

Abstract
(Z)-Endoxifen (4-hydroxy-N-desmethyltamoxifen), an active metabolite generated via actions of CYP3A4/5 and CYP2D6, is a more potent selective estrogen receptor modulator (SERM) than tamoxifen. In the MCF-7 human mammary tumor xenograft model with female athymic mice, (Z)-endoxifen, at an oral dose of 4– 8 mg/kg, significantly inhibits tumor growth. (Z)-Endoxifen's potential as an alternative therapeutic agent independent of CYP2D6 activities, which can vary widely in ER+ breast cancer patients, is being actively evaluated. This paper describes confirmation of the configuration of the active (Z)-isomer through 2D NMR experiments, including NOE (ROESY) to establish spatial proton–proton correlations, and identification of the major impurity as the (E)-isomer in endoxifen drug substance by HPLC/HRMS (HPLC/MS-TOF). Stability of NMR solutions was confirmed by HPLC/UV analysis. For pre-clinical studies, a reverse-phase HPLC–UV method, with methanol/water mobile phases containing 10 mM ammonium formate at pH 4.3, was developed and validated for the accurate quantitation and impurity profiling of drug substance and drug product. Validation included demonstration of linearity, method precision, accuracy, and specificity in the presence of impurities, excipients (for the drug product), and degradation products. Ruggedness and reproducibility of the method were confirmed by collaborative studies between two independent laboratories. The method is being applied for quality control of the API and oral drug product. Kinetic parameters of Z- to E-isomerization were also delineated in drug substance and in aqueous formulation, showing conversion at temperatures above 25 °C.
Keywords: Endoxifen, Isomeric characterization, LC-MS, NMR, Quantitative HPLC-UV analysis
1. Introduction
(Z)-Endoxifen (4-hydroxy-N-desmethyltamoxifen), an active metabolite of the widely used breast cancer drug tamoxifen, is generated via actions of cytochrome P450 (CYP) enzymes CYP3A4/5 and CYP2D6 [1–3]. (Z)-Endoxifen, similar to 4-hydroxytamoxifen, is a more potent selective estrogen receptor modulator (SERM) than tamoxifen, with an IC50 = 0.01–0.10 μM, when tested in the estrogen-stimulated proliferation assay in MCF-7 cells [1]. Additionally, in the MCF-7 human mammary tumor xenograft model with female athymic mice, (Z)-endoxifen, at an oral dose of 4–8 mg/kg, significantly inhibits tumor growth [4]. (Z)-Endoxifen's potential as an alternative therapeutic agent independent of individual CYP2D6 activities, which can vary widely in ER+ breast cancer patients due to genetic polymorphism and/or concomitant medications (e.g., specific selective serotonin reuptake inhibitors, SSSRIs), is being actively evaluated [4,5]. Direct use of (Z)-endoxifen may also avoid undesirable drug interactions with SSSRIs which are often prescribed to ameliorate side effects associated with tamoxifen therapy. Endoxifen exists as the potently anti-estrogenic (Z)-isomer and the lesser known (E)-isomer. The chemical structures are shown in Fig. 1. The (E)-isomer of 4-OH-tamoxifen has been found to be estrogenic or weakly anti-estrogenic, while the (Z)-isomer is a potent antagonist [6]. It is assumed that (E)-endoxifen, structurally related to (E)-4-OH-tamoxifen, will have similar pharmacological properties. The (E)-isomer is an impurity in (Z)-endoxifen drug substance and increases under certain storage conditions. The electron-donating phenolic group in the compound activates the core ethylene toward protonation; addition and subsequent elimination of H+ is the likely course of isomerization. The identity of the (Z)-isomer, especially the configuration of the ethylene in active endoxifen, had not been rigorously established. Previous identification of endoxifen isomers by 1H NMR was empirically based on reported trends for the chemical shifts of the –O-CH2 protons in the sidechain to be upfield in the (Z)- compared to (E)-configuration for similar triarylethylene compounds [7–10].
Fig. 1.
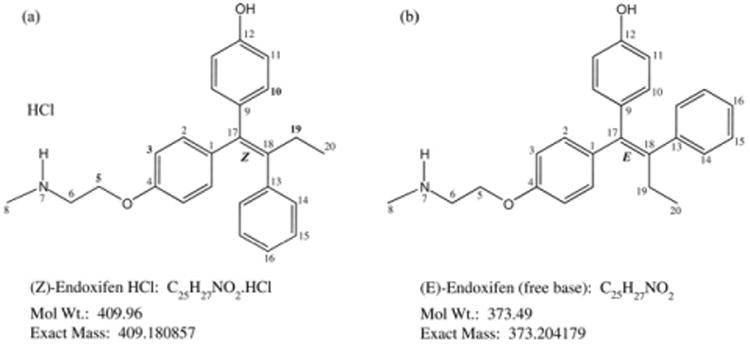
Structures of (a) (Z)-endoxifen HCl and (b) (E)-endoxifen free base with NMR assignment numbering.
