

Abstract
The protonation states of the two active-site lysines (Lys69 and Lys235) of PBP 6 of Escherichia coli were explored to understand the active site chemistry of this enzyme. Each lysine was individually mutated to cysteine, and the resultant two mutant proteins were purified to homogeneity. Each protein was denatured, and its cysteine was chemically modified to produce an S-aminoethylated cysteine (γ-thialysine) residue. Following renaturation, the evaluation of the kinetics of the DD-carboxypeptidase activity of PBP 6 as a function of pH was found consistent with one lysine in its free-base (Lys69) and the other in the protonated state (Lys235) for optimal catalysis. The experimental estimates for their pKa values were compared to the pKa values calculated computationally, using molecular-dynamics simulations and a thermodynamic cycle. Study of the γ-thialysine 69 showed that lysine at position 69 influenced the basic limb of catalysis, consistent with the fact that the two lysine side chains are in proximity to each other in the active site. Based on these observations, a reaction sequence for PBP 6 is proposed, wherein protonated Lys235 serves as the electrostatic substrate anchor and Lys69 as the conduit for protons in the course of the acylation and deacylation half-reactions.
Keywords: pKa, Molecular Dynamics, Thermodynamic integration, DD-carboxypeptidase, pH Profile
INTRODUCTION
A hallmark of enzymatic catalysis is expeditious proton motion. This hallmark is attained by optimization of the positions of the amino-acid side chains, and of the acidity (and basicity) of the titratable functionalities of both protein and substrate, for facile engagement of transition states. Exactly how this engagement is accomplished, however, is a matter of continuing discussion for biological catalysis, as a result of the experimental difficulty in determining the electrostatic environment experienced by the transition states of these catalyzed reactions. The seemingly simple assignment of pKa values to the catalytic residues of enzymes remains one of the most experimentally difficult determinations. An example of this challenge is the penicillin-binding proteins (PBPs), enzymes that are involved in the biosynthetic and remodeling steps of bacterial peptidoglycan (the major constituent of the cell wall), and the enzymes that are the molecular targets of the β-lactam antibiotics. These active sites present an evolutionarily conserved pair of lysines and a pair of serines.1 The conformation of the important active-site residues in Escherichia coli PBP 6 is shown in Fig. 1, as they are observed crystallographically at pH 4.5. The conformations of these residues at the optimal pH for catalysis are highly similar (vide infra). The precise role(s) for these interacting residues in the completion of PBP catalysis is a long-standing debate.2,3 Here, we apply an experimental approach that we have used previously to clarify the roles of the lysines in the PBP and serine β-lactamase mechanisms, that of the replacement of active-site lysines by γ-thialysine residues.4,5 The catalytic effect of the γ-thialysine replacement is evaluated for this PBP both by computation and by kinetic analysis.
Figure 1.

(A) A stereoview ribbon representation of the three-dimensional structure of penicillin-binding protein 6 (PBP 6) from the x-ray structure at pH 4.5 (PDB entry: 3ITB). The catalytic domain is shown in green (the arrow at 3 o’clock identifies the domain boundary), while the stalk domain is shown in light orange. The arrow at the lower left (7 o’clock) points to the C-terminus of the protein, where the anchoring domain (not shown) to the inner membrane of the bacterium is located. The arrow at 1 o’clock points to the active site. (B) Stereoview of the closeup of the active site shown as Connolly surface in green. The two lysines and serines of the active sites are depicted in capped sticks, with atoms colored by atom types (carbons in gray, nitrogens in blue, and oxygens in red).
The central mechanistic pathway used by the PBPs (and also used by their evolutionary descendants, the serine-dependent β-lactamase, antibiotic-resistance enzymes) is promotion of one of the active-site serines as a nucleophile to accomplish an acyl-transfer reaction. The substrate of the PBPs is the terminal -D-Ala-D-Ala motif of the peptide stems of the peptidoglycan. Nucleophilic addition of this serine to the penultimate D-Ala of this motif gives, after tetrahedral collapse, an acyl-enzyme intermediate (Scheme 1). This acyl-enzyme is used by the biosynthetic PBPs for transfer to an amine nucleophile from another strand of the peptidoglycan to complete the transpeptidation event of cell-wall biosynthesis, and by the carboxypeptidase PBPs for transfer to a water molecule during a reaction that moderates the degree of cross-linking of cell wall. As the same serine is acylated by the β-lactam antibiotics during their covalent inhibition of the PBPs by formation of a stable acyl-enzyme, the identity of the nucleophilic serine is known with confidence. Moreover, as this serine is seen by crystallography to be in intimate hydrogen-bond contact with one of the active-site lysines, the premise of a general-base role for this lysine in activating the nucleophilic serine is substantiated by both experimental and computational studies. Among these experiments is the separate replacement of the two active-site lysines of the E. coli PBP 5 carboxypeptidase, a close homolog of the E. coli PBP 6 carboxypeptidase, by a γ-thialysine residue.5 γ-Thialysine is a lysine isostere that is characterized by a shorter (by approximately 1 Å) Cα to Cε distance6 and a diminished Nζ basicity of approximately 10-fold,7 both as a consequence of the replacement of the γ-CH2 by a sulfur atom. Previously, γ-thialysine replacement of the lysine hydrogen-bonded to the nucleophilic serine of the soluble (that is, lacking the membrane-anchor domain) E. coli PBP 5 enzyme showed a diminished kcat further characterized by an acid-shift in the pH optimum of this enzyme, consistent with an unprotonated ionization state for this lysine in the catalytically active form of this enzyme. An identical outcome for thialysine replacement of a catalytic lysine residue is seen with other enzymes.8–10 In contrast, replacement of the other active-site lysine of E. coli PBP 5 showed a reduced kcat without an acid-shift as a function of pH, consistent with a protonated ionization state for this other lysine in catalysis.5
Scheme 1.
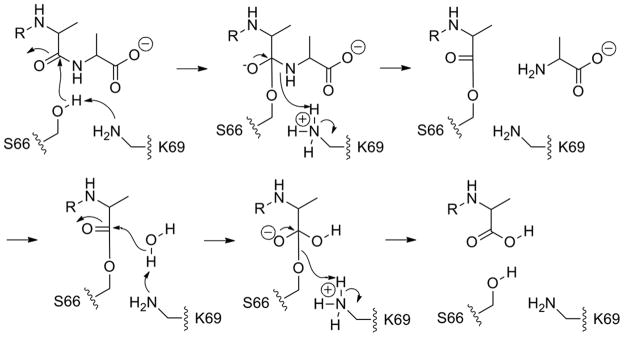
Proton-transfer events in the catalytic event. As Lys69 activates Ser66, it shuttles the proton to the nitrogen of the departing terminal D-Ala, whereby it reverts back to the free-base state. The free-base Lys69 now promotes a water molecule for the deacylation step; hence PBP 6 enjoys symmetry in catalysis. The proton from the water molecule is shuttled to the departing serine Oγ, returning Lys69 to its ground-state free-base form.
