

Abstract
Lung cancer is one of the most deadly diseases worldwide. The current first-line therapies include chemotherapy using epidermal growth factor receptor tyrosine kinase inhibitors and radiotherapies. With the current progress in identifying new molecular targets, acquired drug resistance stands as an obstacle for good prognosis. About half the patients receiving epidermal growth factor receptor-tyrosine kinase inhibitor treatments develop resistance. Although extensive studies have been applied to elucidate the underlying mechanisms, evidence is far from enough to establish a well-defined picture to correct resistance. In the review, we will discuss four different currently developed strategies that have the potential to overcome drug resistance in lung cancer therapies and facilitate prolonged anticancer effects of the first-line therapies.
Keywords: ALK receptors cancer stem cell, chemotherapy, EGFR-TKI, target therapy, pharmacology, molecular biology, biotherapy
Introduction
Lung carcinoma is the leading cause of cancer-related death worldwide.1 It is a disease characterized by uncontrolled cell growth in tissues of the lung. The primary lung cancers are carcinomas that derive from epithelial cells. The main types of lung cancer are small-cell lung carcinoma (SCLC), and non-small-cell lung carcinoma (NSCLC). Approximately 20% of lung cancer cases are SCLC by histology, with the other 80%, which includes adenocarcinoma, squamous cell carcinoma, large-cell carcinoma, and bronchoalveolar cell carcinoma, being lumped together as NSCLC.2 While the primary lung cancer commonly metastasizes to the brain, bones, liver, and adrenal glands, secondary cancers are happening with high incidence and can be derived from various sites.
The poor overall survival rate in lung cancer patients remains a major challenge in the clinical management of lung cancer and underscores the urgent need to develop novel therapeutic approaches that overcome intrinsic drug resistance.
Tumor drug resistance is a major issue in the management of lung cancer as almost all lung tumors are either intrinsically resistant or quickly develop acquired resistance to chemotherapeutic drugs.3 At the intracellular level, resistance may result from decreased drug accumulation (due to decreased drug uptake or increased efflux), increased drug inactivation by detoxifying factors, decreased drug activation or binding to target, increased DNA damage repair, increased tolerance to DNA damage, increased resistance to apoptosis (either by upregulation of antiapoptotic factors or downregulation of proapoptotic factors), or altered cell cycling or transcription factors.3,4 The most cited mechanism for the acquisition of drug resistance is the activation of adenosine triphosphate (ATP)-binding cassette transporters, such as p-glycoprotein, breast cancer resistance protein (ATP-binding cassette sub-family G member 2 [BCRP]), and multidrug resistance proteins, which pump the chemotherapeutic agents toward the outside of tumor cells, thus protecting them from the action of the drugs. However, to date there is no effective treatment strategy to override these transporters for clinical therapy. In addition, several receptor-mediated survival signaling pathways, including mitogen-activated protein kinase (MAPK), protein kinase B (Akt), mammalian target of rapamycin (mTOR), nuclear factor kappa B, and notch pathways, have been associated with lung cancer drug resistance toward conventional chemotherapy.5 Additionally, it has been recently demonstrated that some microRNAs (miRNAs) can also play an important role in lung cancer drug resistance.6–8
Furthermore, resistance to antineoplastic agents has also been recently linked to the existence of a small subpopulation of tumor-cell possessing, whose specific properties are intrinsically more resistant than the remaining tumor cells, the cancer stem-like cells (CSLCs). In order to survive chemotherapy, CSLCs seem to be able to slow down cell cycle kinetics, efficiently repair the DNA, over-express multidrug-resistance membrane transporters, and resist apoptosis.9 The fact that conventional cancer therapeutic regimens do not seem able to effectively eliminate this subpopulation of CSLCs, but only cells constituting the bulk of the tumor, may help to explain why tumor recurrence is often observed several years following an apparently successful first-line of therapy. Nevertheless, it is important to note that, in a great number of cases, lung cancer patients’ tumors are intrinsically resistant to therapy due to one or several of the multiple chemoresistance mechanisms described above, so the extent of contribution of CSLCs toward cancer-drug resistance should be carefully assessed.
