

Abstract
Angiotensin-II production in the subfornical organ acting through angiotensin-II-type-1 receptors is necessary for polydipsia resulting from elevated renin-angiotensin system activity. Protein kinase C and mitogen-activated protein kinase pathways have been shown to mediate effects of angiotensin-II in the brain. We investigated mechanisms that mediate brain angiotensin-II-induced polydipsia. We used double transgenic sRA mice, consisting of human renin controlled by the neuron-specific synapsin promoter crossed with human angiotensinogen controlled by its endogenous promoter, which results in brain-specific overexpression of angiotensin-II, particularly in the subfornical organ. We also employed the deoxycorticosterone acetate-salt model of hypertension which exhibits polydipsia. Inhibition of protein kinase C, but not extracellular signal-regulated kinases, protein kinase A, or vasopressin V1A and V2 erceptors, corrected the elevated water intake of sRA mice. Using an isoform selective inhibitor and an adenovirus expressing dominant negative protein kinase C-α revealed that protein kinase C-α in the subfornical organ was necessary to mediate elevated fluid and sodium intake in sRA mice. Inhibition of protein kinase C activity also attenuated polydipsia in the deoxycorticosterone acetate-salt model. We provide evidence that inducing protein kinase C activity centrally is sufficient to induce water intake in water-replete wildtype mice, and that cell surface localization of PKC-α can be induced in cultured cells from the subfornical organ. These experimental findings demonstrate a role for central protein kinase C activity in fluid balance and further mechanistically demonstrate the importance of protein kinase C-α signaling in the subfornical organ in fluid intake stimulated by angiotensin-II in the brain.
Keywords: angiotensin II, drinking, brain, protein kinases, adenovirus, salt
Introduction
Angiotensin-II (ANG-II) is a known dipsogen which induces drinking in rats when administered peripherally or centrally.1–3 Mice fail to drink water in response to peripheral ANG-II, but do so after ANG-II is injected into the brain.4,5 Central ANG-II-induced fluid intake has been shown to be mediated by an ANG-II type-1 receptor (AT1R)-dependent pathway.6–8 AT1R is a G-protein coupled receptor (GPCR), which upon activation by ANG-II, induces release of Gαq/11 which can activate protein kinase C (PKC) via diacylglycerol and inositol triphosphate-induced calcium release.9 AT1R can also activate extracellular signal-regulated kinases 1 and 2 (ERK1/2) independently from the PKC pathway.10 PKC and ERK1/2 have both been shown to affect central ANG-II-mediated fluid intake and blood pressure, but their effects depend upon their site of action in the brain.11–13
ANG-II injected the lateral ventricle or directly into the subfornical organ (SFO) induces water intake within minutes through AT1R.2 Neuronal excitability is increased upon administration of ANG-II centrally or directly into the SFO.14,15 SFO neurons express outwardly rectifying potassium channels (IKv),16,17 and the excitatory effects of ANG-II were reported to be mediated by AT1R-dependent inhibition of Ikv.17,18 ANG-II activation of AT1R increases the activity of PKC-α centrally, and PKC-α mediates the AT1R-dependent inhibition of Ikv.19,20 Central inhibition of the conventional PKC isoforms, PKC-α/βII, with Gö-6976 attenuates induction of water intake in rats caused by administration of ANG-II within the lateral ventricle.20
Double transgenic (sRA) mice overexpressing human renin and angiotensinogen (AGT) selectively in the brain exhibit both hypertension and a marked increase in fluid intake.7,21 Normal C57BL/6 mice treated with deoxycorticosterone acetate and high salt (DOCA-salt) also exhibit polydipsia.22 It is well recognized that the DOCA-salt model of hypertension is a model of elevated brain angiotensin-II activity. Indeed, we recently showed that many of the cardinal phenotypes observed in the sRA transgenic model (hypertension, polydipsia and elevated resting metabolic rate) are also observed in DOCA-salt mice.22,23 The human angiotensinogen transgene is robustly expressed in the subfornical organ (SFO) in sRA mice, and Cre-recombinase-loxP-mediated ablation of its expression selectively in the SFO blunts fluid intake.7 Selective deletion of AT1AR in the SFO of DOCA-salt treated mice carrying a conditional allele of AT1AR (AT1ARflox) similarly blunts fluid intake.23 These studies highlight the importance of both ANG-II production and AT1R action in the SFO as an important mediator of the polydipsic response. Herein, we tested the hypothesis that PKC-α is an important mediator of ANG-II-dependent fluid intake in sRA and DOCA-salt mice, and that activation of PKC independently of ANG-II is sufficient to rapidly stimulate fluid intake.
Methods
Specific protocols, drug administration and detailed methods are provided in the expanded methods section available in the online-only data supplement.
Animals and surgery
Male and female transgenic mice 12–20 weeks of age were used in this study, and were IACUC approved in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. sRA mice are double transgenic mice derived from a cross of mice expressing human renin under the neuron-specific synapsin promoter (sR) with mice expressing human AGT under the control of its own endogenous promoter (A).7,21 Littermates were used as controls. For acute intracerebroventricular (ICV) injection or placement of a chronic ICV cannula, the lateral ventricle was targeted and injected as described previously.24 Ad5CMV-DN-PKC-α was the generous gift of Dr. Viswanathan Natarajan, University of Illinois, Chicago. Deoxycorticosterone-acetate (DOCA) pellets (50 mg; Sigma) were inserted as described previously.22 ICV mini-pumps (Alzet 2002) were implanted at the start of the second week of DOCA-salt.