A quantitative assay was needed for determination of endoxifen purity. Recent publications on the parent compound tamoxifen have addressed the assay of tamoxifen and its metabolites in biological matrices with detection by mass spectrometry (see [11], specifically b, f, g). The methods either do not fully resolve the (Z)- and (E)-isomers of endoxifen ([11] (f, g)), or are not stability indicating ([11] (b, f, g)). Development and validation of a stability indicating HPLC method with full resolution of the (Z)- and (E)-isomers was pursued with the specific goal of developing a robust HPLC–UV method for assessment of potency and quantification of impurities.
This paper describes the unequivocal determination of the configuration for the (Z)-isomer of endoxifen as the major component in biologically active endoxifen drug substance (HCl salt) by 2D and NOE (nuclear Overhauser enhancement) NMR. Identification of significant impurities by LC–MS/TOF showed (E)-isomer to be the major impurity in endoxifen. Validation of HPLC/UV assay methods developed for quantitation of (Z)-endoxifen in drug substance and 20 mg formulated solid oral drug product is also described. Kinetics of the isomerization of endoxifen in solid drug substance and an aqueous formulation stored under controlled conditions are also presented.
2. Materials and methods
2.1. Chemicals and reagents
Drug substances (Z)-endoxifen hydrochloride (HCl), NSC 750393, and (E)-endoxifen, NSC 750182, were provided by NCI (Bethesda, MD, USA). At NCI's request, the University of Arizona, College of Pharmacy provided endoxifen formulation and placebo for stability testing. Dimethyl sulfoxide (DMSO) was B & J Brand High Purity Solvent, purchased from Honeywell Burdickand Jackson(Muskegon,MI, USA). Solvent for NMR, DMSO-d6, (Isotech, 99.9 atom % D, with 0.05% TMS, v/v), Fluka ammonium formate (HCOONH4) for HPLC, Fluka formic acid (HCOOH), and ACS grade HCOOH were purchased from Sigma– Aldrich (St. Louis, MO, USA). HPLC grade acetonitrile (MeCN) and methanol (MeOH) were purchased from EMD Chemicals USA (Gibbstown, NJ, USA) and from Fisher (Waltham, MA, USA). HPLC-grade water was obtained from a Picosystem UV Plus purification system (Hydro Services & Supplies, Inc., Durham, NC, USA) and purified water from a Labconco WaterPro PS (Kansas City, MO, USA). Reagents used for HPLC method validations and drug substance stability studies included: 97% HCOONH4, scintillation grade silicon dioxide, ACS grade sodium croscarmellose, and Extra Pure microcrystalline cellulose purchased from Acros Organics (New Jersey, USA); certified ACS grade hydrogen peroxide (30% H2O2), NF grade magnesium stearate, and certified ACS Plus hydrochloric acid (HCl) purchased from Fisher; 88% ACS grade HCOOH and Ultra grade ascorbic acid purchased from Sigma–Aldrich; ACS grade sodium hydroxide (NaOH) purchased from VWR/BDH (Radnor, PA, USA).
2.2. Instruments and analytical parameters
2.2.1. NMR analysis
NMR experiments were performed using a 500-MHz Varian Unity Inova NMR Spectrometer, with TMS in the NMR solvent used as a reference. 1H NMR measurements were carried out at 500 MHz, while 13C NMR experiments were performed at 125 MHz.
2.2.2. LC–mass spectrometric analysis
The LC/TOF-MS was an Agilent 1290 UHPLC with 6230 MSD-TOF, UV diode array detector, and MassHunter software (v. B.02.01, SP1) (Wilmington, DE, USA). Electrospray ionization (ESI) was used in positive ion mode with a JetStream source. Data were acquired in a mass range of m/z 100–1700. Except for injection volume (2 μL), LC conditions were the same as for HPLC–UV analyses in Section 2.2.3.