In order to postpone the progress of drug resistance and improve the effects of several chemo- and radiotherapies, many studies have been performed to target single or multiple sites to effect resistance. Extensive efforts have been applied to study the drug resistance mechanism in the first-line epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor (TKI) therapy; there is also accumulating evidence pointing at the cancer stem cells (CSCs) and molecular RNAs as playing an important role in developing resistance. A potential strategy to improve pharmacokinetic progress of the drugs, and, thus, combating resistance, is an oncologic virus delivery system. In the review, we will discuss these molecular mechanisms and new strategies with the possibility of postponing the onset of cancer-cell drug resistance.
EGFR-TKI therapy and the associated resistance
The iterative discovery in various malignancies during the past decades that a number of aberrant tumorigenic processes and signal transduction pathways are mediated by druggable protein kinases has led to a revolutionary change in drug development. In NSCLC, the ErbB family of receptors (eg, EGFR,10 human epidermal growth factor receptor [HER] 211), rat sarcoma gene (RAS), v-raf murine sarcoma viral oncogene homologue B1, MAPK, c-mesenchymal-epithelial transition (c-MET), fibroblast growth factor receptor, discoidin domain receptor 2, phosphatidylinositol-4,5-bisphosphate3-kinase, catalytic subunit alpha (PIK3CA), phosphatase and tensin homologue, Akt, anaplastic lymphoma kinase (ALK), rearranged during transfection (RET), reactive oxygen species 1, and erythropoietin-producing hepatoma are key targets of various agents currently in clinical development. These oncogenic targets exert their selective growth advantage through various intercommunicating pathways, such as through RAS/RAF/MEK, phosphoinositide 3-kinase/Akt/mTOR, and SRC-signal transduction and transcription signaling. In recent clinical studies, EGFR-TKIs and crizotinib were considered as strongly effective targeted therapies in metastatic NSCLC.12 Currently, five molecular targeted agents are approved for treatment of advanced NSCLC: gefitinib; erlotinib and afatinib for positive EGFR mutation; crizotinib for positive echinoderm microtubule-associated protein-like 4 (EML4)-ALK translocation; and bevacizumab (Figure 1).13
Figure 1.

Mechanism of EGFR-related lung cancer drug resistance.
Abbreviations: AKT, protein kinase B; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase; RAF, rapidly accelerated fibrosarcoma; RAS, rat sarcoma gene; EML4-ALK, echinoderm microtubule associated protein like 4 -anaplastic lymphoma kinase.
EGFR-TKI (such as gefitinib or erlotinib) treatment of lung-cancer harboring EGFR gene mutation is one of the prototypes of such therapies. Several clinical trials clearly demonstrated that progression-free survival of patients treated with EGFR-TKI therapy is significantly longer than that of those treated by conventional platinum doublet chemotherapy.14–16 EGFR-TKI therapy dramatically changed the paradigm of lung cancer treatment.11 Which strategy is better in the management of advanced NSCLCs – the inhibition of mutated EGFR with TKIs (L858R mutation increases tyrosine kinase affinity for TKIs) or a combined approach with cetuximab (an anti-EGFR monoclonal antibody) plus first-line chemotherapy – is yet to be determined.17 EGFR-TKI’s associated toxicity is generally well tolerated. The two most common toxicities include dermatologic and gastrointestinal effects, both of which are mild to moderate, easily managed, and reversible.18–20 EGFR-TKI therapy has been carried out in more than half of the patients who are identified to bear EGFR mutations.
In many ways, the experience with TKIs in NSCLC exemplifies the successes and challenges of personalized cancer medicine.15 The finding that EGFR-TKIs were effective and had particular activity in 15%–30% of NSCLC patients with EGFR mutations was a major advance; however, resistance sets in after approximately 1 year of treatment and remains as one of the biggest obstacles in anticancer treatments.
