retention time of the two peaks was the same as that of purchased ral-6-Gluc (peak 1) and ral-4′-Gluc (peak 2) standards (Figure 1C), and were confirmed to be O-glucuronides of raloxifene by their insensitivity to alkali but sensitivity to β-glucuronidase treatment (results not shown). Using MS/MS daughter scan mode, the mass spectrum of both peaks demonstrated a [M+H+] peak at m/z 650 for raloxifene-O-glucuronide, a [M+H]+ peak at m/z 474 for raloxifene after loss of the glucuronide acid moiety (molecular weight = 176 g/mol) (Figure 1G), and a [M+H]+ peak at m/z 474 for raloxifene and a major daughter fragment at m/z 112.2 (Figure 1H).
Figure 1. UPLC-MS/MS analysis of raloxifene glucuronides.

Shown are MRM analysis of (i) raloxifene glucuronides formed by HLM (Panel A) and HJH (Panel B), and observed in the plasma of raloxifene-treated patients (Panel D), (ii) raloxifene in the plasma of raloxifene-treated patients (Panel E), and (iii) the purchased ral-6-Gluc/ral-4′-Gluc (Panel C) and raloxifene (Panel F) standards. UPLC-MS/MS of purchased standards was performed in a standard glucuronidation assay without a protein source (e.g., cell homogenate, HLM or HJH) added. Peak 1, ral-6-Gluc; peak 2, ral-4′-Gluc; peak 3, raloxifene. MS daughter scan spectrum for raloxifene glucuronide and raloxifene are shown in Panels G and H, respectively.
Ral-6-Gluc and ral-4′-Gluc were also observed in plasma samples from subjects treated with raloxifene (Figure 1D), exhibiting the same retention times as the plasma-spiked ral-6-Gluc-d4 and ral-4′-Gluc-d4 internal standards (results not shown), and the same retention times and MS/MS spectrum as the ral-6-Gluc and ral-4′-Gluc standards as well as the in vitro assays with HLM and HJH described above. A third peak detected in plasma from raloxifene-treated subjects exhibited a retention time of 3.61 min (Figure 1E) was the same as that observed for the raloxifene standard (peak 3; Figure 1F). Similar to previous studies (19, 32, 33), a ral-6,4′-diGluc was not observed in the plasma of subjects treated with raloxifene or in vitro with HLM or HJH.
UGTs 1A1, 1A3, 1A7, 1A8 and 1A9 catalyzed both ral-6-Gluc and ral-4′-Gluc formation while UGT1A10 specifically catalyzed the formation of ral-4′-Gluc (Table 1). Representative kinetic analysis curves for ral-6-Gluc and ral-4′-Gluc formation are shown for UGTs 1A1 and 1A8 against raloxifene in Figure 2. After normalizing for UGT1A protein expression as determined by western blot analysis (34), the order of ral-6-Gluc formation based on Vmax/KM was UGT 1A8>1A1>1A7≈1A9>1A3, while the order of ral-4′-Gluc formation based on Vmax/KM was UGT 1A10>1A8>1A9>1A1>1A7>1A3. In addition to UGTs 1A4 and 1A6, none of the UGT2B enzymes screened in this analysis exhibited detectable levels of raloxifene glucuronide formation.
Table 1. Kinetic analysis of the glucuronidation activity of UGTs against raloxifene.
| UGTa | raloxifene-6-Gluc | raloxifene-4′-Gluc | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Vmax pmol·min-1·μg-1 |
KM μM |
Vmax/KM μl·min-1·μg-1 |
Vmax pmol·min-1·μg-1 |
KM μM |
Vmax/KM μl·min-1·μg-1 |
|
| 1A1 | 19.7 ± 2.6 | 8.9 ± 1.9 | 2.2 ± 0.2 | 11.1 ± 1.8 | 11.9 ± 2.5 | 0.95 ± 0.05 |
| 1A3 | 0.41 ± 0.02 | 21.1 ± 1.4 | 0.019 ± 0.002 | 0.48 ± 0.04 | 3.1 ± 0.3 | 0.15 ± 0.02 |
| 1A7 | 1.9 ± 0.2 | 12.7 ± 2.9 | 0.16 ± 0.02 | 6.9 ± 0.4 | 21.6 ± 1.6 | 0.32 ± 0.03 |
| 1A8173Ala/277Cys | 7.0 ± 1.6 | 0.31 ± 0.05 | 22.9 ± 4.1 | 19.2 ± 5.3 | 2.4 ± 0.4 | 7.9 ± 1.0 |
| 1A8173Gly/277Cys | 6.1 ± 1.5 | 0.08 ± 0.04b | 79.9 ± 18.1c | 4.1 ± 1.1c | 0.76 ± 0.11b | 5.3 ± 0.9 |
| 1A8173Ala/277Tyr | No activity detected | |||||
| 1A9 | 3.4 ± 0.5 | 22.1 ± 6.1 | 0.16 ± 0.03 | 8.3 ± 0.9 | 4.5 ± 0.8 | 1.9 ± 0.1 |
| 1A10 | No activity detected | 16.1 ± 1.3 | 0.21 ± 0.03 | 75.8 ± 6.3 | ||
No raloxifene glucuronide formation was observed for homogenates from cells over-expressing UGTs 1A4, 1A6, 2B4, 2B7, 2B10, 2B11, 2B15, or 2B17.
A significant (p<0.005;
p<0.01) difference was observed for 1A8173Gly/277Cys- versus 1A8173Ala/277Cys–over-expressing cell homogenates.
Figure 2. Representative plots for raloxifene glucuronidation kinetics by individual UGTs.