Bacteria express a family of PBP enzymes. Expression of a small ensemble of the biosynthetic PBPs involved in DD-transpeptidase reaction is required for viability.11 Under laboratory conditions, expression of the DD-carboxypeptidase PBPs is not critical for viability, although the broad evolutionary retention of these genes across bacterial genera strongly implies value for these DD-carboxypeptidases.12 Indeed, the most abundant PBP in E. coli (as measured by copies per cell) is the PBP 5 DD-carboxypeptidase13 and its deletion directly correlates to morphological defects (cells showing altered diameters and cells showing a less uniform outer contour) in their sacculus.14–16 The most closely related homolog in E. coli to PBP 5 is PBP 6, and indeed the key residues of their active sites quite nearly superimpose (Fig. S1 and S2). Notwithstanding this high homology, PBP 6 is unable to substitute in vivo for the PBP 5 function in E. coli,17 and is incapable as a DD-carboxypeptidase with respect to the endogenous peptidoglycan.15 Hence, the two homologous DD-carboxypeptidases of E. coli—PBPs 5 and 6—are likely to have distinct functional roles.18 The basis for this dichotomy of close structures, yet dissimilar catalytic capabilities, has been discussed recently in depth.19 This same dichotomy—now also approached from the perspective of our high-resolution structures of PBP 6 as an apo-enzyme, as a β-lactam-inactivated acyl-enzyme, and as a Michaelis (pre-acylation) complex with substrate20—stimulated this further kinetic study of E. coli PBP 6. We undertook this investigation of the catalytic reaction of PBP 6 of E. coli to complement our earlier studies with PBP 5 of the same organism. Our approach is the evaluation of the pKa values of the two lysines within the active site (Fig. 1), further examined by computational studies and culminating with a proposal for the mechanism of its in vitro carboxypeptidase reaction.
MATERIALS AND METHODS
Materials
CH Sepharose 4B resin, D-Ala, horseradish peroxidase (HRP, Type X), FAD, D-amino acid oxidase (DAAO), Triton X-100, ampicillin, kanamycin and Ac2-L-Lys-D-Ala-D-Ala were purchased from Sigma. DNA-modifying enzymes were purchased either from New England Biolabs or Stratagene. The QuantaBlu™ substrate solution was purchased from Pierce Chemical. LB Broth and isopropyl-β-D-thiogalactoside (IPTG) were purchased from Fisher Scientific. Ampicillin-linked CH-Sepharose 4B resin was prepared by mixing 600 mg of ampicillin with 10 mL of an aqueous suspension of 3.5 g of activated CH-Sepharose 4B for 3.5 h at 30 °C, according to the procedure reported previously.21
Cloning of the dacC Gene for Cytoplasmic Expression of Soluble PBP6 and Its Variants
We previously described cloning of a soluble E. coli PBP 6 (sPBP6) lacking both the signal peptide and the anchor domain.20 The requisite mutants for the present study were made using QuikChange® site-directed mutagenesis (Stratagene). The gene for the C137S (serine replacing the wild-type cysteine at position 137) PBP 6 mutant used [5′-GGTAATGACGCCTCTATTGCGCTGGCTG-3′] and [5′-CCATTACTGCGGAGATAACGCGACCGAC-3′] as primers (mutated codons are underlined). This procedure eliminated the cysteine in the wild-type enzyme, which is at a site remote from the active site. For the preparation of the C137S/K69C PBP 6 double mutant, we used [5′-CCCGCGAGCCTGACTTGCATCATGACCAGCTATG-3′] and [5′-GGGCGCTCGGACTGAACGTAGTACTGGTCGATAC-3′] as primers. Similarly, for the preparation of the PBP 6 C137S/K135C double mutant we used [5′-GAATGTTGATGGCATGTGCACAGGAACCACGGCAGG-3′] and [5′-CTTACAACTACCGTACACGTGTCCTTGGTGCCGTCC-3′] as primers. These primers included the recognition sequences for restriction endonucleases NdeI and HindIII. The PCR product was purified by electrophoresis, digested by NdeI and HindIII and cloned into the corresponding sites of the pET24a(+) (Novagen). The nucleotide sequences of the mutant genes, obtained from several transformants, were verified by sequencing of both DNA strands. The plasmids were used to transform E. coli BL21(DE3) for protein expression.5
Expression and Purification of sPBP 6 and Its Mutant Variants
Purification of sPBP 6 and its variants was performed at 4 °C using modifications of our previous procedure.20 E. coli cells transformed with a particular expression plasmid were incubated overnight in 5 mL of LB medium supplemented with 30 μg/mL of kanamycin. This culture was diluted into 0.5 L of fresh kanamycin-supplemented (30 μg/mL) LB medium. Growth was continued with mixing (140 rpm) at 37 °C. When the culture reached an OD600 of 0.8, IPTG was added to a final concentration of 0.4 mM. The culture was incubated at 20 °C for 12 h. Cells were harvested by centrifugation for 15 min at 3000 g. The cell paste was re-suspended in 20 mM NaHCO3 pH 9.5 buffer containing 0.1% Triton X-100. Cells were disrupted by 30 cycles of sonification using 30 s burst and 20 s of rest. The supernatant obtained after removal of cell debris (21,000 g for 40 min) was mixed with 3.5 g of ampicillin-linked CH Sepharose 4B resin.21 This enzyme-binding step was performed on a shaker at room temperature for 30 min for the wild-type sPBP 6 or for 2 h for the PBP 6 variants. Because of high pH of the medium, the reactions of the resin and the mutant enzymes worked well, except the losses (unbound protein) were as much as 60% of the total expressed PBP 6 variant. The resin was transferred to a sintered glass funnel, and was washed over a 30 min interval with 1 L of cold (4 °C) 20 mM Tris pH 9.0 buffer containing 1 M NaCl and 0.1% Triton X-100. PBP 6 was eluted from the resin by gentle stirring of the resin for 20 min at room temperature in 0.5 M Tris pH 9.0 buffer containing 1 M NH2OH. The filtrate, containing PBP 6 (or variants), was dialyzed at 4 °C against 25 mM NaHCO3, pH 9.5 buffer containing 0.15 M NaCl and 0.02% Triton X-100. The protein solution was concentrated using an Amicon concentrator (Mr 5,000 cut-off) to give an approximate enzyme concentration of 10 mg/mL in each case. Glycerol was added to give a final concentration of 20% by volume. The enzymes were stored in this solution at −80 °C. Representative yields from a 0.5 L cell culture were 8–25 mg of each PBP 6 (wild type and mutant variants). Protein concentrations were determined using the BCA Protein Detection Kit (Pierce). The purity of each PBP 6 enzyme as assessed by SDS-PAGE exceeded 95%.