The majority of resistance to EGFR-TKIs are seen with: 1) the second-site exon 20 EGFR gatekeeper mutation plasmid T790M that reduces drug binding in ~50% of all cases of EGFR mutation;21 2) the plasmid T790M mutation plus EGFR amplification in ~8% of cases;22 3) tyrosine kinase switching or receptor dimerization through MET (proto-oncogene that encodes a protein known as hepatocyte growth factor receptor) amplification in ~5%–19% of cases;23 4) overexpression of AXL (encodes tyrosine-protein kinase receptor UFO in human) and its ligand GAS6 in 20% and 25% of cases, respectively;24 and 5) activating mutation of the phosphatidylinositol 3-kinase (PI3K) p110α-encoding gene PIK3CA in ~5% of cases.25–27 There is also evidence of nuclear factor-κB(NF-κB) signaling being implicated as a resistance mechanism to avoid TKI-induced apoptosis, possibly through the low expression of the NF-κB inhibitory protein IκB.25
One straightforward method to overcome drug resistance, the intensification of EGFR inhibition, represents a potential method.28 T790M mutation accounts for 50% of all the EGFR-specific resistance; it facilitates phosphorylation of EGFR despite the presence of a TKI through increasing the affinity of mutant EGFR for ATP.29 Thus, the T790M mutation significantly reduces drug binding and avoids reaching an efficient blood drug concentration. Many trials have studied intensification of EGFR inhibition through use of second-generation TKIs such as neratinib, afatinib, and dacomitinib.30 These inhibitors have the advantage that each forms a covalent irreversible bond with the EGFR protein, and each also inhibits other members of the ErbB family of kinases. However, this broader activity leads to greater toxicity, although with the power to overcome T790M-mediated resistance.30,31 Other mutations in exon 20 are insertions predominantly clustered between codons 767 and 774. Importantly, in contrast to the more classic activating EGFR mutations, these insertions have been associated with de-novo resistance to approved EGFR TKIs (erlotinib and gefitinib)32,33 and to irreversible inhibitors that have recently entered clinical trials (neratinib, afatinib, and dacomitinib).34,35 In vitro studies show that cells harboring some of the most prevalent insertions require an average of 100-fold higher concentrations of these agents for inhibition, well beyond clinically achievable plasma levels. Clinical studies, although limited, confirm the preclinical findings,36–38 but rare cases with better clinical responses have been reported.12,39 Importantly, many of the insertions identified in patient samples have not been tested against these inhibitors. Further understanding of the biology as well as the prognostic and predictive implications of these mutations is needed, but has remained limited by the small number of patients included in clinical trials and the lack of preclinical models, such as patient derived cell lines or genetically engineered mouse models.
Second- and third-generation EGFR-TKIs are developed as part of the strategy to overcome treatment resistance to first-generation EGFR-TKIs. Second-generation agents include the irreversible inhibitors of the ErbB family of receptors: afatinib (also known as BIBW 2992, which targets EGFR, HER2, and HER4), dacomitinib (also known as PF0299804, which targets EGFR, HER2, and HER4), and neratinib (also known as HKI272, which targets EGFR and HER2). These agents have been or are being evaluated in NSCLC-specific clinical trials (for clinical trials see Reungwetwattana and Dy).40 It is thought that primary resistance to these agents will still be encountered for the EGFR T790M mutation as well as exon 20 insertions.40 Thus, an alternate schedule of drug administration, such as intermittent or pulse high-dose therapy using afatinib to determine its activity against T790M (NCT01647711), is under investigation. Intermittent high-dose schedule to attain higher central nervous system (CNS) penetration has demonstrated some efficacy in treating disease progression confined to the CNS, wherein the pathophysiology is different from acquired resistance in extracranial sites of malignancy.
Third-generation EGFR inhibitors designed to inhibit the EGFR T790M mutant include WZ4002, CO-1686, and AZD9291. Poziotinib (also known as HM781-36B), a new potent irreversible inhibitor of EGFR, HER2, HER4, and transient erythroblastopenia of childhood family of kinases inhibitor (BTK, BLK, and BMX), demonstrated preclinical efficacy against T790M mutant at eightfold lower doses compared to afatinib.