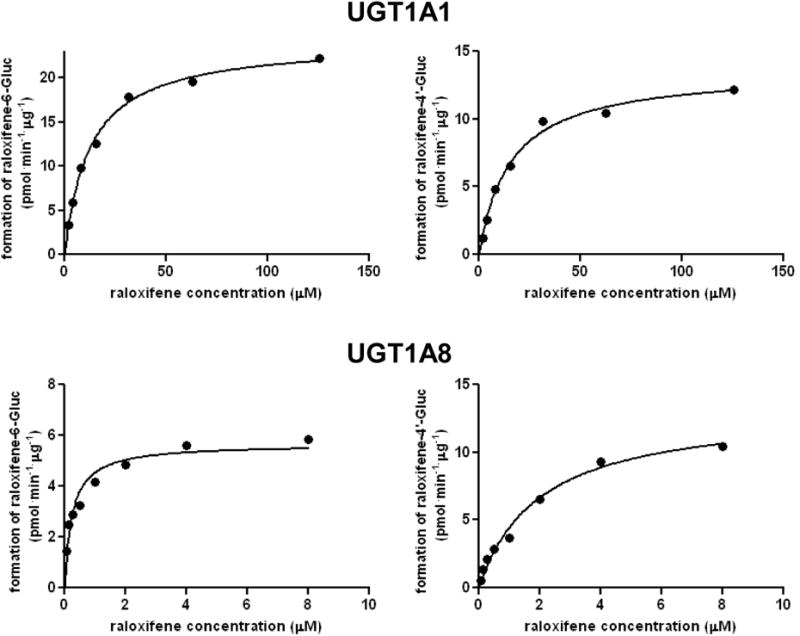
Shown are representative concentration curves for ral-Gluc formation by homogenates from UGT1A1- (Panel A) and UGT1A8- (Panel B) over-expressing cell lines. Left panels are the concentration curves for ral-6-Gluc formation, right panels are the concentration curves for ral-4′-Gluc formation.
Of the hepatic UGT enzymes active against raloxifene, UGT1A1 was the most active UGT for ral-6-Gluc formation and was the second-most active UGT for ral-4′-Gluc formation (Table 1). The UGT1A1*28 allele is a common variant (∼30% frequency in Caucasians) that encodes an A(TA)7TAA repeat in the TATAA-box of the UGT1A1 promoter region instead of the more common A(TA)6TAA repeat encoded by the wild-type UGT1A1*1 allele, leading to lower UGT1A1 expression (18) and a decreased glucuronidation phenotype against a variety of endogenous and exogenous compounds (29, 35, 36). To investigate the possible relationship between raloxifene glucuronidation phenotype and the UGT1A1 TATAA-box polymorphism, a series of 105 HLM were examined in vitro. A concentration of 2 μM raloxifene was chosen for HLM glucuronidation activity assays since this was close to the KM's of 8 μM for ral-6-Gluc formation and 1.5 μM for ral-4′-Gluc formation for three randomly-chosen HLM (data not shown). There was a 16- and 43-fold range in formation observed for ral-6-Gluc and ral-4′-Gluc, respectively, in the 105 HLM specimens. When stratifying the HLM by UGT1A1 genotype, ral-6-Gluc formation was significantly (ptrend=0.005) decreased with increasing numbers of the UGT1A1*28 allele (Figure 3A), with significant decreases observed in UGT1A1 (*1/*28) (26%; p=0.004; n=49) and (*28/*28) (39%; p=0.01; n=11) HLM as compared with HLM with the UGT1A1 (*1/*1) genotype (n=45). Significant differences in the levels of ral-4′-Gluc formation were not observed for HLM after stratification by UGT1A1 genotype (Figure 3A). No significant differences in KM for the formation of ral-4′-Gluc were observed in HLM with the UGT1A1 (*28/*28) or (*1/*28) genotypes versus UGT1A1 (*1/*1) HLM (three HLM examined per genotype group; results not shown).
Figure 3. Importance of UGT1A genotypes in raloxifene glucuronide formation in human tissues and in plasma samples from raloxifene-treated subjects.
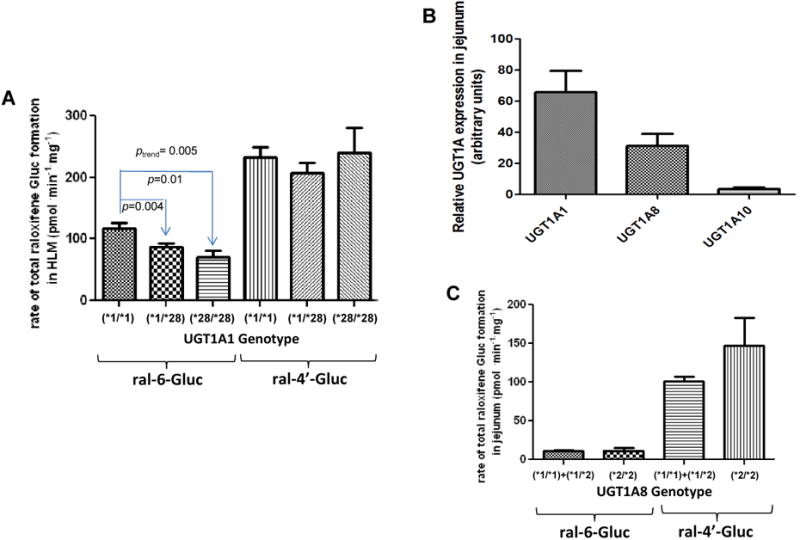
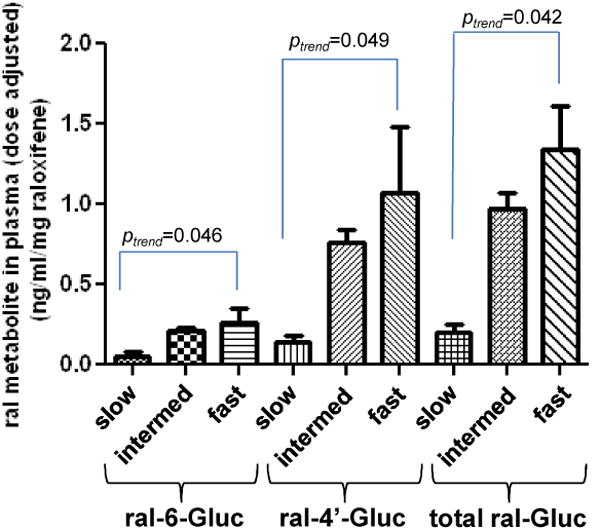
Glucuronidation activity assays were performed by incubation of raloxifene with HLM or HJH, and raloxifene glucuronides were analyzed by UPLC or UPLC/MS/MS as described in Materials and methods. The relative abundance of UGTs 1A1, 1A8 and 1A10 mRNAs in the jejunum was measured in 5 individual jejunum specimens by qPCR using the ΔΔCt method. (A) Rate of raloxifene glucuronide formation in HLM stratified by UGT1A1 genotype; (B) Relative expression levels of UGT1A mRNA in jejunum; (C) Rate of total raloxifene glucuronide formation in HJH stratified by UGT1A8 genotype; (D) Levels of raloxifene glucuronides in plasma stratified by UGT1A8 genotype. Subjects with the UGT1A8 (*1/*3) genotype were defined as slow raloxifene metabolizers (slow), subjects with either the UGT1A8 (*1/*1) or UGT1A8 (*1/*2) genotypes were defined as intermediate raloxifene metabolizers (intermed), and subjects with the UGT1A8 (*2/*2) genotype were defined as fast raloxifene metabolizers (fast). The Student's t-test was used to compare raloxifene Gluc formation in HLM from subjects with UGT1A1 (*1/*28) or (*28/*28) genotypes with the wild type UGT1A1 (*1/*1), and to compare total raloxifene glucuronide formation in HJH from subjects with UGT1A8 (*2/*2) genotype with UGT1A8 (*1/*1+*1/*2) genotypes. The one-way ANOVA trend test was used to examine the overall effect of UGT1A1 genotype on rate of ral-6-Gluc and ral-4′-Gluc formation in HLM. The Jonckheere-Terpstra trend test was used to examine the overall effect of UGT1A8 genotype on ral-6-Gluc, ral-4′-Gluc and total raloxifene glucuronide levels in plasma from women treated with raloxifene. Samples from subjects treated by 60 mg and 30 mg raloxifene daily were combined after plasma raloxifene and glucuronide levels were adjusted for raloxifene dose (mg) for each subject.