2.2.3. HPLC–UV analysis
HPLC–UV analyses were performed on a Waters 2695 Separations Module with Alliance Column Heater and either (1) 996 photodiode array, or (2) 2487 dual absorbance detector (Waters Corporation, Milford, MA, USA) and (3) a Shimadzu LC-2010CHT equipped with a Shimadzu diode array detector (Shimadzu Corporation, Columbia, MD). Acquisition and processing of HPLC/UV data were performed using Waters Empower 2, Build 2154, and Perkin Elmer TotalChrom (Ver 6.3.0) data system software. Separation was performed at 30 °C on a 150 mm × 4.6 mm I.D. Luna Phenyl-Hexyl column, with 3 μm particles (Phenomenex, Torrance, CA, USA). The mobile phase was a combination of mobile phase A: HCOONH4 (pH 4.3; 10 mM) in water–HCOONH4 (10 mM) in MeOH, with equivalent HCOOH as in pH 4.3 aqueous (40:60, v/v), and mobile phase B: HCOONH4 (10 mM) in MeOH, with equivalent HCOOH as in pH 4.3 aqueous. The segmented, linear gradient program went from 0% to 35% mobile phase B in 15 min, then to 90% B in 10 min, returning to 0% B and holding for 5 min between injections. The injection volume was 20 μL. The mobile phase flow rate was 1.0 mL/min, with UV detection at 243 nm. LC/UV analysis of NMR solutions was performed on a Waters 1525 Binary Pump with a 1500 Column Heater, 2996 photodiode array detector, and Empower 2 software (Waters Corporation, Milford, MA, USA) under conditions described above, but using a faster gradient and UV detection at 220 nm. Mobile phase A was HCOONH4 (pH 4.3, 10 mM) in water, and mobile phase B was HCOONH4 (10 mM) in MeOH with equivalent HCOOH as in A. The gradient began with a 10-min hold at 60% B, increased to 70% B in 2 min, then to 95% B in 5 min for a 17 min total gradient time; the gradient was reversed in 5 min, and initial conditions were held for 10 min between injections (32 min run time).
2.3. Sample preparation
For NMR experiments, 11–12 mg of (E)- or (Z)-endoxifen were dissolved in 1 g (∼0.8 mL) of DMSO-d6 just prior to analysis. For LC analysis of NMR solutions, ca. 3 mg of test substance (ca. 240 μL) were diluted to 10 mL with MeCN. This solution was diluted 1:2 with water for a final concentration of ca. 0.15 mg/mL. Fresh comparison samples were prepared by adding ca. 240 μL of DMSO to 3–4 mg of (E)- or (Z)-endoxifen and diluting to 10 mL with MeCN, followed by 1:2 dilution with water for a final concentration of ca. 0.2 mg/mL.
Sample diluent for LC analyses was MeCN–HCOONH4 (pH 4.3, 10 mM) (50:50, v/v). The nominal assay concentration of endoxifen HCl aqueous formulation and drug substance was ∼0.16 mg/mL in diluent prepared by dissolving 4 mg in 25 mL. The drug product solutions were prepared at 0.2 mg/mL from placebo spiked with drug substance (equivalent to the contents of 1 capsule).
For LC/TOF-MS, a stock solution at 0.165 mg/mL was prepared by diluting 4.12 mg of (Z)-endoxifen HCl to 25 mL with diluent. The stock was diluted serially 1:10 and 1:100 with diluent to produce solutions at 0.0165 mg/mL and 0.165 μg/mL. The three solutions were used to obtain optimum MS responses for the major component and impurities in confirmation of identity.
For formulation stability studies, placebo and formulation samples were prepared. The solid formulation contained endoxifen and excipients in a 49:51 ratio; the solid placebo was 100% excipients. A 507-mg aliquot of formulation containing 248 mg of endoxifen was diluted with 6 mL of water to produce a 41.3 mg/mL endoxifen solution. Correspondingly, 256 mg of placebo were diluted with 6 mL of water. Half of each solution was stored at room temperature, the other half at 45 °C. On days 0, 1, 3, 5, 10, and 15, 0.1 mL from each solution was diluted to 25 mL with diluent for HPLC–UV analysis.
For bulk stability studies, ∼140-mg aliquots of (Z)-endoxifen HCl drug substance were stored in tightly capped, clear glass vials at 5 °C and 25 °C/60% RH for 12 months, and at 40 °C/75% RH for 6 months. At 0, 3, 6, 9, and 12 month time points, samples were analyzed for HPLC assay and impurities, moisture by Karl Fischer titration, and appearance.
In forced degradation studies, solutions of approximately 0.32 mg/mL endoxifen HCl were prepared for degradant conditions (1.5 N HCl, 0.75 N NaOH, and 3% H2O2) and a control sample by diluting ∼3.2 mg endoxifen HCl with the appropriate degradant solution in 10-mL flasks. Each condition was stored for ∼24 h at –20 °C, 5 °C, ambient, and 60 °C. Control samples at ambient and 60 °C were stored under laboratory light and in the dark. Prior to analysis, acidic and basic solutions were neutralized and oxidized samples were diluted with an equivalent amount of diluent to yield ∼0.16 mg/mL solutions. Additionally,an ∼0.16 mg/mL solution was prepared for analysis from an aliquot of bulk drug kept at 80 °C under laboratory light for 24 h to evaluate thermal degradation. The HPLC analysis used a Shimadzu diode array detector.
3. Results and discussion
3.1. NMR studies in (Z)- and (E)-isomers
The structure of the (Z)-isomer of endoxifen was confirmed by demonstrating the proximate spatial relationship of key protons on the ethyl group and aromatic rings when in the (Z)-configuration. Structures of (Z)-endoxifen HCl and (E)-endoxifen, annotated with NMR reference numbers, are shown in Fig. 1. The 1H and 13C NMR data, and proposed assignments for the two isomers are presented in Table 1.