A different approach in addressing EGFR-TKI resistance involves the use of combination regimens. Therefore, the combination of erlotinib with cetuximab, and the combination of erlotinib with MM-121 (a fully human mAb that targets HER3), in patients with acquired resistance to EGFR-TKIs did not show sufficient clinical activity for further investigation in this population. Other combination regimens, such as with c-MET inhibitors, heat shock protein 90, and PI3K/mTOR inhibitors will be investigated further.
ALK and leukocyte tyrosine kinase receptors
The EML4-ALK fusion gene (EML4 fused with the ALK) is one of the newer molecular targets elucidated in NSCLC. The ALK is a member of the insulin superfamily of receptor tyrosine kinases normally expressed only in the CNS, small intestine, and testis.10 The translocation of two genes in the short arm of chromosome 2, between the C-terminal kinase domain of ALK and the N-terminal portion of the EML4, was discovered in an NSCLC patient in Japan in 2007. This translocation causes aberrant activation of downstream oncogenic signaling pathways such as the RAS/RAF/MEK, PI3K/Akt/mTOR, and the Janus kinase/signal transducer and activator of transcription signaling pathway, leading to cell proliferation, invasion, and inhibition of apoptosis (Figure 1).18 EML4-ALK translocation is found in 3%–6% of all cases of NSCLC.40 Crizotinib (PF-02341066), an oral dual ALK/MET inhibitor, is currently the only US Food and Drug Administration-approved agent for advanced ALK-positive NSCLC.
Despite the remarkable initial responses, acquired resistance to crizotinib develops within a year.40 Various mechanisms of acquired resistance have been documented, several of which may coexist simultaneously. Multiple secondary mutations have already been identified in patients treated with crizotinib.40 Homologous to the gatekeeper EGFR T790M mutation is the L1196M substitution, which, unlike EGFR T790M, does not appear to confer a growth disadvantage to cells.40 Other secondary mutations such as G1269A, C1156Y, L1152R, and 1151Tins may affect affinity of the mutant ALK for either ATP or drug, and these differences have ramifications on the development of next-generation ALK inhibitors, which have varied mutation-specific efficacy among different agents. Other implicated resistance mechanisms include amplification of ALK gene, aberrant activation of other kinases such as amplification of KIT, or direct MAPK pathway activation as represented by either KRAS mutation upregulation of EGFR or detection of an activating EGFR mutation not seen in the initial tumor tissue.40 Another potential approach that may be effective is dual inhibition of PI3K and MEK pathway, which demonstrated significant activity in an ALK-translocated NSCLC cell line.40
Several second-generation agents against crizotinib-resistant EML4-ALK-positive cancers are being developed. Heat shock protein 90 inhibitors also show preclinical and clinical activity in ALK-rearranged NSCLC and may have broader activity across different ALK mutations.40 The reversible dual ALK/EGFR inhibitor AP26113 is a more potent ALK inhibitor than crizotinib and demonstrates preclinical activity against various secondary mutations resistant to crizotinib, including L1196 and G1269A. In an ongoing Phase I dose-escalation study, it demonstrated activity in up to two-thirds of crizotinib-resistant ALK-positive patients. LDK378 is a selective ALK inhibitor with weak c-MET activity that showed substantial clinical activity, with an overall response rate of 81% at doses >400 mg in ALK-positive NSCLC patients previously treated with crizotinib. Both agents demonstrated tumor responses against crizotinib-resistant brain metastases.