Chemical Modification of the Lys69Cys/Cys137Ser and Lys235Cys/Cys137Ser Mutant Proteins
The procedure of Zhang et al.5 was used for γ-thialysine generation from the individual cysteine-containing proteins, using 5 mg portions of the double-mutant PBP 6 variants. The cysteine-containing mutant protein was denatured by gentle mixing in 200 mM AMPSO, 15 mM EDTA pH 8.5 buffer supplemented with 8 M urea, in a final volume of 10 mL, for 30 min at 25 °C under an argon atmosphere. Removal of a portion of this solution confirmed the presence of 1.0 equiv of cysteine by DTNB titration. A freshly prepared solution of 2-bromoethylamine (final concentration of 50 mM) in the denaturing buffer (200 mM AMPSO, 15 mM EDTA pH 8.5 buffer supplemented with 8 M urea) was added to the remainder of the solution. The reaction mixture was gently stirred for 20 h at 25 °C under an atmosphere of argon, at which point DTNB titration indicated complete cysteine modification. The protein solution was then diluted into the refolding buffer (100 mM sodium phosphate, 100 mM ammonium sulfate, 0.001% Triton X-100 pH 8.5; total volume of 0.5 L). Stepwise removal of urea and ammonium sulfate was carried out by dialysis, first against 25 mM NaHCO3 pH 9.0 buffer containing 0.15 M NaCl, 0.02% Triton X-100, and 60 mM (NH4)2SO4, followed by dialysis against the same buffer containing 30 mM (NH4)2SO4, and finally dialysis against the same buffer without (NH4)2SO4. At the completion of each reaction, the individual γ-thialysine-modified enzyme solutions were concentrated to approximately 2.5 mg/mL for storage as stock solutions.
The two cysteine-mutant proteins were assessed for the completeness of cysteine modification (DTNB titration of 1.0 cysteine) and for purity (SDS-PAGE), as discussed above. Isoelectric focusing of the three proteins further verified their homogeneity. The calculated molecular mass for the two mutants obtained from their respective ESI MS spectra was identical, and within experimental error was the same as the mass obtained for sPBP 6 (Supporting Information). CD analyses of the three enzymes, obtained at three different pH values (7.0, 9.5, and 11.5) showed superimposable spectra (Fig. S3). Time-dependent evaluation of the intensity of the intrinsic protein fluorescence at pH 11.5 for the sPBP 6 and for the γ-thialysine69 PBP 6 mutant showed partial loss over a 2 h interval (Fig. S4), indicating that denaturation of these proteins at pH 11.5 is not rapid, and supporting the validity of the kinetics measured over a shorter time interval (30 min).
The pH Profile for the DD-Carboxypeptidase Reaction
DD-Carboxypeptidase activity was measured using a fluorescence-based assay at the different buffered pH values as was described previously.5,22 The buffers used were 100 mM Tris·HCl (pH 7.0–9.0), 100 mM NaHCO3 (pH 9.0–11.0), and 100 mM NaPi (pH 11.0–12.0), each containing 150 mM NaCl and 0.5 mg/mL BSA. The standard assay mixture (40 μL) consisted of Ac2-L-Lys-D-Ala-D-Ala (0.7 to 17 mM) as the DD-carboxypeptidase substrate. The concentration of PBP 6 used in the assay was 0.21 μM. Reaction time points were obtained following 30 min incubation at 25 °C. Assay values were obtained by addition to the assay mixture to the detection reagent (10 μL QuantaBlu™ substrate solution and 0.5 mg/mL ampicillin, 1.0 unit of HRP, 0.06 units of D-amino acid oxidase, and 2.8 μg/mL FAD in of 350 μL of 0.1 M Tris·HCl pH 8.5 buffer), to give a total volume of 400 μL. After a 60 min development period, the fluorescence intensity of each point was measured using an excitation wavelength of 325 nm and an emission wavelength of 410 nm. The fluorescence intensity was converted to D-Ala concentration by the use of a standard curve. Control measurements (substrate in the absence of enzyme, and enzyme in the absence of substrate) were also performed. We performed non-linear regression of the kinetic data to the Michaelis-Menten equation using GraFit 4.0 (Erithacus Software). The resulting data for the pH dependency of kcat and kcat/Km for γ-thialysine69 sPBP 6 and γ-thialysine 235 sPBP 6 were fit to the following equation for a double ionization-dependency of the reaction velocity as a function of pH:
| Eq. 1 |
The data for wild-type sPBP 6 were fit to a triple-ionization model:
| Eq. 2 |
Computational Methods
Initial atomic coordinates for the PBP 6 structure were obtained by extracting monomer A from the 1.8 Å-resolution crystal structure of the PBP 6 apo-enzyme (PDB entry: 3ITB), using SYBYL 8.1 (Tripos).23 Hydrogen atoms were added using the AMBER (2008) program. The system, including the 98 crystallographic water molecules found within 2.5 Å of the protein surface, was fully solvated within a rectangular solvent box (TIP3P water model). The final system contained 67,941 atoms. AMBER ff99 forcefield provided the molecular mechanical parameters. The water molecules were equilibrated initially, while protein atom coordinates were restrained. Then, the system was heated to 300 K over 20 ps and equilibrated for 500 ps, prior to the beginning of the pKa calculation cycle.
The pKa was calculated using a thermodynamic cycle as described elsewhere.24–26 The free-energy change associated with Lys69 deprotonation (K69+ → K69 + H+) constitutes one half of the thermodynamic cycle, and the deprotonation of Lys331 as a surface lysine residue (K331+ → K331 + H+) constitutes the other half.
Using the relationship between different free-energy quantities of the thermodynamic cycle, the pKa difference between Lys69+ and Lys331+ can be written as in Eq. 2,27 where ΔΔGhyd(X − X+) = ΔGhyd(X) − ΔGhyd(X+) and X represents either lysine. The calculated value of 1/2.303RT is 0.729. Thus, evaluation of ΔΔGhyd(K69 − K69+) and ΔΔGhyd(K331 − K331+) give the pKa difference between the two lysines. By assigning to Lys331+ an experimentally determined consensus value of 10.4 for the pKa of the Nζ amine of a lysine within a peptide,28,29 the pKa of Lys69 can be obtained. The ΔΔGhyd values are determined using a biasing potential,30 followed by a thermodynamic integration methodology implemented within the AMBER program. The biasing potential V(λ) is defined by Eq. 3. The biasing potential evaluates the change in the system going from the protonated (P) to the unprotonated (U) state in terms of λ, the coupling parameter, where 0 ≤ λ ≤ 1. Thus, λ is varied stepwise between 0 and 1. At each λ value, a simulation was carried out using the last set of coordinates from the previous λ. This method samples multiple conformations and thus it is largely independent of the starting set of crystallographic coordinates.