Statistics
We analyzed the data with one- or two-way analysis of variance (ANOVA), with repeated measurements as appropriate. Bonferroni multiple comparisons procedures were used to further explore treatment effects. If equal variance or normality failed, we used non-parametric analysis of our data, such as Mann-Whitney U or Wilcoxon tests. We considered differences significant at P<0.05, and all data plotted are mean ± standard error of the mean (SEM).
Results
ICV Drug Panel in sRA mice
As a first pass to screen mechanisms mediating brain ANG-II-induced polydipsia, a panel of drugs was individually injected ICV into sRA mice and water intake after each administration was measured (Figure 1A). Sufficient time was allowed for recovery after each drug, and this was validated by measuring water intake after ICV injection of aCSF the day before the next drug was tested. Consistent with our previous study, water intake was elevated in sRA mice compared with controls (Figure 1B).21 The increase in water intake in sRA mice was corrected by BIM, a non-selective PKC inhibitor, which acts by blocking the activation domain (genotype x BIM interaction P=0.01; Figure 1B). FR180204, a direct inhibitor of ERK1/2, significantly decreased water intake of both littermate control and sRA mice; however, there was no significant interaction between genotype and drug (P=0.63). Water intake of sRA mice was still elevated compared to littermate controls after FR180204 (P<0.01). Neither conivaptan, an antagonist of arginine vasopressin V1A and V2 receptors, nor H-89, an inhibitor of PKA, had any effect upon water intake in sRA and controls. Consistent with the actions of BIM, inhibiting the regulatory domain of PKC with calphostin C corrected the elevated water intake in sRA mice such that it was not significantly different from controls (P=0.83). Polydipsia in sRA mice was retained at the end of the experiment although it was blunted. These data suggest that the multiple testing procedure was not deleterious and the washout periods were sufficient. Thus, this initial screen identified the PKC pathway as an important mechanism regulating brain ANG-II-induced polydipsia in sRA mice.
Figure 1. Initial Pharmacological Screen in sRA Mice.

Twenty-four hour water intake in sRA (n=4–7) and littermate control mice (n=4–8) was examined pharmacologically using the protocol shown (A). sRA mice drank more water than littermate controls with aCSF, FR180204 (4 μg in 2 μL DMSO), conivaptan (0.8 μg in 2 μL of aCSF), and H-89 (4 μg in 2 μL DMSO). sRA water intake was not significantly different from littermate controls when given BIM (32 μg in 2 μL DMSO) or Calphostin C (0.4 μg in 2 μL DMSO; B). *P<0.05, †P<0.05=genotype x drug interaction.
Conventional PKCs are necessary to elevate water intake due to the brain-RAS
A dose response-study was performed to verify that central activation of PKC mediates brain ANG-II-induced polydipsia. BIM had no significant effect at a dose of 32 pg. Starting at 3.2 ng, BIM significantly decreased 24 hour water intake in sRA compared to control mice (Figure 2A). There was a significantly greater maximum change in water intake from vehicle in sRA compared with control mice (Figure 2B). Starting at 1 ng, Gö-6976, a selective PKC-α/βII inhibitor, significantly decreased water intake in sRA mice (Figure 2C), and the maximum change in water intake due to Gö-6976 was significantly greater in sRA than littermate controls (Figure 2D). Thus, conventional isoforms of PKC mediate brain ANG-II-mediated polydipsia in this model.
Figure 2. Drinking Response to PKC Blockade in sRA Mice.
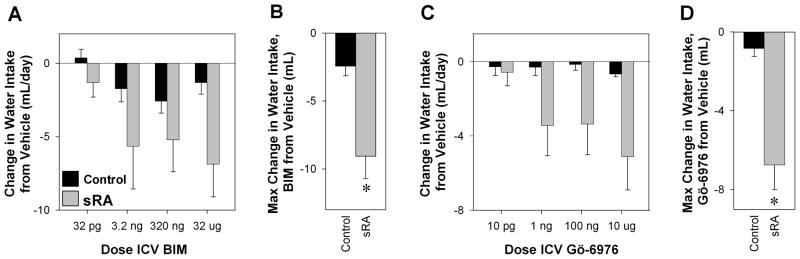
Change in 24 hour water intake of sRA and littermate control mice due to ICV aCSF to increasing doses of BIM (A; sRA n=7, control n=9) or Gö-6976 (C; sRA n=6, control n=6). There are significant interactions between genotype and dose of BIM and Gö-6976. The maximum response to a change in water intake due to ICV BIM (B) or Gö-6976 (D) is shown. *P<0.01.