Of the extra-hepatic UGT enzymes shown to be active against raloxifene in vitro, UGTs 1A8 and 1A7 exhibit missense SNPs with prevalences of >3% in the population. Two coding region SNPs resulting in Ala to Gly at codon 173 (encoded by the UGT1A8*2 allele) and Cys to Tyr at codon 277 (encoded by the UGT1A8*3 allele) are present in the UGT1A8 gene (prevalences of 0.24 and 0.036, respectively, in Caucasians according to HapMap (37)). In an in vitro analysis of UGT1A8-over-expressing HEK293 cell homogenates, the UGT1A8173Gly/277Cys variant (encoded by the UGT1A8*2 allele) exhibited a significantly lower KM (p<0.005) and higher overall activity as determined by Vmax/KM (p<0.01) for ral-6-Gluc formation, as compared with wild type UGT1A8173Ala/277Cys (Table 1). While this variant also exhibited a significantly lower KM (p<0.005) for ral-4′-Gluc formation, a similar Vmax/KM was observed. No detectable glucuronidation activity was observed for the UGT1A8173Ala/277Tyr variant (encoded by the UGT1A8*3 allele) against raloxifene in vitro.
UGT1A7 exhibits four major alleles differing at residues 129, 131, and 208 (prevalences of 0.36 for UGT1A7*1, 0.26 for UGT1A7*2, 0.36 for UGT1A7*3, and 0.017 for UGT1A7*4 in Caucasians; (26, 38). In an in vitro analysis of UGT1A7-overexpressing HEK293 cell homogenates, no difference in glucuronidation activity was observed from UGT1A7 variants vs wild-type UGT1A7 against raloxifene (results not shown).
Previous reports have shown that small intestine is an important contributor to raloxifene glucuronidation and clearance in vivo (13, 14, 39) and that several UGTs including UGT1A8 are well-expressed in tissues of the digestive tract (40). However, previous studies examining the expression of intestinal UGTs have been relatively non-quantitative (41). Using real-time PCR, UGT1A1 was shown to be expressed at the highest levels in jejunum (Figure 3B). While UGT1A8 was expressed in jejunum at levels that were ∼2-fold lower than UGT1A1, UGT1A8 was expressed at levels that were 10-fold higher than UGT1A10. The mRNA expression level of UGT1A9 was not quantifiable in all five jejunum specimens analyzed.
As UGT1A8 was among the two most active UGTs against raloxifene and was shown to exhibit high levels of relative expression in human jejunum, a potential role for UGT1A8 genotypes on raloxifene glucuronidation phenotype was examined using a series of 46 HJH specimens. A concentration of 0.8 μM raloxifene was used for HJH glucuronidation activity assays, which approximated the KM of 0.75 μM for the major raloxifene metabolite in HJH, ral-4′-Gluc (as measured by kinetic analysis of 3 randomly chosen HJH; data not shown). There was a 64-fold range in formation observed for total raloxifene glucuronide formation in the 46 HJH specimens. When stratifying the HJH specimens by UGT1A8 codon 173 genotype, a significant (p=0.018) 1.8-fold increase in total raloxifene glucuronide formation was observed in HJH from subjects with the UGT1A8 (*2/*2) genotype (n=3) as compared to subjects with at least one UGT1A8*1 allele (n=40; Figure 3C). No difference in total raloxifene glucuronide formation was observed in HJH specimens that exhibited the UGT1A8 (*1/*3) genotype [n=3; there were no specimens with the UGT1A8 (*3/*3) genotype]. There was a near-significant (p=0.058) decrease in KM for ral-4′-Gluc formation in HJH with the UGT1A8 (*2/*2) genotype (0.46 ± 0.11 μmol/L) than UGT1A8 (*1/*1) HJH (0.75 ± 0.16 μmol/L); the KM of UGT1A8 (*1/*3) HJH (0.80 ± 0.39 μmol/L) was similar to that observed for HJH with the UGT1A8 (*1/*1) genotype.
The levels of raloxifene and its glucuronides were determined simultaneously in the plasma of subjects treated with either 30 or 60 mg/day raloxifene. Validation of the analytical method utilized for this analysis demonstrated high assay recovery of a range of plasma raloxifene/raloxifene glucuronide levels (88-110%). In the UPLC-MS/MS system utilized for this analysis, the quantification limit (signal/noise > 10) was 0.08 ng/ml for raloxifene, 0.625 ng/ml for ral-6-Gluc, and 0.78 ng/ml for ral-4′-Gluc. The intra-day and inter-day precision (CV), respectively, for raloxifene were 9.4% and 12.3% at 0.32 ng/ml, 4.1% and 6.5% at 2.5 ng/ml, and 0.6% and 1.4% at 160 ng/ml. For ral-6-Gluc, the CV were 8.0% and 9.6% at 1.28 ng/ml, 6.2% and 7.0% at 20 ng/ml, and 1.6% and 4.1% at 640 ng/ml. For ral-4′-Gluc the CV were 3.8% and 8.8% at 1.28 ng/ml, 2.7% and 4.0% at 20 ng/ml, 1.3% and 2.3% at 640 ng/ml. The levels of plasma raloxifene and its metabolites showed extensive variability between subjects at all blood draw times examined (6-, 12-, 18-, and 24-month blood draws following initiation of the trial). However, the range of raloxifene metabolite levels was similar for all blood draw times within individuals. The range of the plasma ral-4′-Gluc for subjects from the 30 mg daily treatment group was 2.7-95 ng/ml for the month 6 visit (n=35), 3.3-96 ng/ml for the month 12 visit (n=27), 5.1-66 ng/ml for the month 18 visit (n=14) and 13-89 ng/ml for the month 24 visit (n=3). A similar pattern was observed for ral-4′-Gluc for the 60 mg daily treatment group as well as ral-6-Gluc for both treatment groups (results not shown).