The free-energy difference between the two states is given by Eq. 4, where the integral can be approximated by an nth-order Gaussian quadrature formula and where the weight of the ith quadrature is wi.31 We used a 12th order Gaussian quadrature. Thus twelve simulations of 1.5 ns duration each were carried out for each individual lysine, according to the AMBER formulation. Due to non-convergence, data from the first 500 ps of each simulation were excluded from the free-energy calculations. The λ = 0 state represented the protonated state of Lys69 and λ = 1 represented the unprotonated state of Lys69. These two states were each simulated for an additional 40 ns and averaged structures were extracted from the coordinates of the final 1 ns interval. Subsequently, each averaged structure was subjected to conjugated-gradient energy minimization. The same procedure was then followed to calculate pKa of Lys235 with Lys331 serving as the one with the known pKa. For this case, Lys69 was kept in the free-base form.
| Eq. 3 |
| Eq. 4 |
| Eq. 5 |
RESULTS AND DISCUSSION
γ-Thialysine Replacements for the PBP 6 Active-Site Lysines
The protonation states of the two active-site lysines at optimal catalysis by PBP 6 were evaluated by the replacement of each with a γ-thialysine residue. Substitution of γ-thialysine for lysine was done by performing a specific lysine to cysteine mutation, purification of the mutant protein to homogeneity, followed by protein denaturation to allow modification of the cysteine thiol to produce S-aminoethylated cysteine (γ-thialysine). Protein refolding gave the requisite γ-thialysine-containing enzyme. As the native structure of PBP 6 has a non-catalytic cysteine away from the active site, conservative replacement of this cysteine with a serine concurrent with the active-site lysine to cysteine mutations was required to enable selective formation of γ-thialysine only within the active site. Hence, the PBP 6 proteins used for this study were triple-mutant constructs. The first mutation deleted the PBP 6 membrane-anchor, so as to render the enzyme water-soluble. The second mutation replaced the non-active-site cysteine with a serine, a substitution that we documented to have no effect on enzyme activity, and consistent with the earlier observation of Nicholas and Strominger.21 The third mutation involved separate replacement of each active-site lysine to cysteine. Accordingly, two mutant PBP 6 enzymes were made. Both mutants are without the membrane anchor and have a C137S exchange. The unmodified Lys69Cys and Lys235Cys mutants of sPBP 6 were evaluated as a prelude to mechanistic study of the γ-thialysine variants. Both unmodified variants were essentially devoid of activity (0.8% and 1.1% of the wild-type sPBP 6 activity, respectively), considering that the in vitro activity that we measure for PBP 6 is already modest (discussed further below). Transformation of the cysteine of the Cys69 mutant and of the Cys235 mutant to γ-thialysine restored significant activity (18% activity for the γ-thialysine69 enzyme and 37% activity for the γ-thialysine235 enzyme, as measured by kcat), compared to the wild-type sPBP 6 at the respective optimal pH values. These activity differences are consistent with our previous observations on γ-thialysine substitution for the active-site lysines of the TEM-1 β-lactamase and of PBP 5.4,5
The pH Dependence of Kinetics of the γ-Thialysine PBP 6 Variants
The pH dependence of the DD-carboxypeptidase activities of sPBP 6 and its γ-thialysine69 and γ-thialysine235 mutants were evaluated over the pH range of 7 to 12 using Ac2-L-Lys-D-Ala-D-Ala as substrate (Fig. 2 and Fig. 3). The optimal pH was determined for this substrate with respect to kcat (providing a pKa of the enzyme-substrate complex) and to kcat /Km (providing a pKa of the free enzyme or free substrate). Optimal catalysis for the wild-type enzyme with respect to kcat was at pH 10.5 and with respect to kcat/Km was at pH 10. Optimal catalysis for the both γ-thialysine mutants, with respect to both kcat and kcat/Km, occurred at pH 9.2–9.5. The bell-shaped kinetics for the γ-thiaLys69 PBP 6 mutant were governed by two pKa values of approximately 8.4 and 10.3 (8.1 and 10.4 as determined by kcat/Km; 8.7 and 10.3 as determined by kcat), giving an approximately pH 9.3 optimum. The bell-shaped kinetics for the γ-thiaLys235 PBP 6 mutant were governed by two pKa values of approximately 8.7 and 10.0 (8.4 and 10.1 as determined by kcat/Km; 8.9 and 10.0 as determined by kcat), giving an approximately pH 9.4 optimum. The determined pKa values are summarized in Table I.
Figure 2.

The pH profiles of kcat/Km (A) and kcat (B) of γ-thialysine69 PBP 6 compared to sPBP 6. The data for γ-thialysine69 PBP 6 are depicted as open squares with respect to the values of the right y-axis. The data for sPBP 6 are shown using closed circles with respect to the values of the left y-axis. The error bars represent the standard errors for each data point. Data points are the average of two to four determinations. γ-Thialysine69 PBP 6 data are fit to Equation 1, while the wild-type data are fit to equation 2.32 The lines indicate the results of data fitting.
Figure 3.

The pH profiles of kcat/Km (A) and kcat (B) of γ-thialysine235 PBP 6 (closed circles with respect to the values of the left y-axis) compared to sPBP 6 (open rhombus with respect to the values of the right y-axis). Data points are the average of two to four determinations. The data for sPBP 6 shown in Fig. 2 are redrawn to facilitate comparison. γ-Thialysine235 PBP 6 data are fit to Equation 1, while the wild-type data are fit to equation 2. The lines indicate the results of data fitting.
Table I.
Summary of experimentally estimated pKa values using the respective kcat/Km and kcat (within parenthesis) profiles.
| sPBP 6 Species | Acidic Limb pKa | Basic Limb pKa |
|---|---|---|
| Wild-typea | 8.3 (8.4), 9.0 (9.4) | 11.1 (11.4) |
| γ-thialysine69b | 8.1 (8.7) | 10.4 (10.3) |
| γ-thialysine235b | 8.4 (8.9) | 10.1 (10.0) |
Fit to the triple ionization model.
Fit to the double ionization model.
The kinetics for sPBP 6 were not cleanly bell-shaped (Fig. 2). Analysis of the sPBP 6 kinetics required a fit according to the model of Thomas et al. for three ionizable groups,32 coinciding to two residues influencing the acid limb (pKa values of 8.3 and 9.0) and a single pKa of 11.1 for the basic limb. We observed previously a similar behavior for sPBP 5.5 The values of the slopes of the two limbs for log(kcat) or log(kcat/Km) versus pH plots reflect the number of residues that contribute to the titration.33 The data of Figure 4 show slopes of 1.5 and 2.0 for the acidic limb (two titrating functional groups), and the higher-pH limb slopes show slopes of 2.4 and 2.5 (two to three contributors). These slopes indicate the titration of at least one additional residue, likely outside of the active site, for each limb of catalysis. For the acidic limb of the wild-type sPBP 6 kcat/Km kinetics, it is reasonable to assume that the pKa of 8.3 corresponds to Lys69 titrating, as supported by the computational pKa determination (vide infra). The stability of the enzyme at pH 11.5 over the 30 min interval required for the kinetic determinations (Supporting Information) supports the conclusion that the decreasing activity at the highest pH values arises from catalytically unfavorable titration as opposed to enzyme denaturation. Zhang et al.5 suggested that a water molecule coordinated to the lysine that is activated for the deacylation step could contribute to the basic-limb of the pH profile. This ionization, and additional residues of the protein, may explain the steeper slope for the second limb compared to the first.