PKC-α from the SFO mediates brain-RAS polydipsia
We and others have demonstrated that an ICV injection of adenovirus targets the SFO. 23,25,26 To confirm this in the current cohort, adenovirus encoding Cre-recombinase (AdCRE) was first injected ICV into a reporter model that specifically detects Cre-mediated recombination (ROSA-CAG-LSL-tdTomato mice). One-month after ICV AdCRE, recombination was only found within the SFO (Figure 3B) and tanycytes of the central canal (Figure 3F). Nuclei important for cardiovascular control, such as the median preoptic nucleus (Figure 3A), organum vasculosum of the lamina terminalis, supraoptic nucleus (Figure 3C), paraventricular nucleus (Figure 3D), rostral ventrolateral medulla (Figure 3E), area postrema, and nucleus tractus solitarius (Figure 3F), were largely devoid of evidence of recombination. Thus, ICV adenovirus selectively infects cells within the SFO.
Figure 3. SFO Selectivity of Adenoviral Infection.

TdTomato fluorescence as an indicator of cre-recombination in CAG-ROSA-LSL-tdTomato mice after ICV AdCRE as shown in the MnPO (A), SFO (B), SON (C), PVN (D), RVLM (E), and brainstem (E; 1: AP, 2: NTS, and 3: Central Canal) (F). Arrows indicate location of the indicated nucleus. The scale bar is 100 μm.
An adenovirus expressing dominant negative PKC-α (Ad-DN-PKC-α) was used to determine if PKC-α activation mediates brain ANG-II-induced polydipsia, and to determine if this occurs within the SFO. In this experiment, we provided sRA mice with a two-bottle choice of de-ionized water and isotonic saline. Nine- to 10-days after ICV Ad-DN-PKC-α the intakes of water (Figure 4A–B), 0.15M NaCl (Figure 4C–D), total fluid (Figure 4E–F), and total sodium (Figure S1A) of sRA mice were attenuated while control mice were unaffected. There was no difference between the genotypes in salt preference (Figure S1B). It is important to note that the decrease in fluid intake after SFO-selective Ad-DN-PKC-α was approximately half that observed after ICV BIM or Gö-6976. An 11% decrease in food intake (Figure S1C) contributed to an attenuation of total sodium consumption in sRA mice; however, there was no change in body weight (Figure S1D). These data suggest that the increase in fluid and sodium intake in response to increased brain ANG-II requires PKC-α activity within the SFO.
Figure 4. Drinking Response to Dominant Negative PKC-α.
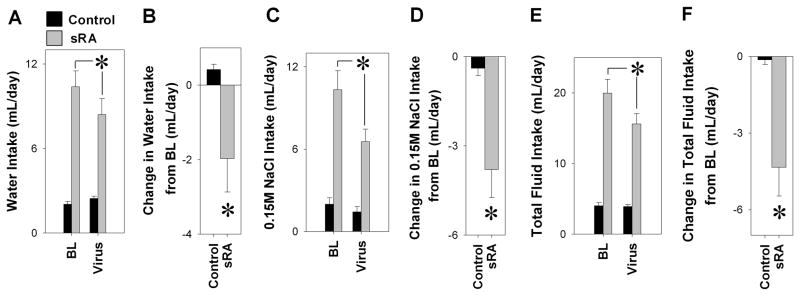
Intakes of water (A–B), 0.15M NaCl (C–D), and total fluid (E–F) were attenuated by ICV Ad-DN-PKC-α from baseline (BL) in sRA (grey, n=13) but not littermate control mice (black, n=14). There are statistically significant interactions between genotype and virus for intakes of water, 0.15M NaCl, and total fluid. *P<0.05, genotype x virus interaction.
PKC activity is necessary for DOCA-salt polydipsia
We next tested the role of central PKC in an independent model of hypertension. ICV osmotic mini-pumps containing aCSF or BIM were placed into DOCA- or sham-treated mice at the start of the second week after initiation of DOCA-salt treatment. Chronic ICV BIM blunted the increase in fluid intake caused by DOCA-salt (Figure 5A). DOCA-salt mice treated with aCSF drank more water and 0.15M NaCl than sham mice, and consumed more sodium (Figure 5B–D), effects blunted by BIM treatment. DOCA-salt mice did not exhibit a significantly different preference for saline or food intake compared to littermate controls, and BIM had no effect upon these measurements (Figure 5E–F). Thus, as observed in sRA mice, central PKC also plays a role in fluid intake caused by DOCA-salt.
Figure 5. Role of PKC in Mediating the Drinking Response to DOCA-Salt.
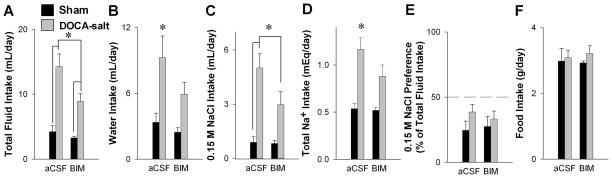
Total fluid (A), water (B), saline (C), total sodium (D), sodium preference (E), and food (F) intakes in sham (aCSF n= 5; BIM n=5) and 3-week DOCA-salt mice (aCSF n=10; BIM n=12) with chronic ICV aCSF (Alzet) or BIM (100 μg/day in aCSF). Preference for 0.15 M NaCl in sham and DOCA-salt mice with either chronic ICV aCSF or BIM (D). *P<0.05 Bonferroni post-hoc comparison between genotypes or treatments.