Table 1.
Proton and carbon chemical shifts and structural assignments for (Z)-endoxifen HCl and (E)-endoxifen.
| Assignment | (Z)-Endoxifen HCl Chemical shift (ppm) | (E)-Endoxifen Chemical shift (ppm) | ||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Carbon | Proton | No. of protons | Multiplicitya | Carbon | Proton | No. of protons | Multiplicitya | |
| 20 | 13.41 | 0.85 | 3H | t, (J = 7.3 Hz) | 13.45 | 0.85 | 3H | t, (J = 7.3 Hz) |
| 19 | 28.59 | 2.41 | 2H | q, (J = 7.3 Hz) | 28.49 | 2.39 | 2H | q, (J = 7.3 Hz) |
| 8 | 32.70 | 2.56 | 3H | s | 35.97 | 2.35 | 3H | s |
| 6 | 47.24 | 3.23 c | 2H | t, (J = 4.9 Hz) | 50.16 | 2.85 | 2H | t, (J = 5.6 Hz) |
| 5 | 62.98 | 4.11 c | 2H | t, (J = 4.9 Hz) | 66.86 | 4.03 | 2H | t, (J = 5.6 Hz) |
| 3 | 113.48 | 6.64 | 2H | d, (J = 8.7 Hz) | 114.06 | 6.92 | 2H | d, (J = 8.7 Hz) |
| 11 | 115.01 | 6.76 | 2H | d, (J = 8.7 Hz) | 114.29 | 6.40 | 2H | d, (J = 8.7 Hz) |
| 16 | 126.02 | 7.10 | 1H | m | 125.91 | 7.09 | 3H | m |
| 15 | 127.93 | 7.17 | 2H | t, (J = 7.0 Hz) | 127.85 | 7.18 | 2H | t, (J = 7.3 Hz) |
| 14 | 129.40 | 7.10 | 2H | m | 129.39 | 7.09 | 3H | m |
| 10 | 130.11 | 6.97 | 2H | d, (J = 8.7 Hz) | 130.13 | 6.60 | 2H | d, (J = 8.7 Hz) |
| 2 | 131.49 | 6.74 | 2H | d, (J = 9.4 Hz) | 131.38 | 7.09 | 2H | d, (J = 9.4 Hz) |
| 9 | 133.78 | – | 133.62 | – | ||||
| 1 | 136.24 | – | 135.72 | – | ||||
| 17 | 137.76 | – | 137.89 | – | ||||
| 18 | 140.18 | – | 139.72 | – | ||||
| 13 | 142.18 | – | 142.24 | – | ||||
| 4 | 155.54 | – | 155.23 | – | ||||
| 12 | 156.19 | – | 157.24 | – | ||||
| 12-OH | – | 9.50 | 1H | s | – | 9.19 | 1H | br s |
| 7-NH | – | 9.03 | 2Hb | br s | – | – | ||
With coupling constant, J, if applicable.
Integral presumably includes proton from HCl.
NMR results for these methylene protons in Z-endoxifen (free base) have been reported by Ali et al.: δ 2.74 ppm (t, J = 5.6 Hz, 2H, CH2-N); 3.86 ppm (t, J = 5.6 Hz, 2H, CH2-O) [12].
Unequivocal assignment of ethyl (H19, H20), ethanolamine (H5, H6), and N-methyl (H8) protons was made starting from the characteristic chemical shifts of their adjacent carbons in the 13C spectrum. Correlations between directly attached protons and carbons in the HSQC (heteronuclear single quantum correlation) spectrum, Fig. 2, were then used to determine corresponding protons. The five protons of the mono-substituted aromatic ring (H14–H16) were identified as the multiplet at 7.10 ppm and a multiplet at 7.17 ppm.
Fig. 2.
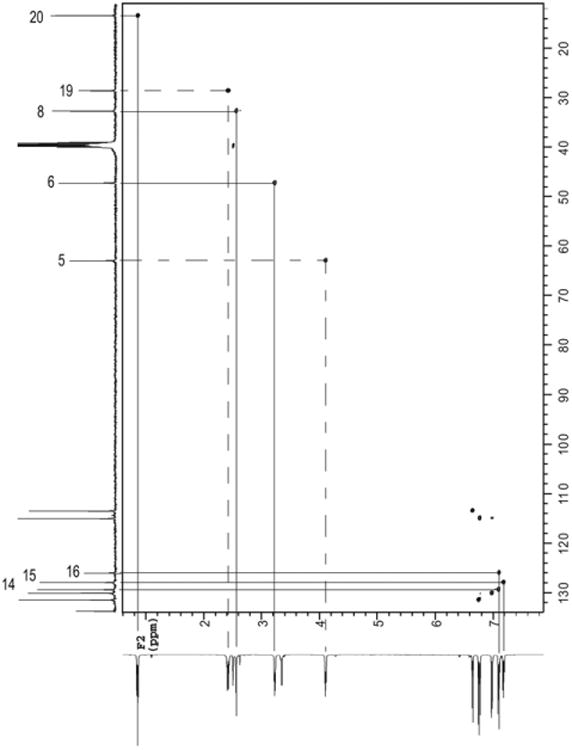
HSQC NMR spectrum of (Z)-endoxifen HCl in DMSO- d6.