Using bloods drawn from the first available visiting time for each raloxifene-treated subject, there was extensive metabolism to ral-6-Gluc and ral-4′-Gluc, with unchanged raloxifene comprising only 0.98 and 0.88% of the total plasma raloxifene metabolite profile (raloxifene + ral-6-Gluc + ral-4′-Gluc) in subjects taking either 30 or 60 mg/day raloxifene, respectively (Table 2). The level of ral-4′-Gluc was ∼3-4 fold higher than that of ral-6-Gluc in both groups. There were 26- and 23-fold differences in the level of plasma ral-6-Gluc, 35-and 32-fold differences in the level of plasma ral-4′-Gluc, and 4.8- and 13-fold differences in the level of plasma raloxifene between subjects from the 30 and 60 mg/day treatment groups, respectively. While the mean ratios of both plasma ral-4′-Gluc/ral-6-Gluc and total plasma ral-Gluc/raloxifene were similar between groups, the levels of ral-6-Gluc, ral-4′-Gluc and raloxifene increased by 2.4-, 2.3-, and 2.1-fold, respectively, in subjects from the 60 mg/day group as compared to subjects from the 30 mg/day group. Similar raloxifene metabolite profiles were observed when examining bloods drawn from subjects at other time points or when using an average of all time points (results not shown).
Table 2. Raloxifene metabolite profiles in plasma from subjects treated with raloxifene.
| Treatment group | ral-4′-Gluc (ng/ml plasma) | ral-6-Gluc (ng/ml plasma) | ral-4′-Gluc/ral-6-Gluc | raloxifene (ng/ml plasma) | total ral-Gluc/raloxifene |
|---|---|---|---|---|---|
| Raloxifene 30 mg (n=39) | 22 (2.7-95) | 6.3 (0-26) | 3.3 | 0.28 (0.12-0.58) | 114 |
| Raloxifene 60 mg (n=42) | 50 (5.9-188) | 15 (2.6-59) | 3.8 | 0.58 (0.18-2.3) | 133 |
Based on the cell line data obtained in this study, it was predicted that individuals with a UGT1A8*3 allele would exhibit lower raloxifene glucuronidation capacities while subjects with a UGT1A8*2 allele would exhibit higher raloxifene glucuronidation capacities. When stratifying by UGT1A8 genotype, we found essentially no difference in the levels of plasma ral-6-Gluc, ral-4′-Gluc or raloxifene in subjects who were either UGT1A8 (*1/*2) vs UGT1A8 (*1/*1) (results not shown), so they were combined into one group. To best compare the levels of plasma raloxifene and its glucuronides in the two treatment groups, the ratios of ral-6-Gluc/raloxifene, ral-4′-Gluc/raloxifene, and total raloxifene glucuronide/raloxifene were examined in subjects after stratifying by UGT1A8 genotype. Since the two raloxifene glucuronides comprise >99% of the total plasma raloxifene metabolites in subjects talking raloxifene, other ratios including total ral-Gluc/(total ral-Gluc + raloxifene) were not informative. As shown in Table 3, increases in the ratios of ral-6-Gluc/raloxifene, ral-4′-Gluc/raloxifene, and total ral-Gluc/raloxifene were observed for plasma specimens from subjects who were UGT1A8 (*1/*3) vs subjects who were either UGT1A8 (*1/*1) or UGT1A8 (*1/*2). A similar pattern was observed when comparing plasma ral-6-Gluc/raloxifene, ral-4′-Gluc/raloxifene, and total ral-Gluc/raloxifene ratios from subjects who were either UGT1A8 (*1/*1) or UGT1A8 (*1/*2) vs subjects who were UGT1A8 (*2/*2). This trend was significant (ptrend=0.020 for ral-6-Gluc/raloxifene, ptrend=0.003 for ral-4′-Gluc/raloxifene and ptrend=0.005 for total ral-Gluc/raloxifene) when subjects from both treatment groups were combined. The levels of dose-adjusted plasma ral-6-Gluc, ral-4′-Gluc, and total ral-Gluc in the combined group were significantly (ptrend=0.0025, 0.001, and 0.001, respectively) increased in predicted slow metabolizers [UGT1A8 (*1/*3)] vs intermediate metabolizers [UGT1A8 (*1/*1) or UGT1A8 (*1/*2)] vs fast metabolizers [UGT1A8 (*2/*2); Panel D, Figure 3). No difference in the levels of ral-6-Gluc or ral-4′-Gluc were observed in plasma from subjects with either the UGT1A1 (*1/*1) (n=34), UGT1A1 (*1/*28) (n=39) or UGT1A1 (*28/*28) (n=9) genotypes (results not shown). Unfortunately, there was insufficient power to examine combined UGT1A1/UGT1A8 genotypes versus plasma raloxifene metabolites in this study.
Table 3. Raloxifene metabolites in plasma stratified by UGT1A8 genotype.
| Group | Metabolite | UGT1A8 (*1/*3)a | UGT1A8 (*1/*1)+(*1/*2)a | UGT1A8(*2/*2)a |
|---|---|---|---|---|
| 60 mg raloxifene | ral-6-Gluc/raloxifene | 13 (1) | 31 ± 27 (36) | 43 ± 21 (5) |
| ral-4′-Gluc/raloxifene | 42 | 94 ± 74 | 146 ± 62 | |
| total ral-Gluc/raloxifine | 55 | 125 ± 96 | 189 ± 77 | |
| 30 mg raloxifeneb | ral-6-Gluc/raloxifene | 10 ± 14 (2) | 27 ± 24 (37) | |
| ral-4′-Gluc/raloxifene | 18 ± 9 | 93 ± 97 | ||
| total ral-Gluc/raloxifine | 27 ± 23 | 120 ± 120 | ||
| Combined treatment groups | ral-6-Gluc/raloxifenec | 11 ± 10 (3) | 29 ± 26 (73) | 43 ± 21 (5) |
| ral-4′-Gluc/raloxifened | 26 ± 15 | 93 ± 86 | 146 ± 62 | |
| total ral-Gluc/raloxifenee | 37 ± 23 | 123 ± 101 | 189 ± 77 |
Values are ng/ml. Numbers in parenthesis represent the number of subjects analyzed in each group.