Central PKC activity is sufficient to increase water intake
Given the potential role of PKC-α, we next determined if PKC-α becomes activated in primary cells cultured from the SFO.17,27 Cells were treated with vehicle or PMA for 15 min and phosphorylated PKC-α was visualized by immunofluorescence. Approximately 12% of the cells exhibited a positive signal. PKC-α was observed as punctate staining throughout the cytoplasm in vehicle-treated cells (Figure 6A–B), whereas in PMA-treated cells, the PKC-α signal was localized primarily at the plasma membrane (Figure 6C–D).
Figure 6. PKC-α Activity in Cultured Cells from SFO.

Immunofluorescent images detecting nuclei (blue nuclei stained with DAPI) and phosphorylated-PKC-α in primary cells cultured from the SFO of newborn rats. Cells were treated with PMA for 15 min. Scale bar = 100 μm.
Finally, we tested whether a direct pharmacological activator of conventional and novel PKCs (PMA) when injected into the lateral ventricle of water-replete C57BL/6 mice would be sufficient to induce water intake. PMA induction of water intake occurred after 5 minutes, and most of the cumulative water intake occurred within 15 minutes of injection (Figure 7A). The PMA-induced water intake over a 5 hour period, as measured by the area under the curve, was markedly elevated compared to aCSF (Figure 7B). Whereas PMA induced water intake acutely, it did not affect 24 hour total consumption of water compared to aCSF suggesting the agent is rapidly cleared (aCSF: 3.02 ± 0.16 mL/day vs. PMA: 2.08 ± 0.58 mL/day; P=0.195). Blocking central PKC activity by pretreating mice with ICV BIM almost completely blunted the PMA induction of water intake over 30 minutes (Figure 7C,D). Thus, central PKC activity is sufficient to induce water intake in water-replete mice.
Figure 7. Central PKC Activation Stimulates Drinking.

Cumulative water intake in water-replete C57BL/6 mice over 5 hours (A; n=5) after ICV vehicle (aCSF) or PMA (40 ng in aCSF). The area under the curve (AUC) for cumulative water intake over time was calculated by the trapezoid method (B). Cumulative water intake over 30-minutes (C, n=6) after ICV pre-treatment with BIM (32 ng in aCSF) and treatment with PMA or vehicle, and after ICV pre-treatment with vehicle and treatment with PMA (aCSF) or vehicle. This is quantified by the AUC (D). There is a significant interaction between pre- and post-drug in C and D (P=0.01). *P<0.01, independent t-test. †P<0.01, Bonferroni post-hoc comparison.
Discussion
There are several findings from this study. First, the activity of central PKC, but not ERK or PKA, is required to mediate polydipsia in sRA mice. Second, the action of central PKC in mediating drinking in sRA mice is through the PKC-α/βII isoform. Third, blocking the activity of PKC-α selectively in the SFO blunts, but does not fully abolish, increased water and sodium intake in sRA mice. Fourth, as observed in sRA mice, inhibition of central PKC activity blunts increased water intake in DOCA-salt mice. Last, inducing PKC activity with PMA caused an increase cell surface localization of phosphorylated PKC-α in cultured cells from the rat SFO, and central PKC activity is sufficient to increase water intake in water-replete mice. Together these findings highlight an important role for central PKC-α as a regulator of fluid homeostasis in two different models of that exhibit increased brain RAS activity, and suggest that PKC-α activity specifically in the SFO plays a significant role in this response.
Both sRA and DOCA-salt models exhibit increased brain angiotensin system activity, and we showed that the drinking responses in both models were blocked by AT1R antagonist losartan.7,22,23 We recognize that sRA is an over-expression model that serves as a genetic tool enabling the demonstration of what occurs in response to endogenous Ang-II formation within the CNS. That both sRA and DOCA-salt mice exhibit very similar phenotypes, and DOCA-salt is recognized as a model which has an central ANG-II component validates their use herein.21,22 Moreover, we demonstrated that ablation of ANG-II over-production selectively in the SFO attenuates polydipsia in sRA transgenic mice,7 and selective ablation of endogenous AT1AR in the SFO attenuates DOCA-salt-induced polydipsia.23 These findings therefore demonstrate that both ANG-II production and AT1R action in the SFO are mechanistically necessary for polydipsia induced by the brain RAS. The importance of the SFO for water and saline intake has been shown by lesion studies, and by measurements of c-fos expression as an indicator of neuronal activity in the SFO in response to various dipsogenic stimuli.2,28–32
Herein we show that targeting PKC-α in the SFO through an ICV injection of a virus expressing dominant negative PKC-α attenuates the elevated fluid and sodium intake in sRA mice. These data extend our previous finding to suggest that PKC-α activity downstream of AT1R signaling in the SFO partially mediates ANG-II-induced polydipsia. Compared with the non-selective inhibitor of PKC, BIM, and the selective PKC-α/βII inhibitor, Gö-6976, SFO-selective Ad-DN-PKC-α was less effective in blunting fluid intake in sRA mice. First, through ICV administration, the pharmacological inhibitors likely gain access to many nuclei in the brain and the effects on fluid intake are the sum of PKC inhibition at all of these sites. This would suggest that PKC-α signaling in the SFO regulates a portion of the total dipsogenic response. Second, Ad-DN-PKC-α infection of the SFO may have been incomplete, and as previously shown, adenoviral infection targets both glia