Direct assignments for protons of the two para-disubstituted aromatic rings (C1–4, C9–12) were not possible (HSQC, Fig. 2) due to the high degree of similarity in the proton and carbon chemical shifts. In the COSY (correlated spectroscopy) spectrum (Fig. 3a), the two doublets associated with each ring were paired (H2, H3 and H10, H11) using proton-proton vicinal couplings.
Fig. 3.

(a) COSY NMR spectrum and (b) ROESY NMR spectrum, both (Z)-endoxifen HCl in DMSO- d6.
To differentiate the two rings, the ring with the ethanolamine substituent was first assigned by through-space correlation of H5 in the ethanolamine with the doublet for the aromatic proton at 6.64 ppm in the ROESY (Rotating frame enhancement spectroscopy) spectrum (Fig. 3b), identifying this proton as H3; and therefore, the doublet at 6.74 ppm as H2. The remaining two doublets were then assigned to H10 and H11.
With all protons assigned, the clear and definitive correlations between H10 and H19 and somewhat stronger correlation between H10 and H20 in the ROESY spectrum provided the most conclusive evidence for the proximate locations of these protons confirming the (Z) orientation. A weaker supporting correlation was observed between H2 and H14 and also suffered from overlap of the H2 and H11 resonances.
In (E)-endoxifen, resolution of the two pairs of aromatic proton signals was unsuccessful due to extensive overlap of both carbon and proton signals of H2 and H10 with other resonances in all spectra, especially in the COSY spectrum, in which this critical distinction had been made for the (Z)-isomer. Therefore, NMR alone could not confirm the isomeric configuration for (E)-endoxifen; however, comparison with published NMR data ([10], p. 2008, cis-endoxifen) and determination of exact mass by HRMS (Section 3.2, below) verified its identity.
HPLC/UV analysis based on %total area showed that NMR samples were stable and that results were not affected by conversion during the experiments. The fresh (Z)-isomer solution was 95.7% Z and 3.7% E vs. 92.0% Z and 7.4% E after NMR analysis. Fresh (E)-isomer solution was 97.1% E and 2.9% Z vs. 96.9% E and 3.1% Z after NMR analysis.
3.2. LC/TOF-MS studies
The accurate mass was determined for the major component and (non-blank) impurities present at >0.1% of total area (UV detection) in the concentrated sample. The major component and larger impurity had the same molecular formula, consistent with the structure of endoxifen, but LC/MS could not distinguish the (E)- and (Z)-isomers. The molecular formula of a smaller, earlier-eluting impurity at RRT 0.85 (RT 8.94 min) was consistent with loss of a methyl group (desmethyl-endoxifen). Thus, LC/MS results confirmed the major peak and the impurity at RRT 0.89 (RT9.40 min) as endoxifen isomers;NMR studies with corresponding HPLC analyses confirmed the major component as (Z)-endoxifen, and the minor component as the (E)-isomer. Table 2 shows LC/MS data with formula assignments for impurities and the major component.
Table 2.
LC/MS accurate mass determination for endoxifen and impurities.
| Peak no. | Retention time (min)a | Area% of major component | Sample conc. | Retention time (min)b | Exact mass of [M + H] + Ion (amu) | Formula assignment of neutral species | Mass error of assigned formula (ppm) |
|---|---|---|---|---|---|---|---|
| 1 | 8.940 | 0.12 | 0.165 mg/mL | 8.940 | 360.1970 | C24H25NO2 | −3.32 |
| 2 | 9.400 | 2.28 | 0.0165 mg/mL | 9.393 | 374.2118 | C25H27NO2 | −0.75 |
| 3 | 10.540 | 100 | 0.165 μg/mL | 10.533 | 374.2123 | C25H27NO2 | −2.11 |
In 0.165 mg/mL sample, UV detection.
For sample used for accurate mass determination, UV detection.
3.3. HPLC method validation
3.3.1. Method development
A validated HPLC method was sought for analysis of (Z)-endoxifen drug substance and formulation. Optimization included stationary phases scouting, which showed the Luna phenyl-hexyl column to provide excellent chromatography and resolution. Likewise, exploration of mobile phase conditions indicated that a mildly acidic (pH 4.3) ammonium formate buffer/methanol mobile phase with gradient elution was ideal.
3.3.2. System suitability
The system suitability requirements for this method were established as shown in Fig. 4, which also shows a representative chromatogram for endoxifen analysis.
Fig. 4.