There were no subjects with the UGT1A8 (*2/*2) genotype in this group.
ptrend=0.020,
ptrend=0.003;
ptrend=0.005.
The mechanism of raloxifene action for the prevention of breast cancer is to compete with estrogen for binding to the estrogen receptor to prevent the stimulation of proliferation of breast cancer cells. To examine the relative binding affinity to the estrogen receptor of the two raloxifene glucuronides versus raloxifene, cytosolic fractions of MCF-7 cells were used as an estrogen receptor source as previously described (31). The IC50 for raloxifene, ral-6-Gluc and ral-4′-Gluc was (4.0 ± 3.5) × 10-10 M, (2.9 ± 0.8) × 10-7 M, and (3.7 ± 1.9) × 10-8 M, respectively.
Discussion
While raloxifene does not undergo significant P450-dependent oxidation (10), it is extensively glucuronidated by first-pass metabolism. Similar to that observed in previous studies (14), several UGT1A enzymes were found to exhibit raloxifene glucuronidating activity in the current study, with UGTs 1A1 and 1A9 the most active hepatic UGTs, and the extra-hepatic UGTs 1A8 and 1A10 exhibiting the highest levels of activity of any UGT screened in this study. UGT1A8 exhibited the highest overall activity for ral-6-Gluc formation and the second-highest activity for ral-4′-Gluc formation, and UGT1A10 exhibited the lowest KM and highest overall activity for ral-4′-Gluc formation. The KM values reported in the current study are 25- and 19-fold lower for ral-6-Gluc and ral-4′-Gluc formation by UGT1A8, and 23-fold lower for ral-4′-Gluc formation by UGT1A10, compared to previous studies (14), discrepancies that are likely due to differences in assay conditions. The KM values for UGT1A1 against raloxifene were ∼10 μM for both ral-6-Gluc and ral-4′-Gluc formation in our study while the KM for UGT1A1 could not be determined in previous studies due to solubility limitations as indicated in that study (14). UGTs 1A3 and 1A7 were also shown to be active in the present study. While this previous study did not test the activity of UGT1A3, no glucuronidation activity was previously observed for UGT1A7. This is likely due to the fact that UGT overexpressing baculosomes, which have been found to exhibit significant differences in substrate specificities as compared to UGT-over-expressing human cell homogenates (42), were used in this previous study. While the present study is the first to examine UGT2B enzyme activities against raloxifene, none were found to be active.
Previous studies have demonstrated that the level of UGT1A1 in human liver is 2-fold higher than that of UGT1A9 (43). There was only a ∼2.5-fold difference in the KM's for ral-6-Gluc and ral-4′-Gluc formation between enzymes, suggesting that both UGTs 1A1 and 1A9 may be important in the hepatic glucuronidation of raloxifene. However, while UGT1A1 exhibits 2- and 10-fold higher levels of expression in jejunum than UGTs 1A8 and 1A10, respectively, it exhibits a ∼29-fold higher KM and a ∼10-fold lower Vmax/KM for ral-6-Gluc formation as compared to UGT1A8, and ∼5- and ∼57-fold higher KM, and ∼8- and 80-fold lower Vmax/KM, for ral-4′-Gluc formation activity as compared to UGTs 1A8 and 1A10, respectively, suggesting a lesser role for UGT1A1 in jejunum raloxifene glucuronidation activity. The barely detectable level of expression for UGT1A9 in the small intestine in the present study is consistent with that observed previously (44) and suggests a minimal role for UGT1A9 in raloxifene glucuronidation in this tissue. Given the very low activity of UGTs 1A3 and 1A7 against raloxifene, it is likely that these UGTs play only a marginal role in raloxifene glucuronidation in either liver or jejunum.
Previous studies have shown that the UGT1A1*28 allele is associated with altered glucuronidation activity against a variety of endogenous and exogenous substrates (29, 35, 36). The association observed between UGT1A1 genotype and HLM raloxifene glucuronidation in the present study is consistent with the likely importance of UGT1A1 in overall hepatic raloxifene glucuronidation activity. The fact that this was observed specifically for the formation of ral-6-Gluc is consistent with UGT1A1 cell homogenates exhibiting the highest overall activity of any hepatic UGT for this metabolite. The fact that this pattern was not observed for hepatic ral-4′-Gluc formation is consistent with UGT1A9 playing a more important role in the formation of this metabolite given the higher Vmax/KM exhibited by UGT1A9 vs UGT1A1 in vitro. The fact that no differences in raloxifene glucuronide KM's were observed in HLM stratified by UGT1A1 genotypes is consistent with UGTs 1A9 and 1A1 exhibiting similar KM's for both raloxifene metabolites in vitro. The fact that no association was observed between HJH raloxifene glucuronidation activities and UGT1A1 genotype is consistent with UGT1A1 playing a more minor role in jejunum raloxifene glucuronidation. As high-prevalence coding SNPs are not observed for UGT1A9 (45), a similar hepatic phenotype-genotype study was not performed for this enzyme.
The vast majority of circulating raloxifene in the plasma of subjects treated with raloxifene was in the form of a glucuronide conjugate, with unchanged raloxifene comprising approximately 1% of total plasma raloxifene in subjects treated with either 30 or 60 mg raloxifene/day. Considerable variation in raloxifene glucuronide levels were observed in plasma between individuals taking raloxifene. Previous studies focusing on the role of the UGT1A1*28 allele on raloxifene glucuronidation gave conflicting results (19, 46, 47). The fact that UGT1A1 genotype did not contribute to variation in plasma raloxifene glucuronide levels in vivo in the present study suggests that, despite it contributing to ral-6-Gluc formation variation in HLM, potential effects by the UGT1A1*28 allele were overcome in vivo by glucuronidation contributions of other UGTs in both liver (UGT1A9) and jejunum (UGTs 1A8 and 1A10).