and neurons in the SFO.25 Whereas our studies using ICV AdCRE in Cre-reporter mice indicates effective infection of the SFO, the number of neurons infected is a variable.25 In this study, Ad-DN-PKC-α did not contain an independent marker, such as GFP. We did not co-inject Ad-eGFP as a surrogate marker for Ad-DN-PKC-α infection because we considered that by the time the experiment was completed and the mice would be euthanized, adenoviral expression of eGFP would likely be waning.25 The effectiveness of the Ad-DN-PKC-α has been previously validated.33 Third, other PKC isoforms that are effectively inhibited by BIM, but not by Gö-6976 or Ad-DN-PKC-α, may also play a role in mediating the ANG-II response. For example, ANG-II-stimulation of human glioma cells activates PKC-α and PKC-ε.19 Inhibition of PKC-ε blocks ANG-II-inhibition of Na+/K+ ATPase in cardiomyocytes and ANG-II-induced activation of ERK1/2 in human adenocarcinoma cells.34,35 Finally, it is possible that some of the effects of pharmacologically inhibiting PKC are mediated by off-target inhibition of calmodulin-dependent kinase II (CAMKII) as Gö-6976 (a derivative from BIM) decreases the activity of CAMKII-α and -β in vitro.36 In hypothalamic and brainstem cultures, the excitatory effects of ANG-II through AT1R are attenuated with inhibition of either PKC or CAMKII, but are corrected if both are inhibited.37 Furthermore, ANG-II increases the expression of CAMKII-α in cultures of rat septum and hypothalamus. Inhibition of CAMKII via KN-93 attenuates central ANG-II-induction of water intake. Attenuation of water intake via inhibition of CAMKII and PKC is not additive. CAMKII and PKC appear to specifically mediate ANG-II-induction of water intake since their inhibition did not attenuate carbachol-induced water intake.20
It is notable that when the two bottle choice paradigm was used for the Ad-DN-PKC-α studies, we observed a larger effect on saline rather than water intake in sRA mice. Prior studies implied that central PKC-α does not mediate intake of saline due to central ANG-II, which appears to contradict our results.11,20 However, in the previous studies saline was not given as a choice with water, saline intake at a non-aversive concentration was not evaluated, and specifically PKC-α in the SFO was not inhibited. Interestingly, BIM blunted total fluid intake and saline intake in DOCA-salt mice, and although the decrease in water intake was not significant, there was no significant difference between water intake in DOCA-salt and Sham mice after BIM. Thus, we cannot rule out that the effects of specifically inhibiting PKC-α in the SFO may differ from the effects of blocking all PKC isoforms throughout the brain. Also, the mechanisms of fluid intake in DOCA-salt mice are likely a sum of the effects of increased local ANG-II signaling in the brain, the stimulation of central mineralocorticoid receptors, and possibly an interaction between both. Indeed, evidence suggests that central MR receptors can sensitize rats to the pressor effects of ANG-II.27 It is unclear if pressor responses to ANG-II can be extended to the dipsogenic responses, although it is interesting to note that aldosterone increases calcium signaling in cultured SFO neurons in response to ANG-II.27
In the initial drug screen we observed that direct inhibition of ERK1/2 with FR180204 attenuated water intake in both sRA and littermate controls. The expression of ERK1/2 in the SFO increases in fluid-restricted mice.38 Daniels and colleagues have shown that inhibition of ERK1/2 attenuates the acute increase in sodium appetite, but not water intake of rats injected with ANG-II into the lateral ventricle.11 Thus, ERK1/2 may affect fluid intake by increasing the intake of a normally aversive concentration of saline. Since sodium appetite is not elevated in sRA mice and we measured only the intake of water in the initial screening experiment, the lack of a robust effect of FR180204 upon water intake by sRA mice was not entirely unexpected.21
We previously showed that hypertension in sRA mice is attenuated by a chronic, peripheral infusion of conivaptan or tolvaptan,39 but in the current study we did not observe an effect on water intake. This suggests that central V1A and V2R (expressed in the hypothalamus, hippocampus, cortex, and cerebellum; GenSat.org) do not mediate the water intake due to elevated brain ANG-II. While the V1BR is not inhibited with conivaptan, it is unlikely that they mediate the polydipsia of sRA mice. Indeed, we previously showed that adrenal steroids mediate polydipsia in sRA mice as adrenalectomy completely blocks polydipsia.21
While prior studies and our present data demonstrate the necessity of central PKC, specifically PKC-α, for central ANG-II-induction of water intake, we also show that activity of conventional or novel isoforms of PKC in the brain is sufficient to induce the intake of water in water-replete mice.11,20 This may occur as a result of PKC-α activation within the SFO. This conclusion is based on the observations that the induction of water intake in response to PMA follows a similar time-course to a central injection of ANG-II,2 that lesion of the SFO attenuates central ANG-II-induced polydipsia,40 our data showing that PKC-α in the SFO is necessary to mediate the full extent of polydipsia in sRA mice, and our data showing cell surface-associated phosphorylated PKC-α in cultured cells from the rat SFO. We conclude that local production and action of ANG-II within the SFO increases PKC-α activity, which is necessary and sufficient for the elevated intake of water and non-aversive saline.