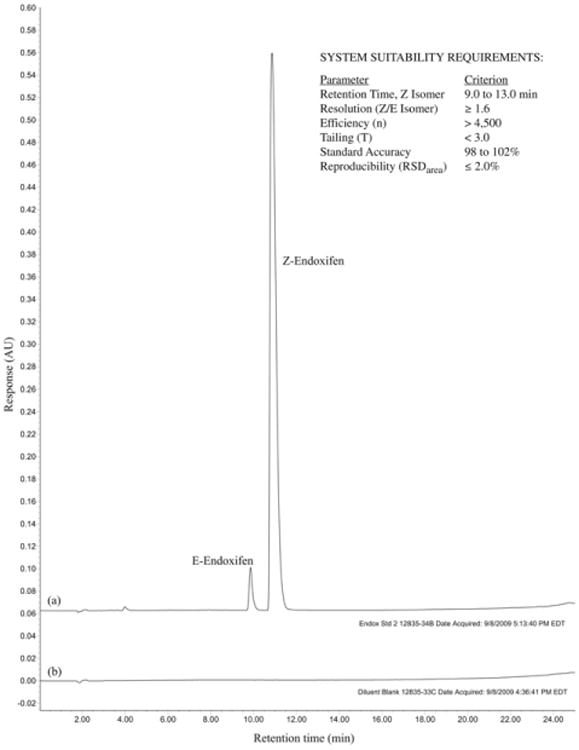
Representative HPLC chromatogram for assay of (Z)-endoxifen in (a) a 0.165 mg/mL solution of (Z)-endoxifen HCl and (b) blank diluent.
3.3.3. Selectivity and sensitivity
Forced degradation studies on solutions of acid, base, and peroxide, and on the bulk drug substance under photo and thermal exposure, revealed that (E)-endoxifen and various degradation products were well separated from (Z)-endoxifen and demonstrated peak homogeneity via diode array analysis of (Z)-endoxifen for all degradation conditions. Significant degradation (≤54% remaining vs. fresh (Z)-endoxifen sample) occurred in acid and base under all conditions and in peroxide with light and heat (Supplementary Table 1). (E)-endoxifen was identified as the primary degradant. Five replicate injections of a 0.016-μg/mL solution (0.01% of the target concentration) generated a %RSD ≤ 15% and a signal-to-noise (S/N) value of eight; therefore, the 0.01% solution was assigned as the LOQ, and the LOD was determined to be 6 ng/mL.
3.3.4. Linearity
The linearity of the test sample response was evaluated by analysis of five solutions ranging from 50% to 150% of the target concentration of 0.16 mg/mL. A first-order least squares linear regression analysis of the peak responses versus concentrations was used to assess linearity, which showed y = 55,493,345x + 45,290 (r = 0.999) for the drug substance. To evaluate the response of related impurities, a 0.1% solution was also examined, and the result was found to be linear when averaged and included with the standard curve points (r = 0.999). For the drug product, three solutions ranging from 75% to 125% of the target concentration of 0.2 mg/mL were used to assess linearity. The results showed y = 55,082,874x – 55,579 (r = 0.999). Linearity plots are shown in Supplementary Fig. 1.
3.3.5. Method precision and accuracy (recovery)
For the drug substance, nine sample solutions were prepared containing approximately 0.16 mg/mL endoxifen HCl dissolved in sample diluent. Method precision, evaluated as the %RSD of the relative response factors (RRF), was 1.2% (Supplementary Table 2). Accuracy was found to be 101.2%, evaluated by determining the average RRF of three randomly selected sample solutions and comparing to the average RRF of the remaining six sample solutions.
Likewise, for the drug product, three solutions were prepared (placebo spiked with drug substance) at 0.2 mg/mL Method precision was 2.0% RSD and accuracy was found to be 99% (results are shown in Supplementary Tables 3 and 4).
3.3.6. Stability of analytical solution
Sample solution stability was determined from solutions containing approximately 0.16 mg/mL endoxifen HCl dissolved in the sample diluting solution. The solution was stable at ambient temperature through 48 h (assay values of 99.3 and 98.5% for 24 and 48 h, respectively) and at 5 °C through 72 h (assay values of 99.7,100.2, and 100.0% for 24, 48, and 72 h, respectively). The solution stability for the drug product similarly showed stability for up to 72 h at both ambient and refrigerated conditions.
3.3.7. Intermediate and inter-lab precision
The chromatographic method was evaluated by a second analyst at MRIGlobal as well as by an analyst at RTI International. All results were comparable to those obtained by the original analyst. Results are provided in Supplementary Table 5.
3.3.8. Robustness
The robustness of the chromatographic system was determined by changing the mobile phase, including organic content (± 2.5%) for the starting gradient, the pump flow rate (± 0.2 mL/min), and the pH of the aqueous portion of the mobile phase (± 0.25 units).
The chromatographic system was shown to be relatively robust, with no significant variations in retention time, efficiency, asymmetry, resolution, or relative standard deviation (%RSD) for any of the conditions; system suitability criteria (Fig. 4) for these parameters were met in all cases (Supplementary Table 6).