Figure 4.

The plots of log(kcat/Km) (A) and log(kcat) (B) of sPBP 6 versus pH.
Notwithstanding the challenge to the correlation of enzyme kinetics to discrete residue ionization, the specific replacement of Lys69 by γ-thialysine69 shifts the pH dependency of both limbs of catalysis: to pKa values of 8.7 and 10.3 from the kcat profile and to pKa values of 8.1 and 10.4 from the kcat/Km profile (Fig. 2). These values represent an approximately 0.9 pH unit downward change for the acidic pH limb and a lesser 0.7 pH unit downward change for the basic limb with respect to the kcat/Km profile. An alteration of approximately one pKa unit is the expectation for substitution of a γ-thialysine for a lysine, and the direction of the change in the acidic limb is entirely consistent with the diminished basicity that accompanies a thialysine for lysine exchange. The conclusion that Lys69 in its free-base form promotes catalysis (activation of Ser66 during acylation and of the hydrolytic water molecule during deacylation) is fully consistent with this observation and with the conclusions that we and others have made concerning the cognate active-site lysine of PBP 5.5,34 The basis for the smaller effect of the γ-thialysine69 substitution on the basic limb is less obvious. The suggestion that this change reflects an alteration of the electrostatic environment of Lys235 so as to lower its own pKa is reasonable (the two Nζ atoms of these lysines are separated by only 7 Å), but is speculative.
The γ-thialysine235 substitution likewise affects the pKa value(s) of the basic limb (a downward change of 0.6 pH unit) and acidic limb (a downward change of 1 pH unit), as shown by the kcat/Km data of Fig. 3. This result presents a similar interpretive dilemma: while the kinetic change for the basic limb is fully consistent with the loss of a protonated Lys235 to catalysis, an explanation for the effect of this mutation is less obvious. γ-Thialysine contrasts with lysine in terms of both structure and basicity. Given the extraordinary intimacy of the relationship between the dielectric of the active site and the pKa of residues within the active site,35 we can only reiterate that the experimental outcome is plausible. Both sets of mutagenesis experiments reveal influences on both the acidic and the basic limbs, indicative of the spatial proximity of the side chains of these two residues within the active site.
Computational Evaluation of the pKa Values
In order to substantiate our interpretation of the kinetics, a computational pKa evaluation of the two lysines of the PBP 6 active site was undertaken. In the simulations, the state of Lys69 was perturbed by the introduction of a proton, using a biasing potential V(λ) (Eq. 3). To keep this perturbation minimal, a series of simulations were carried out by varying the λ parameter between 0 and 1. Simulations at each λ were confirmed to have converged cumulative averages of ∂V/∂λ before the corresponding <∂V/∂λ> was evaluated. The computed difference between ΔΔGhyd(K69 − K69+) and ΔΔGhyd(K331 − K331+) was 2.88 kcal/mol. According to Eq. 2, this energy difference corresponds to a pKa difference of 2.1 units. Using the consensus value for the pKa of a solvent-exposed lysine (pKa = 10.4) in a peptide for the pKa Lys331, the pKa of Lys69 is computationally estimated as 8.3. This computed Lys69 pKa is in excellent agreement with the experimentally estimated pKa value of 8.3 for the catalytically dominant free-enzyme state obtained from the kcat/Km determination.
Longer simulations of the λ = 0 and λ = 1 states generated averaged structures from the stable phases of the simulations. These two states serve as the “reactant” and “product” states for the proton transfer. For these two states, we compared the calculated root-mean-square deviation (rmsd) for the protein backbone of the catalytic domain. The λ = 0 and λ = 1 (averaged and energy-minimized) states showed 1.1 and 1.6 Å backbone rmsd compared to the crystal structure. The rmsd between the λ = 0 and λ = 1 structures was 1.4 Å. Thus, the change of Lys69 from a protonated to an unprotonated state does not result in a large-scale adjustment of the protein backbone of the catalytic domain. This observation supports the assertion that the difference in the pKa of Lys69 (thialysine69, in the case of the γ–thialysine69 mutant) relative to Lys331 arises from the different hydrogen-bonding network of Lys69 in the two states, rather than a significant adjustment of conformations within the PBP 6 active site. An additional set of four implicit-solvent simulations with protonated/unprotonated lysine/thialysine variants of PBP6 verified this (Fig. S5). Protonated Lys69 interacts with Asn134 (Fig. 5A), which could be attributed to the acquired charge on the Lys69 Nζ. Whereas in the case of neutral Lys69 (Fig. 5B), this interaction is less obvious (Fig. 5C). When Lys69 is in the free-base form, the side-chain amine remains largely in hydrogen-bonding interaction with the hydroxyl of Ser66 for the duration of simulations (Fig. 5D).
Figure 5.

(A) Stereoview of the simulated λ = 0 (protonated Lys69) state of PBP 6 active site. Residues are shown in capped-stick representation (carbons in gray, nitrogens in blue and oxygens in red) in the same perspective as in Fig. 1. Asparagine134 (at 8 o’clock) interacts with the protonated side chain of Lys69, but not with its free-base form. The remaining residues of the enzyme are depicted as a Connolly surface, with a few surface residues hidden in order to expose the cavity. (B) The stereoview of the λ = 1 (free-base Lys69) state. (C) The interaction between the side chains of Lys69 and Asn134 throughout simulation, when Lys69 is in the free-base form (in red) and when protonated (in black). The respective running averages of the distance d1 are in yellow and green. The hydrogen-bonding interaction is stable when Lys69 is in the protonated form, but it is not when in the free-base form. (D) The interaction between the side chains of Lys69 and Ser66 throughout simulation, when Lys69 is in the free-base form (in red) and when protonated (in black). The respective running averages of the distance d2 are in yellow and green; hence when Lys69 is in the free-base form, the distance to Ser66 is both largely constant and within a hydrogen-bonding separation.