Perspectives
Polydipsia occurs in, and can aggravate, type 2 diabetes mellitus, heart failure, chronic kidney disease, chronic psychosis, and adverse reactions to drugs. We show that polydipsia due to hyperactivity of brain angiotensin activity occurs through the activity of PKC-α within the SFO. We also show that central PKC activity (presumably PKC-α) is sufficient to induce water intake in water-replete mice. Understanding the molecular mechanisms of fluid intake will allow us to pharmacologically treat polydipsia.
Supplementary Material
Novelty and Significance.
What is New?
Central activity of conventional or novel PKCs is sufficient to induce water intake in water-replete mice.
Fluid intake of both water and saline (0.15M NaCl) due to hyperactivity of the brain-RAS is mediated through PKC-α within the SFO.
Central ERK1/2, PKA, or vasopressin receptors (V1A and V2) appear to not mediate elevated water intake due to hyperactivity of the brain angiotensin system.
What is Relevant?
Central PKC is both necessary and sufficient to induce water intake.
Polydipsia due to increased activity of the brain angiotensin system is mediated through PKC-α within the SFO.
Summary
We show that hyperactivity of the brain-RAS increases intake of both water and saline. PKC-α activity within the SFO is necessary for this polydipsia, and central ERK1/2, PKA, and vasopressin receptors V1A and V2R do not appear to mediate polydipsia due to hyperactivity of the brain-RAS. Furthermore, induction of central PKC is sufficient on its own to induce water intake.
Acknowledgments
The authors would like to thank Dr. Viswanathan Natarajan, University of Illinois, Chicago for the gift of Ad-DN-PKC-α; and Deborah R. Davis for assistance with mice. We also thank Dr. L. Philip Sanford, Norma Sinclair, JoAnne Schwarting, and Patricia Yarolem for genotyping mice. The University of Iowa Central Microscopy Facility was used for confocal imaging, and adenoviruses were generated at the University of Iowa Vector Core. Transgenic mice were generated at the University of Iowa Transgenic Animal Facility supported in part by grants from the NIH and from the Roy J. and Lucille A. Carver College of Medicine.
Sources of Funding: This work was supported through research grants from the NIH to CDS (HL048058, HL061446, HL062984), to CDS and JLG (HL084207), to AKJ (HL014388, HL098207, and MH08024) and to JLG (HL098276). The authors also gratefully acknowledge the generous research support of the Roy J. Carver Trust.
Footnotes
Disclosures: None.
References
- 1.Rowland NE, Fregly MJ. Comparison of the effects of the dipeptidyl peptidase inhibitors captopril, ramipril, and enalapril on water intake and sodium appetite of Sprague-Dawley rats. Behav Neurosci. 1988;102:953–960. doi: 10.1037//0735-7044.102.6.953. [DOI] [PubMed] [Google Scholar]
- 2.Simpson JB, Epstein AN, Camardo JS., Jr Localization of receptors for the dipsogenic action of angiotensin II in the subfornical organ of rat. J Comp Physiol Psychol. 1978;92:581–601. doi: 10.1037/h0077503. [DOI] [PubMed] [Google Scholar]
- 3.Simpson JB, Routtenberg A. Subfornical organ: a dipsogenic site of action of angiotensin II. Science. 1978;201:379–381. doi: 10.1126/science.663664. [DOI] [PubMed] [Google Scholar]
- 4.Rowland NE, Goldstein BE, Robertson KL. Role of angiotensin in body fluid homeostasis of mice: fluid intake, plasma hormones, and brain Fos. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1586–R1594. doi: 10.1152/ajpregu.00730.2002. [DOI] [PubMed] [Google Scholar]
- 5.Denton DA, Blair-West JR, McBurnie M, Osborne PG, Tarjan E, Williams RM, Weisinger RS. Angiotensin and salt appetite of BALB/c mice. Am J Physiol. 1990;259:R729–R735. doi: 10.1152/ajpregu.1990.259.4.R729. [DOI] [PubMed] [Google Scholar]
- 6.Park CG, Leenen FH. Effects of centrally administered losartan on deoxycorticosterone-salt hypertension rats. J Korean Med Sci. 2001;16:553–557. doi: 10.3346/jkms.2001.16.5.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakai K, Agassandian K, Morimoto S, Sinnayah P, Cassell MD, Davisson RL, Sigmund CD. Local production of angiotensin II in the subfornical organ causes elevated drinking. J Clin Invest. 2007;117:1088–1095. doi: 10.1172/JCI31242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pereira DT, Menani JV, De Luca LAJ. FURO/CAP: a protocol for sodium intake sensitization. Physiol Behav. 2010;99:472–481. doi: 10.1016/j.physbeh.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Daniels D, Yee DK, Fluharty SJ. Angiotensin II receptor signalling. Exp Physiol. 2007;92:523–527. doi: 10.1113/expphysiol.2006.036897. [DOI] [PubMed] [Google Scholar]