In previous studies, the polymorphic variants of UGT1A8 at codons 173 and 277 have been associated with altered glucuronidation activity (48) and cancer risk (49). A functional effect by these variants was also observed in the present study. UGT1A8 genotype was significantly correlated with raloxifene glucuronide formation in HJH in vitro and plasma raloxifene glucuronide levels in subjects treated with raloxifene, with the plasma levels of both ral-6-Gluc and ral-4′-Gluc significantly lower in subjects with the predicted UGT1A8 slow metabolizer genotype as compared with subjects with intermediate or fast metabolizer genotypes. These data were also consistent with the results from cell lines over-expressing UGT1A8 variants demonstrating functional effects by UGT1A8 variants on raloxifene glucuronidation capacity. The fact that no association was observed between HJH raloxifene glucuronidation and the UGT1A8*3 allele may have been due to low power due to low UGT1A8*3 prevalence (allelic frequency=2.2%) and the fact that all of the HJH specimens with an UGT1A8*3 allele examined in this study were from subjects who were heterozygous (*1/*3) for that allele. As high-prevalence coding SNPs are not observed for UGTs 1A9 (45) or 1A10 (50), a similar phenotype-genotype study in jejunum was not performed for these enzymes. In addition to suggesting that UGT1A8 coding SNPs may play an important role in the glucuronidation of raloxifene in vivo, these data support an important role for jejunum in overall raloxifene metabolism.
Ral-4′-Gluc was the major form of raloxifene metabolite observed in the plasma of subjects treated with raloxifene. The ER-binding affinity of ral-4′-Gluc was shown to exhibit an IC50 value that was approximately 100-fold less than raloxifene itself. However, as >70% of total circulating plasma raloxifene is in the form of ral-4′-Gluc and because the levels of ral-4′-Gluc are >70-fold higher than parent unconjugated raloxifene, variations in the level of ral-4′-Gluc could potentially have an important effect on overall patient response to raloxifene. Therefore, UGT1A8 genotype could potentially impact overall patient response to raloxifene by altering circulating levels of raloxifene glucuronides, particularly the levels of ral-4′-Gluc. Clinical studies with a larger population size will be required to further examine the role of UGT1A8 genotype on the preventive effect of raloxifene for breast cancer.
Acknowledgments
We thank the Molecular Genetics Core facility at Penn State University College of Medicine for performing genotyping and DNA sequencing, and Dr. Gang Chen for helpful discussions. These studies were supported by a Penn State Center for Pharmacogenetics award to D. Sun, a Public Health Service (PHS) R01-CA164366 (National Cancer Institute) from the National Institutes of Health, Department of Health and Human Services to P. Lazarus, and a Promise Grant KG081632 from Susan G. Komen for the Cure to A. Manni.
Abbreviations
- UGT
uridinediphosphate glucuronosyltransferase
- SERM
selective estrogen receptor modulator
- RT-PCR
reverse transcription polymerase chain reaction
- RQ
relative quantification
- UPLC
ultra-performance liquid chromatography
- HPLC
high-performance liquid chromatography
- UTR
untranslated region
- ral-6-Gluc
raloxifene-6-glucuronide
- ral-4′-Gluc
raloxifene-4′-glucuronide
Footnotes
We declare that all authors have no conflict of interest in the authorship or publication of this contribution.
References
- 1.Estimated new cases and deaths from breast cancer in the United States in 2012. 2012 Available from: http://www.cancer.gov/cancertopics/types/breast.
- 2.Fisher B, Costantino JP, Wickerham DL, Cecchini RS, Cronin WM, Robidoux A, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. Journal of the National Cancer Institute. 2005;97:1652–62. doi: 10.1093/jnci/dji372. [DOI] [PubMed] [Google Scholar]
- 3.Raloxifene hydrochloride. 2010 Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2007/ucm108981.htm.
- 4.Black LJ, Sato M, Rowley ER, Magee DE, Bekele A, Williams DC, et al. Raloxifene (LY139481 HCI) prevents bone loss and reduces serum cholesterol without causing uterine hypertrophy in ovariectomized rats. The Journal of clinical investigation. 1994;93:63–9. doi: 10.1172/JCI116985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freedman AN, Yu B, Gail MH, Costantino JP, Graubard BI, Vogel VG, et al. Benefit/risk assessment for breast cancer chemoprevention with raloxifene or tamoxifen for women age 50 years or older. J Clin Oncol. 29:2327–33. doi: 10.1200/JCO.2010.33.0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, et al. Update of the National Surgical Adjuvant Breast and Bowel Project Study of Tamoxifen and Raloxifene (STAR) P-2 Trial: Preventing breast cancer. Cancer Prev Res (Phila) 2010;3:696–706. doi: 10.1158/1940-6207.CAPR-10-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstein SR, Scheele WH, Rajagopalan SK, Wilkie JL, Walsh BW, Parsons AK. A 12-month comparative study of raloxifene, estrogen, and placebo on the postmenopausal endometrium. Obstetrics and gynecology. 2000;95:95–103. doi: 10.1016/s0029-7844(99)00502-5. [DOI] [PubMed] [Google Scholar]
- 8.Morrello K, W GT, DeGregorio MW. Pharmacokinetics of selective estrogen receptor modulators. Clin Pharmacokinet. 2003;42:361–72. doi: 10.2165/00003088-200342040-00004. [DOI] [PubMed] [Google Scholar]
- 9.Snyder KR, Sparano N, Malinowski JM. Raloxifene hydrochloride. Am J Health Syst Pharm. 2000;57:1669–75. quiz 76-8. [PubMed] [Google Scholar]
- 10.Hochner-Celnikier D. Pharmacokinetics of raloxifene and its clinical application. European journal of obstetrics, gynecology, and reproductive biology. 1999;85:23–9. doi: 10.1016/s0301-2115(98)00278-4. [DOI] [PubMed] [Google Scholar]
- 11.EVISTA (raloxifene) Tablet. 2007 Available from: http://pi.lilly.com/us/evista-pi.pdf.