For Lys235, the computed difference between ΔΔGhyd(K235 − K235+) and ΔΔGhyd(K331 − K331+) was 1.10 kcal/mol. This energy corresponds to a pKa difference of 0.8 units, amounting to a calculated pKa for Lys235 of 9.6. The different pKa of Lys235 from that of a fully solvent-exposed lysine is presumably due to its electrostatic environment. Lysine235 is surrounded by other amino acids within the binding site, including the side chains of Lys69 and Ser131, and the backbone carbonyl oxygens of Ile129 and Ile130. Thus, its electrostatic environment is different from that of a surface fully solvent-exposed lysine, likely altering its pKa. We add that we did not observe a significant difference in the positioning of Lys235 side chain or its interactions with surrounding residues, between the free-base and the protonated forms. The energy-minimized averaged structure for the free-base form of Lys235 produced a backbone rmsd of 1.6 Å for the catalytic domain.
Given the two calculated lysine pKa values, the important inference here is that out of the two conserved lysines in the PBP 6 active site, Lys69 of the S-X-X-K motif is poised to serve as a catalytic base for the promotion of serine in the acylation event (and of water in the deacylation process). While the calculated pKa for Lys235 is somewhat lower than that of the consensus pKa of a fully solvent-exposed lysine and that of the experimentally determined value for Lys235, it is yet more than one pKa unit higher than that of Lys69. Additionally, as it has been observed frequently, we note that computational enzymatic pKa determination has its caveats and the generated pKa values must be treated with vigilance.36–38 Atypical environments such as is found for Lys235 may specifically challenge the computational pKa calculation.35 While the calculated pKa of Lys235 does not match to an experimentally assignable value, it certainly corroborates the assertion that Lys69 is the better poised to function as a base in the acylation half-reaction of PBP 6 (Scheme 1).
CONCLUSIONS
The context for a discussion of the mechanistic implications is framed through comparison of the E. coli PBP 5 and 6 enzymes. This study and our earlier study on PBP 5 represent a side-by-side comparison of two DD-carboxypeptidases from the same organism. Indeed, one of the most important questions with respect to the low-molecular-mass PBPs such as these two from E. coli is their respective functional roles.12 The two peptidoglycan DD-carboxypeptidases of E. coli, PBP 5 and 6, represent both profound similarity and profound contrast. The conformational states of their catalytic residues are virtually identical.1 Their cellular location—along the lateral cell wall, but with greater sequestration at the nascent division septum—is identical,16 but with PBP 5 two- to four-fold more abundant.13 These observations would seem to suggest catalytic redundancy—in nature catalytic redundancy often exists to safeguard important biological functions—but this suggestion is incorrect. The seminal work of Young and colleagues shows that PBP 6 is unable to substitute for PBP 5 for the regulation of the cell diameter and for the uniformity of the E. coli rod shape.14,39,40 This difference may reflect in part the temporal difference in their expression during planktonic growth (PBP 5 during early log phase and PBP 6 early stationary phase).41 Overproduction of PBP 5 in E. coli causes spherical morphology that is ultimately lethal, but this was not seen as a result of overproduction of PBP6.39 The key structural difference may be the size of the active-site cleft that governs substrate access to their active sites. Crystallographic structures show a smaller cleft for PBP 6 compared to PBP 5, and indeed PBP 6 accepts a more limited set of small substrates, such as X-D-Ala- D-Ala tripeptides, as compared to PBP 5.17 The spatial presentation of the catalytic amino acids in the active site of the two enzymes is virtually identical.16 In contrast, PBP 5 exerts DD-carboxypeptidase activity toward -D-Ala-D-Ala stem termini, regardless of whether this motif is encountered in small muropeptides or the much larger substrates Lipid II, or the cell wall.16,42 While this conclusion must be tempered by the recognition that these PBPs permit substantial domain motion,43 based on these (and other) observations the conclusion that might appear to hold is that PBP 5 has specific role(s) in cell-wall biosynthesis relating to the stem peptide -D-Ala-D-Ala DD-carboxypeptidase activity, whereas PBP 6 might not. Although the functional role of PBP 6 is important—the retention of the genes for all of the low-molecular-mass PBPs throughout time allows no other conclusion12—these other roles must be different from its documented DD-carboxypeptidase activity.
Accordingly, further comparison must be at the molecular level. The structures of both enzymes are uncannily similar, as mentioned above. The active sites have the canonical assembly of the two serines and two lysines found in other PBPs, all in spatial contact. Both enzymes undergo acylation by the peptidoglycan mimetic at one serine as the first step of catalysis. This catalytic serine for PBP 6 is Ser66, located on a helix dipole, a position expected to lower the pKa of its side-chain hydroxyl. This hydroxyl is also in hydrogen-bonding contact with the amine of Lys69, located one turn of the helix away. Our analysis disclosed in this report reveals that Lys69 exists in the free-base form for optimal catalysis, which is required for its general-base promotion of Ser66 as a nucleophile for acylation. Upon transfer of the proton from Ser66, the now-protonated Lys69 is spatially predisposed to donate the proton to the scissile nitrogen of the departing D-Ala.44,45 This proton transfer returns the lysine to its free-base state (Scheme 1). Approach of a water molecule (using the reverse trajectory of the departed D-Ala) so as to engage both the free-base Lys69 and acyl-enzyme sets in place the deacylation event (Scheme 1). Again, the transiently protonated Lys69 acts as the conduit for protonation of the ester oxygen to complete deacylation. Our studies indicate that Lys235 remains protonated throughout catalysis. Its location within the active site would preclude it from serving as a proton conduit for catalysis, but positions it so as to interact with the terminal D-Ala carboxylate of the substrate. Its function is likely to be the electrostatic anchor that positions the substrate into the active site. Indeed, this study complements our earlier studies with the E. coli PBP 5: an enzyme that has a virtually identical active site compared to PBP 6, and where the outcome of γ-thialysine substitution of the two active site lysines is essentially identical.1 A comprehensive discussion of the mechanistic breadth within the active sites of the PBP/β-lactamase amidohydrolase family is provided by Pratt and McLeish.3
Whereas the DD-carboxypeptidase activity of PBP 6 is quantifiable in vitro, it is decidedly low. The basis for the poor in vitro activity for PBP 6 (and for PBP 5) is not known. The greater activity suggested to occur for PBP 5 in vivo with respect to cell-wall biosynthesis may reflect further organization of its active site (not yet precedented by crystal structure) as a consequence of the proven interaction of PBP 5 with other enzymes,16 or influenced by the interaction of its membrane anchor or membrane-proximal surface with lipid. These same possibilities may hold for PBP 6. Alternatively, the primary function of E. coli PBP 6 might not be cell-wall biosynthesis. Certain low-molecular-mass PBPs in other Gram-negative bacteria participate in β-lactamase expression as a resistance response.46 In addition, the emerging role of alternative D-amino acids47–49 suggests that a solitary focus on the customary Ac2-L-Lys-D-Ala-D-Ala-type substrate might be incorrect. The exceptional crystallinity of the soluble E. coli PBP 6,20 foundational to further computational study, positions E. coli PBP 6 for these studies. These possibilities are subjects for future discovery.