- 10.Hines J, Fluharty SJ, Yee DK. Structural determinants for the activation mechanism of the angiotensin II type 1 receptor differ for phosphoinositide hydrolysis and mitogen-activated protein kinase pathways. Biochem Pharmacol. 2003;66:251–262. doi: 10.1016/s0006-2952(03)00257-0. [DOI] [PubMed] [Google Scholar]
- 11.Daniels D, Mietlicki EG, Nowak EL, Fluharty SJ. Angiotensin II stimulates water and NaCl intake through separate cell signalling pathways in rats. Exp Physiol. 2009;94:130–137. doi: 10.1113/expphysiol.2008.044446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daniels D, Yee DK, Faulconbridge LF, Fluharty SJ. Divergent behavioral roles of angiotensin receptor intracellular signaling cascades. Endocrinology. 2005;146:5552–5560. doi: 10.1210/en.2005-0774. [DOI] [PubMed] [Google Scholar]
- 13.Yu Y, Xue BJ, Zhang ZH, Wei SG, Beltz TG, Guo F, Johnson AK, Felder RB. Early interference with p44/42 mitogen-activated protein kinase signaling in hypothalamic paraventricular nucleus attenuates angiotensin II-induced hypertension. Hypertension. 2013;61:842–849. doi: 10.1161/HYPERTENSIONAHA.111.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moellenhoff E, Blume A, Culman J, Chatterjee B, Herdegen T, Lebrun CJ, Unger T. Effect of repetitive icv injections of ANG II on c-Fos and AT(1)-receptor expression in the rat brain. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1095–R1104. doi: 10.1152/ajpregu.2001.280.4.R1095. [DOI] [PubMed] [Google Scholar]
- 15.Xu Z, Xinghong J. Drinking and Fos-immunoreactivity in rat brain induced by local injection of angiotensin I into the subfornical organ. Brain Res. 1999;817:67–74. doi: 10.1016/s0006-8993(98)01251-7. [DOI] [PubMed] [Google Scholar]
- 16.Anderson JW, Smith PM, Ferguson AV. Subfornical organ neurons projecting to paraventricular nucleus: whole-cell properties. Brain Res. 2001;921:78–85. doi: 10.1016/s0006-8993(01)03093-1. [DOI] [PubMed] [Google Scholar]
- 17.Johnson RF, Beltz TG, Jurzak M, Wachtel RE, Johnson AK. Characterization of ionic currents of cells of the subfornical organ that project to the supraoptic nuclei. Brain Res. 1999;817:226–231. doi: 10.1016/s0006-8993(98)01224-4. [DOI] [PubMed] [Google Scholar]
- 18.Du JQ, Sun CW, Tang JS. Effect of angiotensin II type 1 receptor on delayed rectifier potassium current in catecholaminergic CATH.a cells. Acta Pharmacol Sin. 2004;25:1145–1150. [PubMed] [Google Scholar]
- 19.Greenland KJ, Mukhopadhyay AK. Selective activation of protein kinase C isoforms by angiotensin II in neuroblastoma X glioma cells. Mol Cell Endocrinol. 2004;213:181–191. doi: 10.1016/j.mce.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 20.Fleegal MA, Sumners C. Drinking behavior elicited by central injection of angiotensin II: roles for protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Am J Physiol Regul Integr Comp Physiol. 2003;285:R632–R640. doi: 10.1152/ajpregu.00151.2003. [DOI] [PubMed] [Google Scholar]
- 21.Grobe JL, Grobe CL, Beltz TG, Westphal SG, Morgan DA, Xu D, de Lange WJ, Li H, Sakai K, Thedans DR, Cassis LA, Rahmouni K, Mark AL, Johnson AK, Sigmund CD. The Brain Renin-Angiotensin System Controls Divergent Efferent Mechanisms to Regulate Fluid and Energy Balance. Cell Metabolism. 2010;12:431–442. doi: 10.1016/j.cmet.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grobe JL, Buehrer BA, Hilzendeger AM, Liu X, Davis DR, Xu D, Sigmund CD. Angiotensinergic signaling in the brain mediates metabolic effects of deoxycorticosterone (DOCA)-salt in C57 mice. Hypertension. 2011;57:600–607. doi: 10.1161/HYPERTENSIONAHA.110.165829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hilzendeger AM, Cassell MD, Davis DR, Stauss HM, Mark AL, Grobe JL, Sigmund CD. Angiotensin type 1a receptors in the subfornical organ are required for deoxycorticosterone acetate-salt hypertension. Hypertension. 2013;61:716–722. doi: 10.1161/HYPERTENSIONAHA.111.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davisson RL, Yang G, Beltz TG, Cassell MD, Johnson AK, Sigmund CD. The brain renin-angiotensin system contributes to the hypertension in mice containing both the human renin and human angiotensinogen transgenes. Circ Res. 1998;83:1047–58. doi: 10.1161/01.res.83.10.1047. [DOI] [PubMed] [Google Scholar]