3.4. Stability studies and kinetics
3.4.1. Aqueous formulation
In an aqueous (Z)-endoxifen formulation monitored for 15 days at room temperature and 45 °C along with a placebo, the compound remained essentially unchanged at room temperature through out the period, but degraded significantly at the elevated temperature (from 98% to 75% of total area on day 15). The (E)-isomer was the only impurity observed on day 0 (2.3% of total area) and was the largest impurity observed after 15 days at 45 °C, accounting for 25.2% of total area. Six other minor impurities were observed which together accounted for 1.5% of total area.
3.4.2. Bulk drug
Bulk drug stability studies of (Z)-endoxifen stored at 5 °C and 25 °C/60% RH for 12 months, and 40 °C/75% RH for 6 months showed % total chromatographic area (starting at 98%) for the major component to be stable at 5 °C (97%), with slow degradation to 96% at 25 °C/60% RH. The compound showed rapid degradation to 87% at 40 °C/75% RH in 3 months, and little additional change at 6 months (also 87%). The decrease in (Z)-endoxifen was accompanied by a corresponding increase in % (E)-endoxifen. As in stored aqueous formulation, (E)-endoxifen was the largest impurity at all conditions throughout the storage periods; three other minor impurities were observed.
3.4.3. Kinetics of the formation of E-isomer in solution and in bulk drug
Kinetic profiles for the (E)-isomer in aqueous (Z)-isomer formulation, stored at room temperature and 45 °C, and bulk drug, stored at 25 and 40 °C/75% RH, were generated from the stability data. The (E)-isomer kinetic equations (ln of % total area vs. time) for room temperature, ln % area = 0.0099t + 0.8310 (r2 = 0.9006), and for 45 °C, ln % area = 0.17351t + 0.7328 (r2 = 0.9810), were used to calculate predicted t90 values for storage at room temperature (149 days) and 45 °C (9.0 days). These intervals represent the time for 10% conversion to (E)-isomer in the active drug, (Z)-isomer, as an aqueous solution.
Arrhenius equations (ln k vs. 1/absolute temperature, K) were generated from solid drug substance stability data as: y = −12162x + 34.648, r2 = 0.976 for degradation of (Z)-isomer (at 5 °C and 25 °C/60% RH for 12 months, and 40 °C/75% RH for 6 months) and y = −15662x + 50.601, r2 =1, for formation of (E)-isomer (at 25 °C/60% RH for 12 months, and 40 °C/75% RH for 6 months). From (E)-isomer formation data, predicted t90 values for 10% conversion to (E)-isomer in the active drug, (Z)-isomer, in the solid state were determined to be 52.7 months at 25 °C/60% RH and 4.3 months at 40 °C/75% RH. The intervals were calculated as: shelf-life (t90) = ([D] – [D0])/A exp(Ea/RT), where [D] = 10, [D0] = 2.29,and R = 1.987 cal mol−1 K−1, with calculated values A = 9.46 × 1021%/ month and Ea = 31.1 kcal/mol.
4. Conclusions
The (Z)-configuration in the active drug endoxifen HCl was confirmed independently by 2D NMR, using ROESY to establish key spatial proton-proton correlations. The (E)-isomer, though not fully resolved by NMR, was confirmed by comparison to the published data and accurate mass determination (HRMS). An accurate, precise, linear, specific, and sensitive HPLC method was developed and validated for the analyses of endoxifen HCl. The method is being applied for the quality control of the API and oral drug product. Determination of kinetic parameters for the isomerization in solution and in solid state, showed facile conversion of (Z)- to (E)-isomer at temperatures above 25 °C.
Supplementary Material
Acknowledgments
This work was funded by the Pharmaceutical Research Branch, DTP, DCTD, NCI, NIH, DHHS under Contract Nos. N02-CM-72201 and -72202.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.jpba.2013.07.010.
Contributor Information
Phyllis Elkins, Email: elkins@rti.org.
Jonathan White, Email: jwhite@mriglobal.org.
Paul Liu, Email: liup@dtpepn.nci.nih.gov.