Supplementary Material
Acknowledgments
NIH grant AI090348 provided financial support.
ABBREVIATION USED
- AMPSO
N-(1,1-dimethyl-2-hydroxyethyl)-3-amino-2-hydroxypropanesulfonic acid
- BSA
bovine serum albumin
- DTNB
5,5′-dithiobis-2-nitrobenzoic acid
- HRP
horseradish peroxidase
- IPTG
isopropyl-β-D-thiogalactoside
- MD
molecular dynamics
- PBP
penicillin-binding protein
- RMSD
root mean square deviation
- sPBP 6
the soluble Cys137Ser mutant of PBP 6
Footnotes
A comparison of the crystallographic structures for sPBP 5 and sPBP 6 used for the computational experiments; experimental characterization of the three folded PBP 6 enzymes and implicit-solvent simulations of the wild-type and thiaLys69 PBP 6.
References
- 1.Bobba S, Gutheil W. Multivariate geometrical analysis of catalytic residues in the penicillin-binding proteins. Int J Biochem Cell Biol. 2011;43(10):1490–1499. doi: 10.1016/j.biocel.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 2.Dzhekieva L, Rocaboy M, Kerff F, Charlier P, Sauvage E, Pratt R. Crystal structure of a complex between the Actinomadura R39 DD-peptidase and a peptidoglycan-mimetic boronate inhibitor: interpretation of a transition state analogue in terms of catalytic mechanism. Biochemistry. 2010;49(30):6411–6419. doi: 10.1021/bi100757c. [DOI] [PubMed] [Google Scholar]
- 3.Pratt R, McLeish M. Structural relationship between the active sites of β-lactam-recognizing and amidase signature enzymes: convergent evolution? Biochemistry. 2010;49(45):9688–9697. doi: 10.1021/bi1012222. [DOI] [PubMed] [Google Scholar]
- 4.Golemi-Kotra D, Meroueh S, Kim C, Vakulenko S, Bulychev A, Stemmler A, Stemmler T, Mobashery S. The importance of a critical protonation state and the fate of the catalytic steps in class A β-lactamases and PBPs. J Biol Chem. 2004;279(33):34665–34673. doi: 10.1074/jbc.M313143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang W, Shi Q, Meroueh SO, Vakulenko SB, Mobashery S. Catalytic mechanism of penicillin-binding protein 5 of Escherichia coli. Biochemistry. 2007;46(35):10113–10121. doi: 10.1021/bi700777x. [DOI] [PubMed] [Google Scholar]
- 6.Ammon H, Prasad S, Gerlt J. Structure of thialysine hydrochloride. Acta Cryst. 1991;C47:1476–1478. doi: 10.1107/s0108270190012835. [DOI] [PubMed] [Google Scholar]
- 7.Hopkins C, Hernandez G, Lee J, Tolan D. Aminoethylation in model peptides reveals conditions for maximizing thiol specificity. Arch Biochem Biophys. 2005;443:1–10. doi: 10.1016/j.abb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 8.Li C, Gershon PD. pKa of the mRNA cap-specific 2′-O-methyltransferase catalytic lysine by HSQC NMR detection of a two-carbon probe. Biochemistry. 2006;45(3):907–917. doi: 10.1021/bi051736h. [DOI] [PubMed] [Google Scholar]
- 9.Timms N, Windle CL, Polyakova A, Ault JR, Trinh CH, Pearson AR, Nelson A, Berry A. Structural insights into the recovery of aldolase activity in N-acetylneuraminic acid lyase by replacement of the catalytically active lysine with γ-thialysine by using a chemical mutagenesis strategy. ChemBioChem. 2013;14(4):474–481. doi: 10.1002/cbic.201200714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yakovleva L, Shuman S. Chemical mutagenesis of vaccinia DNA topoisomerase lysine 167 provides insights to the catalysis of DNA transesterification. Biochemistry. 2013;52(5):984–991. doi: 10.1021/bi301643h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denome S, Elf P, Henderson T, Nelson D, Young K. Escherichia coli mutants lacking all possible combinations of eight PBPs: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol. 1999;181(13):3981–3993. doi: 10.1128/jb.181.13.3981-3993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh A, Chowdhury C, Nelson D. Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends Microbiol. 2008;16(7):309–317. doi: 10.1016/j.tim.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Dougherty T, Kennedy K, Kessler R, Pucci M. Direct quantitation of the number of individual penicillin-binding proteins per cell in Escherichia coli. J Bacteriol. 1996;178(21):6110–6115. doi: 10.1128/jb.178.21.6110-6115.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson D, Young K. Penicillin binding protein 5 affects cell diameter, contour, and morphology of Escherichia coli. J Bacteriol. 2000;182(6):1714–1721. doi: 10.1128/jb.182.6.1714-1721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh A, Young K. Sequences near the active site in chimeric PBP 5 and 6 affect uniform morphology of Escherichia coli. J Bacteriol. 2003;185(7):2178–2186. doi: 10.1128/JB.185.7.2178-2186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potluri L, Karczmarek A, Verheul J, Piette A, Wilkin J, Werth N, Banzhaf M, Vollmer W, Young K, Nguyen-Disteche M, den Blaauwen T. Septal and lateral wall localization of PBP5, the major D,D-carboxypeptidase of Escherichia coli, requires substrate recognition and membrane attachment. Mol Microbiol. 2010;77(2):300–323. doi: 10.1111/j.1365-2958.2010.07205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chowdhury C, Nayak T, Young K, Ghosh A. A weak DD-carboxypeptidase activity explains the inability of PBP 6 to substitute for PBP 5 in maintaining normal cell shape in Escherichia coli. FEMS Microbiol Lett. 2010;303(1):76–83. doi: 10.1111/j.1574-6968.2009.01863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laubacher ME, Melquist AL, Chandramohan L, Young KD. Cell sorting enriches Escherichia coli mutants that rely on peptidoglycan endopeptidases to suppress highly aberrant morphologies. J Bacteriol. 2013;195(4):855–866. doi: 10.1128/JB.01450-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chowdhury C, Ghosh A. Differences in active-site microarchitecture explain the dissimilar behaviors of PBP5 and 6 in Escherichia coli. J Mol Graph Model. 2011;29(5):650–656. doi: 10.1016/j.jmgm.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Zhang W, Shi Q, Hesek D, Lee M, Mobashery S, Shoichet B. Crystal structures of penicillin-binding protein 6 from Escherichia coli. J Am Chem Soc. 2009;131(40):14345–14354. doi: 10.1021/ja903773f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholas RA, Strominger JL. Site-directed mutants of a soluble form of penicillin-binding protein 5 from Escherichia coli and their catalytic properties. J Biol Chem. 1988;263(4):2034–2040. [PubMed] [Google Scholar]