- 25.Sinnayah P, Lindley TE, Staber PD, Cassell MD, Davidson BL, Davisson RL. Selective gene transfer to key cardiovascular regions of the brain: comparison of two viral vector systems. Hypertension. 2002;39:603–608. doi: 10.1161/hy0202.103295. [DOI] [PubMed] [Google Scholar]
- 26.Sinnayah P, Lazartigues E, Sakai K, Sharma RV, Sigmund CD, Davisson RL. Genetic Ablation of Angiotensinogen in the Subfornical Organ of the Brain Prevents the Central Angiotensinergic Pressor Response. Circ Res. 2006;99:1125–1131. doi: 10.1161/01.RES.0000250259.66683.f5. [DOI] [PubMed] [Google Scholar]
- 27.Xue B, Zhang Z, Roncari CF, Guo F, Johnson AK. Aldosterone acting through the central nervous system sensitizes angiotensin II-induced hypertension. Hypertension. 2012;60:1023–1030. doi: 10.1161/HYPERTENSIONAHA.112.196576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Starbuck EM, Fitts DA. Influence of salt intake, ANG II synthesis and SFO lesion on thirst and blood pressure during sodium depletion. Subfornical organ. Appetite. 1998;31:309–331. doi: 10.1006/appe.1998.0169. [DOI] [PubMed] [Google Scholar]
- 29.Starbuck EM, Wilson WL, Fitts DA. Fos-like immunoreactivity and thirst following hyperosmotic loading in rats with subdiaphragmatic vagotomy. Brain Res. 2002;931:159–167. doi: 10.1016/s0006-8993(02)02273-4. [DOI] [PubMed] [Google Scholar]
- 30.Morris MJ, Wilson WL, Starbuck EM, Fitts DA. Forebrain circumventricular organs mediate salt appetite induced by intravenous angiotensin II in rats. Brain Res. 2002;949:42–50. doi: 10.1016/s0006-8993(02)02963-3. [DOI] [PubMed] [Google Scholar]
- 31.Fitts DA, Freece JA, Van Bebber JE, Zierath DK, Bassett JE. Effects of forebrain circumventricular organ ablation on drinking or salt appetite after sodium depletion or hypernatremia. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1325–R1334. doi: 10.1152/ajpregu.00391.2004. [DOI] [PubMed] [Google Scholar]
- 32.Rowland NE, Fregly MJ, Li BH, Han L. Angiotensin-related induction of immediate early genes in rat brain. Regul Pept. 1996;66:25–29. doi: 10.1016/0167-0115(96)00054-7. [DOI] [PubMed] [Google Scholar]
- 33.Park JW, Kim HP, Lee SJ, Wang X, Wang Y, Ifedigbo E, Watkins SC, Ohba M, Ryter SW, Vyas YM, Choi AM. Protein kinase C alpha and zeta differentially regulate death-inducing signaling complex formation in cigarette smoke extract-induced apoptosis. J Immunol. 2008;180:4668–4678. doi: 10.4049/jimmunol.180.7.4668. [DOI] [PubMed] [Google Scholar]
- 34.White CN, Figtree GA, Liu CC, Garcia A, Hamilton EJ, Chia KK, Rasmussen HH. Angiotensin II inhibits the Na+-K+ pump via PKC-dependent activation of NADPH oxidase. Am J Physiol Cell Physiol. 2009;296:C693–C700. doi: 10.1152/ajpcell.00648.2008. [DOI] [PubMed] [Google Scholar]
- 35.LeHoux JG, Lefebvre A. Angiotensin II activates p44/42 MAP kinase partly through PKCepsilon in H295R cells. Mol Cell Endocrinol. 2007;265–266:121–125. doi: 10.1016/j.mce.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 36.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun C, Sumners C, Raizada MK. Chronotropic action of angiotensin II in neurons via protein kinase C and CaMKII. Hypertension. 2002;39:562–566. doi: 10.1161/hy0202.103057. [DOI] [PubMed] [Google Scholar]
- 38.Hiyama TY, Yoshida M, Matsumoto M, Suzuki R, Matsuda T, Watanabe E, Noda M. Endothelin-3 expression in the subfornical organ enhances the sensitivity of Na(x), the brain sodium-level sensor, to suppress salt intake. Cell Metab. 2013;17:507–519. doi: 10.1016/j.cmet.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 39.Littlejohn NK, Siel RB, Jr, Ketsawatsomkron P, Pelham CJ, Pearson NA, Hilzendeger AM, Buehrer BA, Weidemann BJ, Li H, Davis DR, Thompson AP, Liu X, Cassell MD, Sigmund CD, Grobe JL. Hypertension in mice with transgenic activation of the brain renin-angiotensin system is vasopressin dependent. Am J Physiol Regul Integr Comp Physiol. 2013;304:R818–R828. doi: 10.1152/ajpregu.00082.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thunhorst RL, Beltz TG, Johnson AK. Effects of subfornical organ lesions on acutely induced thirst and salt appetite. Am J Physiol. 1999;277:R56–R65. doi: 10.1152/ajpregu.1999.277.1.r56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.