References
- 1.Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P, et al. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. Journal of the National Cancer Institute. 2003;95:1758–1764. doi: 10.1093/jnci/djg108. [DOI] [PubMed] [Google Scholar]
- 2.Johnson MD, Zuo H, Lee KH, Trebley JP, Rae JM, Weatherman RV, et al. Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Research and Treatment. 2004;85:151–159. doi: 10.1023/B:BREA.0000025406.31193.e8. [DOI] [PubMed] [Google Scholar]
- 3.Lim YC, Desta Z, Flockhart DA, Skaar TC. Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemotherapy and Pharmacology. 2005;55:471–478. doi: 10.1007/s00280-004-0926-7. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad A, Ali SM, Ahmad MU, Sheikh S, Ahmad I. Orally administered Endoxifen is a new therapeutic agent for breast cancer. Breast Cancer Research and Treatment. 2010;122:579–584. doi: 10.1007/s10549-009-0704-7. [DOI] [PubMed] [Google Scholar]
- 5.Goetz MP, Rae JM, Suman VJ, Safgren SL, Ames MM, Visscher DW, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. Journal of Clinical Oncology. 2005;23:9312–9318. doi: 10.1200/JCO.2005.03.3266. [DOI] [PubMed] [Google Scholar]
- 6.Katzenellenbogen BS, Norman MJ, Eckert RL, Peltz SW, Mangel WF. Bioactivities, estrogen receptor interactions, and plasminogen activator-inducing activities of tamoxifen and hydroxytamoxifen isomers in MCF-7 human breast cancer cells. Cancer Research. 1984;44:112–119. [PubMed] [Google Scholar]
- 7.Bedford GR, Richardson DN. Preparation and identification of cis and trans isomers of a substituted triarylethylene. Nature. 1966;5063:733–734. [Google Scholar]
- 8.Shani J, Gazit A, Livshitz T, Biran S. Synthesis and receptor-binding affinity of fluorotamoxifen, a possible estrogen-receptor imaging agent. Journal of Medicinal Chemistry. 1985;28:1504–1510. doi: 10.1021/jm00148a022. [DOI] [PubMed] [Google Scholar]
- 9.Ogawa K, Matsushita Y, Yamawaki I, Kaneda M, Shibata J, Toko T, et al. Synthesis and antiestorgenic activity of the compounds related to the metabolites of (Z)-4-[1-[4-[2-(dimethylamino)ethoxy]phenyl]-2-4-isopropylphenyl)-1-butenyl]phenyl monophosphate (TAT-59) Chemical and Pharmaceutical Bulletin. 1991;39:911–916. doi: 10.1248/cpb.39.911. [DOI] [PubMed] [Google Scholar]
- 10.Sun D, Sharma AK, Dellinger RW, Blevins-Primeau AS, Balliet RM, Chen G, et al. Glucuronidation of active tamoxifen metabolites by the human UDP glucuronosyltransferases. Drug Metabolism and Disposition. 2007;35:2006–2014. doi: 10.1124/dmd.107.017145. [DOI] [PubMed] [Google Scholar]
- 11.For related analytical methods see Fauq AH, Maharvi GM, Sinha D. A convenient synthesis of (Z)-4-hydroxy-N-desmethyltamoxifen (endoxifen) Bioorganic & Medicinal Chemistry Letters. 2010;20:3036–3038. doi: 10.1016/j.bmcl.2010.03.117.; Teunissen SF, Rosing H, Koornstra RHT, Linn SC, Schellens JHM, Schinkel AH, et al. Development and validation of a quantitative assay for the analysis of tamoxifen with its four main metabolites and the flavonoids daidzein, genistein and glycitein in human serum using liquid chromatography coupled with tandem mass spectrometry. Journal of Chromatography B. 2009;877:2519–2529. doi: 10.1016/j.jchromb.2009.06.029.; Lee KH, Ward BA, Desta Z, Flockhart DA, Jones DR. Quantification of tamoxifen and three metabolites in plasma by high-performance liquid chromatography with fluorescence detection: application to a clinical trial. Journal of Chromatography B. 2003;791:245–253. doi: 10.1016/s1570-0232(03)00218-6.; Li XF, Carter S, Dovichi NJ, Zhao JY, Kovarik P, Sakuma T. Analysis of tamoxifen and its metabolites in synthetic gastric fluid digests and urine samples using high-performance liquid chromatography with electrospray mass spectrometry. Journal of Chromatography A. 2001;914:5–12. doi: 10.1016/s0021-9673(01)00538-6.; Berthou F, Dreano Y. High-performance liquid chromatographic analysis of tamoxifen, toremifene and their major human metabolites. Journal of Chromatography. 1993;616:117–127. doi: 10.1016/0378-4347(93)80478-m.; Gjerde J, Kisanga ER, Hauglid M, Holm PI, Mellgren G, Lien EA. Identification and quantification of tamoxifen and four metabolites in serum by liquid chromatography–tandem mass spectrometry. Journal of Chromatography A. 2005;1082:6–14. doi: 10.1016/j.chroma.2005.01.004.; Jones RM, Yuan ZX, Lamb JH, Lim CK. On-line high-performance liquid chromatographic-electrospray ionization mass spectrometric method for the study of tamoxifen metabolism. Journal of Chromatography A. 1996;722:249–255. doi: 10.1016/0021-9673(95)00794-6.
- 12.Ali SM, Ahmad A, Shahabuddin S, Ahmad MU, Sheikh S, Ahmad I. Endoxifen is a new potent inhibitor of PKC: a potential therapeutic agent for bipolar disorder. Bioorganic & Medicinal Chemistry Letters. 2010;20:2665–2667. doi: 10.1016/j.bmcl.2010.02.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.