- 22.Gutheil WG, Stefanova ME, Nicholas RA. Fluorescent coupled enzyme assays for D-alanine: application to penicillin-binding protein and vancomycin activity assays. Anal Biochem. 2000;287(2):196–202. doi: 10.1006/abio.2000.4835. [DOI] [PubMed] [Google Scholar]
- 23.Tripos. Sybyl 8.1. St. Louis, MO: Tripos Inc; 2008. [Google Scholar]
- 24.Warshel A, Russell S. Calculations of electrostatic interactions in biological systems and in solutions. Q Rev Biophys. 1984;17(3):283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- 25.Russell S, Warshel A. Calculations of electrostatic energies in proteins. The energetics of ionized groups in bovine pancreatic trypsin inhibitor. J Mol Biol. 1985;185(2):389–404. doi: 10.1016/0022-2836(85)90411-5. [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen W, Briggs J, Gao J. A priori calculations of pKa’s for organic-compounds in water–the pKa of ethane. J Am Chem Soc. 1987;109:6857–6858. [Google Scholar]
- 27.Merz K. Determination of the pKa’s of ionizable groups in proteins: the pKa of Glu-7 and Glu-35 in hen egg-white lysozyme and Glu-106 in human carbonic anhydrase-II. J Am Chem Soc. 1991;113:3572–3575. [Google Scholar]
- 28.Thurlkill RL, Grimsley GR, Scholtz JM, Pace CN. pKa values of the ionizable groups of proteins. Protein Sci. 2006;15(5):1214–1218. doi: 10.1110/ps.051840806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pace CN, Grimsley GR, Scholtz JM. Protein ionizable groups: pKa values and their contribution to protein stability and solubility. J Biol Chem. 2009;284(20):13285–13289. doi: 10.1074/jbc.R800080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warshel A. Computer modeling of chemical reactions in enzymes and solutions. New York: John Wiley & Sons, Inc; 1991. [Google Scholar]
- 31.Simonson T, Carlsson J, Case D. Proton binding to proteins: pKa calculations with explicit and implicit solvent models. J Am Chem Soc. 2004;126(13):4167–4180. doi: 10.1021/ja039788m. [DOI] [PubMed] [Google Scholar]
- 32.Thomas B, Wang Y, Stein RL. Kinetic and mechanistic studies of penicillin-binding protein 2x from Streptococcus pneumoniae. Biochemistry. 2001;40(51):15811–15823. doi: 10.1021/bi011368r. [DOI] [PubMed] [Google Scholar]
- 33.Tipton K, Dixon H. Effects of pH on enzymes. Methods Enzymol. 1979;63:183–234. doi: 10.1016/0076-6879(79)63011-2. [DOI] [PubMed] [Google Scholar]
- 34.Stefanova ME, Davies C, Nicholas RA, Gutheil WG. pH, inhibitor, and substrate specificity studies on Escherichia coli penicillin-binding protein 5. Biochim Biophys Acta. 2002;1597(2):292–300. doi: 10.1016/s0167-4838(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 35.Chimenti MS, Khangulov VS, Robinson AC, Heroux A, Majumdar A, Schlessman JL, Garcia-Moreno B. Structural reorganization triggered by charging of Lys residues in the hydrophobic interior of a protein. Structure. 2012;20(6):1071–1085. doi: 10.1016/j.str.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mongan J, Case DA. Biomolecular simulations at constant pH. Curr Opin Struct Biol. 2005;15(2):157–163. doi: 10.1016/j.sbi.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Warshel A, Dryga A. Simulating electrostatic energies in proteins: perspectives and some recent studies of pKas, redox, and other crucial functional properties. Proteins. 2011;79(12):3469–3484. doi: 10.1002/prot.23125. [DOI] [PubMed] [Google Scholar]
- 38.Alexov E, Mehler EL, Baker N, Baptista AM, Huang Y, Milletti F, Nielsen JE, Farrell D, Carstensen T, Olsson MH, Shen JK, Warwicker J, Williams S, Word JM. Progress in the prediction of pKa values in proteins. Proteins. 2011;79(12):3260–3275. doi: 10.1002/prot.23189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson D, Young K. Contributions of PBP 5 and DD-carboxypeptidase PBPs to maintenance of cell shape in Escherichia coli. J Bacteriol. 2001;183(10):3055–3064. doi: 10.1128/JB.183.10.3055-3064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson D, Ghosh A, Paulson A, Young K. Contribution of membrane-binding and enzymatic domains of PBP 5 to maintenance of uniform cellular morphology of Escherichia coli. J Bacteriol. 2002;184(13):3630–3639. doi: 10.1128/JB.184.13.3630-3639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buchanan CE, Sowell MO. Synthesis of penicillin-binding protein 6 by stationary-phase Escherichia coli. J Bacteriol. 1982;151(1):491–494. doi: 10.1128/jb.151.1.491-494.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sauvage E, Powell A, Heilemann J, Josephine H, Charlier P, Davies C, Pratt R. Crystal structures of complexes of bacterial DD-peptidases with peptidoglycan-mimetic ligands: the substrate specificity puzzle. J Mol Biol. 2008;381(2):383–393. doi: 10.1016/j.jmb.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicola G, Fedarovich A, Nicholas R, Davies C. A large displacement of the SXN motif of Cys115-modified penicillin-binding protein 5 from Escherichia coli. Biochem J. 2005;392(Pt 1):55–63. doi: 10.1042/BJ20050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi Q, Meroueh S, Fisher J, Mobashery S. Investigation of the mechanism of the cell wall DD-carboxypeptidase reaction of penicillin-binding protein 5 of Escherichia coli by QM/MM calculations. J Am Chem Soc. 2008;130(29):9293–9303. doi: 10.1021/ja801727k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi Q, Fisher J, SM Transpeptidase reaction of penicillin-binding protein 1b of Streptococcus pneumoniae and cross-linking of the cell wall. J Am Chem Soc. 2011;133:5274–5283. doi: 10.1021/ja1074739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mark B, Vocadlo D, Oliver A. Providing β-lactams a helping hand: targeting the AmpC β-lactamase induction pathway. Future Microbiol. 2011;6(12):1415–1427. doi: 10.2217/fmb.11.128. [DOI] [PubMed] [Google Scholar]
- 47.Vollmer W, Blanot D, de Pedro M. Peptidoglycan structure and architecture. FEMS Microbiol Rev. 2008;32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 48.Lam H, Oh D, Cava F, Takacs C, Clardy J, de Pedro M, Waldor M. D-Amino acids govern stationary phase cell wall remodeling in bacteria. Science. 2009;325(5947):1552–1555. doi: 10.1126/science.1178123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cava F, de Pedro M, Lam H, Davis B, Waldor M. Distinct pathways for modification of the bacterial cell wall by non-canonical D-amino acids. EMBO J. 2011;30(16):3442–3453. doi: 10.1038/emboj.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.