- 12.Trdan T, Roskar R, Trontelj J, Ravnikar M, Mrhar A. Determination of raloxifene and its glucuronides in human urine by liquid chromatography-tandem mass spectrometry assay. Journal of chromatography. 879:2323–31. doi: 10.1016/j.jchromb.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 13.Dalvie D, Kang P, Zientek M, Xiang C, Zhou S, Obach RS. Effect of intestinal glucuronidation in limiting hepatic exposure and bioactivation of raloxifene in humans and rats. Chemical research in toxicology. 2008;21:2260–71. doi: 10.1021/tx800323w. [DOI] [PubMed] [Google Scholar]
- 14.Kemp DC, Fan PW, Stevens JC. Characterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearance. Drug metabolism and disposition: the biological fate of chemicals. 2002;30:694–700. doi: 10.1124/dmd.30.6.694. [DOI] [PubMed] [Google Scholar]
- 15.Mizuma T. Intestinal glucuronidation metabolism may have a greater impact on oral bioavailability than hepatic glucuronidation metabolism in humans: a study with raloxifene, substrate for UGT1A1, 1A8, 1A9, and 1A10. International journal of pharmaceutics. 2009;378:140–1. doi: 10.1016/j.ijpharm.2009.05.044. [DOI] [PubMed] [Google Scholar]
- 16.Kim AR, L S, Lee BJ. Metabolic inhibition and kinetics of raloxifene by pharmaceutical excipients in human liver microsomes. Int J Pharm. 2009;368:37–44. doi: 10.1016/j.ijpharm.2008.09.049. [DOI] [PubMed] [Google Scholar]
- 17.Knadler M, L RJ, Gillespie TA, Allerheiligen SR, Henry DP. The disposition and metabolism of [14C]-raloxifene in humans. Pharm Res. 1995;12:S–372. [Google Scholar]
- 18.Bosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. N Engl J Med. 1995;333:1171–5. doi: 10.1056/NEJM199511023331802. [DOI] [PubMed] [Google Scholar]
- 19.Trontelj J, Marc J, Zavratnik A, Bogataj M, Mrhar A. Effects of UGT1A1*28 polymorphism on raloxifene pharmacokinetics and pharmacodynamics. British journal of clinical pharmacology. 2009;67:437–44. doi: 10.1111/j.1365-2125.2009.03363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiener D, Doerge DR, Fang JL, Upadhyaya P, Lazarus P. Characterization of N-glucuronidation of the lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human liver: importance of UDP-glucuronosyltransferase 1A4. Drug metabolism and disposition: the biological fate of chemicals. 2004;32:72–9. doi: 10.1124/dmd.32.1.72. [DOI] [PubMed] [Google Scholar]
- 21.Ren Q, Murphy SE, Zheng Z, Lazarus P. O-Glucuronidation of the lung carcinogen 4-(methylnitrosamino)-1- (3-pyridyl)-1-butanol (NNAL) by human UDP-glucuronosyltransferases 2B7 and 1A9. Drug metabolism and disposition: the biological fate of chemicals. 2000;28:1352–60. [PubMed] [Google Scholar]
- 22.Dellinger RW, Fang JL, Chen G, Weinberg R, Lazarus P. Importance of udp-glucuronosyltransferase 1a10 (ugt1a10) in the detoxification of polycyclic aromatic hydrocarbons: decreased glucuronidative activity of the ugt1a10139lys isoform. Drug metabolism and disposition: the biological fate of chemicals. 2006;34:943–9. doi: 10.1124/dmd.105.009100. [DOI] [PubMed] [Google Scholar]
- 23.Chen G, Blevins-Primeau AS, Dellinger RW, Muscat JE, Lazarus P. Glucuronidation of nicotine and cotinine by UGT2B10: loss of function by the UGT2B10 Codon 67 (Asp>Tyr) polymorphism. Cancer Res. 2007;67:9024–9. doi: 10.1158/0008-5472.CAN-07-2245. [DOI] [PubMed] [Google Scholar]
- 24.Sun D, Sharma AK, Dellinger RW, Blevins-Primeau AS, Balliet RM, Chen G, et al. Glucuronidation of active tamoxifen metabolites by the human UDP glucuronosyltransferases. Drug metabolism and disposition: the biological fate of chemicals. 2007;35:2006–14. doi: 10.1124/dmd.107.017145. [DOI] [PubMed] [Google Scholar]
- 25.Huang YH, Galijatovic A, Nguyen N, Geske D, Beaton D, Green J, et al. Identification and functional characterization of UDP-glucuronosyltransferases UGT1A8*1, UGT1A8*2 and UGT1A8*3. Pharmacogenetics. 2002;12:287–97. doi: 10.1097/00008571-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Balliet RM, Chen G, Gallagher CJ, Dellinger RW, Sun D, Lazarus P. Characterization of UGTs active against SAHA and association between SAHA glucuronidation activity phenotype with UGT genotype. Cancer Res. 2009;69:2981–9. doi: 10.1158/0008-5472.CAN-08-4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Signori C, DuBrock C, Richie JP, Prokopczyk B, Demers LM, Hamilton C, et al. Administration of omega-3 fatty acids and Raloxifene to women at high risk of breast cancer: interim feasibility and biomarkers analysis from a clinical trial. European journal of clinical nutrition. 2012;66:878–84. doi: 10.1038/ejcn.2012.60. [DOI] [PubMed] [Google Scholar]
- 28.Sun D, Chen G, Dellinger RW, Sharma AK, Lazarus P. Characterization of 17-dihydroexemestane glucuronidation: potential role of the UGT2B17 deletion in exemestane pharmacogenetics. Pharmacogenet Genomics. 2010;20:575–85. doi: 10.1097/FPC.0b013e32833b04af. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang JL, Lazarus P. Correlation between the UDP-glucuronosyltransferase (UGT1A1) TATAA box polymorphism and carcinogen detoxification phenotype: significantly decreased glucuronidating activity against benzo(a)pyrene-7,8-dihydrodiol(-) in liver microsomes from subjects with the UGT1A1*28 variant. Cancer Epidemiol Biomarkers Prev. 2004;13:102–9. doi: 10.1158/1055-9965.epi-03-0070. [DOI] [PubMed] [Google Scholar]
- 30.Jones NR, Sun D, Freeman WM, Lazarus P. Quantification of hepatic UGT1A splice variant expression and correlation of UGT1A1 variant expression with glucuronidation activity. J Pharmacol Exp Ther. 2012 doi: 10.1124/jpet.112.192658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Y, Sun D, Sharma AK, Chen G, Amin S, Lazarus P. Elimination of antiestrogenic effects of active tamoxifen metabolites by glucuronidation. Drug metabolism and disposition: the biological fate of chemicals. 2007;35:1942–8. doi: 10.1124/dmd.107.016279. [DOI] [PubMed] [Google Scholar]
- 32.Jeong EJ, Liu Y, Lin H, Hu M. Species- and disposition model-dependent metabolism of raloxifene in gut and liver: role of UGT1A10. Drug metabolism and disposition: the biological fate of chemicals. 2005;33:785–94. doi: 10.1124/dmd.104.001883. [DOI] [PubMed] [Google Scholar]
- 33.Trdan Lusin T, Trontelj J, Mrhar A. Raloxifene glucuronidation in human intestine, kidney, and liver microsomes and in human liver microsomes genotyped for the UGT1A1*28 polymorphism. Drug metabolism and disposition: the biological fate of chemicals. 39:2347–54. doi: 10.1124/dmd.111.041897. [DOI] [PubMed] [Google Scholar]
- 34.Dellinger RW, Chen G, Blevins-Primeau AS, Krzeminski J, Amin S, Lazarus P. Glucuronidation of PhIP and N-OH-PhIP by UDP-glucuronosyltransferase 1A10. Carcinogenesis. 2007;28:2412–8. doi: 10.1093/carcin/bgm164. [DOI] [PubMed] [Google Scholar]
- 35.Strassburg CP. Gilbert-Meulengracht's syndrome and pharmacogenetics: is jaundice just the tip of the iceberg? Drug Metab Rev. 2010;42:162–75. doi: 10.3109/03602530903209429. [DOI] [PubMed] [Google Scholar]
- 36.Iyer L, Hall D, Das S, Mortell MA, Ramirez J, Kim S, et al. Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther. 1999;65:576–82. doi: 10.1016/S0009-9236(99)70078-0. [DOI] [PubMed] [Google Scholar]
- 37.Winer EP, Hudis C, Burstein HJ, Wolff AC, Pritchard KI, Ingle JN, et al. American Society of Clinical Oncology technology assessment on the use of aromatase inhibitors as adjuvant therapy for postmenopausal women with hormone receptor-positive breast cancer: status report 2004. J Clin Oncol. 2005;23:619–29. doi: 10.1200/JCO.2005.09.121. [DOI] [PubMed] [Google Scholar]
- 38.Guillemette C, Ritter JK, Auyeung DJ, Kessler FK, Housman DE. Structural heterogeneity at the UDP-glucuronosyltransferase 1 locus: functional consequences of three novel missense mutations in the human UGT1A7 gene. Pharmacogenetics. 2000;10:629–44. doi: 10.1097/00008571-200010000-00006. [DOI] [PubMed] [Google Scholar]
- 39.Cubitt HE, Houston JB, Galetin A. Relative importance of intestinal and hepatic glucuronidation-impact on the prediction of drug clearance. Pharmaceutical research. 2009;26:1073–83. doi: 10.1007/s11095-008-9823-9. [DOI] [PubMed] [Google Scholar]
- 40.Cheng Z, Radominska-Pandya A, Tephly TR. Cloning and expression of human UDP-glucuronosyltransferase (UGT) 1A8. Arch Biochem Biophys. 1998;356:301–5. doi: 10.1006/abbi.1998.0781. [DOI] [PubMed] [Google Scholar]
- 41.Tukey RH, Strassburg CP. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 42.Dellinger RW, Fang JL, Chen G, Weinberg R, Lazarus P. Importance of UDPglucuronosyltransferase 1A10 (UGT1A10) in the detoxification of polycyclic aromatic hydrocarbons: decreased glucuronidative activity of the UGT1A10 139Lys isoform. Drug Metabolism and Disposition. 2006;34:943–9. doi: 10.1124/dmd.105.009100. [DOI] [PubMed] [Google Scholar]
- 43.Jones NR, S D, Freeman WM, Lazarus P. Quantification of hepatic UGT1A splice variant expression and correlation of UGT1A1 variant expression with glucuronidation activity. J Pharmacol Exp Ther. 2012 doi: 10.1124/jpet.112.192658. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohno S, Nakajin S. Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug metabolism and disposition: the biological fate of chemicals. 2009;37:32–40. doi: 10.1124/dmd.108.023598. [DOI] [PubMed] [Google Scholar]
- 45.Olson KC, Dellinger RW, Zhong Q, Sun D, Amin S, Spratt TE, et al. Functional characterization of low-prevalence missense polymorphisms in the UDP-glucuronosyltransferase 1A9 gene. Drug metabolism and disposition: the biological fate of chemicals. 2009;37:1999–2007. doi: 10.1124/dmd.108.024596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lusin T, Trontelj J, Mrhar A. Raloxifene glucuronidation in human intestine, kidney, and liver microsomes and in human liver microsomes genotyped for the UGT1A1*28 polymorphism. Drug metabolism and disposition: the biological fate of chemicals. 2011;39:2347–54. doi: 10.1124/dmd.111.041897. [DOI] [PubMed] [Google Scholar]
- 47.Rauchschwalbe SK, Zuhlsdorf MT, Schuhly U, Kuhlmann J. Predicting the risk of sporadic elevated bilirubin levels and diagnosing Gilbert's syndrome by genotyping UGT1A1*28 promoter polymorphism. Int J Clin Pharmacol Ther. 2002;40:233–40. doi: 10.5414/cpp40233. [DOI] [PubMed] [Google Scholar]
- 48.Blevins-Primeau AS, Sun D, Chen G, Sharma AK, Gallagher CJ, Amin S, et al. Functional Significance of UDP-Glucuronosyltransferase Variants in the Metabolism of Active Tamoxifen Metabolites. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-08-3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dura P, S J, Te Morsche RH, Roelofs HM, Kristinsson JO, Wobbes T, Witteman BJ, Tan AC, Drenth JP, Peters WH. High enzyme activity UGT1A1 or low activity UGT1A8 and UGT2B4 genotypes increase esophageal cancer risk. Int J Oncol. 2012 doi: 10.3892/ijo.2012.1385. [DOI] [PubMed] [Google Scholar]
- 50.Elahi A, Bendaly J, Zheng Z, Muscat JE, Richie JP, Jr, Schantz SP, et al. Detection of UGT1A10 polymorphisms and their association with orolaryngeal carcinoma risk. Cancer. 2003;98:872–80. doi: 10.1002/cncr.11587. [DOI] [PubMed] [Google Scholar]