miRNA is a promising target to overcome drug resistance in chemotherapies
miRNA is a small noncoding RNA molecule that functions in transcriptional and posttranscriptional regulation of gene expression. miRNA genes are evolutionarily conserved and are located within the introns or exons of protein-coding genes, as well as in intergenic areas.41,42 miRNA has been implicated with cancer development. An interesting observation is that, for the widespread miRNA downregulation observed in human cancers, p53 is often dysfunctional.43 Moreover, deletion of Dicer abrogates the production of mature miRNAs,44 and conditional deletion of Dicer1 enhances lung tumor development in a K-ras-induced lung cancer mouse model.45 There are a variety of miRNAs, including the let-7 miRNA which is a cancer suppressor. In humans, the let-7 family is a cluster of miRNAs whose genes map to different chromosomal regions that are frequently deleted in lung cancer.46 All members of the miR-17-29 cluster and miR-31 are oncogenes. A recent in vitro study by Galluzzi et al47 showed that miR-630 inhibits p53-regulated proapoptotic signaling pathways that are specifically induced by cisplatin and carboplatin. The fact that miRNAs may involve chemosensitivity/chemoresistance determination, especially in EGFR-TKI treatments, suggests the possibility that manipulating miRNAs may be potentially useful to modulate the cancer chemoresistance. These novel therapeutic interventions of miRNA include drug-induced apoptosis by sensitizing tumor cells as well as inhibiting tumor proliferation and invasive capabilities.
Oncomirs in lung cancer
Oncomirs have higher basal expression in malignant, as compared to adjacent, normal lung tissues. Some are potential prognostic biomarkers, as in the case of miR-92a-2* in SCLC48 or the examples of miR-155, miR-130a, let-7f, and miR-30e-3p in NSCLC.49,50 To date, few candidate oncomirs have had mechanistic validation or identification of the target genes that would exert their oncogenic effects.
One of these oncomirs is miR-21; it plays a key functional role in several cancers,51–53 including lung cancer.54 Prior work revealed that miR-21 promotes cellular growth and augments tumor invasion and metastasis39 by reducing expression of specific sets of target genes that exert tumor suppressive effects. For example, in K-ras-dependent mouse lung cancers, there is an increase in miR-21 expression; miR-21 targets multiple negative regulators of the RAS/MEK/extracellular regulated MAP kinase pathway. Also, miR-21 inhibits apoptosis by reducing expression of proapoptotic gene products.
It is also reported that miR-31 acts as an oncomir in murine and human lung cancers.55 That miR-31 and other miRNAs were overexpressed in transgenic cyclin E-driven murine lung cancers implied that a similar miRNA expression profile would occur in human lung cancers.56 This was the case when human lung cancers (versus adjacent normal lung tissues) were found to have a similar expression pattern for several of the miRNAs that were highlighted in previously described transgenic lung cancers (Figure 2).56
Figure 2.
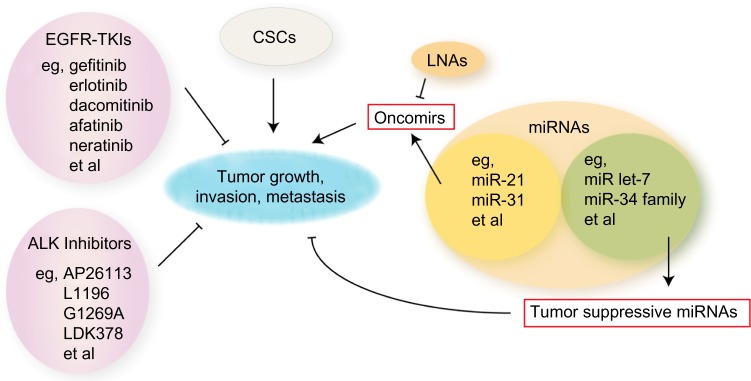
Schematic diagram of potential therapies combating resistance in lung cancer.
Notes: EGFR-TKIs and ALK inhibitors help postpone drug resistance and suppress tumor progression. CSCs may be an obstacle for therapies. Though oncomirs promote tumor growth, tumor suppressive micro (mi)RNAs play positive roles in tumor inhibition.
Abbreviations: ALK, anaplastic lymphoma kinase; CSCs, cancer stem cells; EGFR-TKIs, epidermal growth factor receptor-tyrosine kinase inhibitors; LNAs, locked nucleic acids.
Tumor suppressive miRNAs in lung cancer
In contrast to oncomirs, expression profiles of candidate tumor-suppressive miRNAs are repressed in cancers versus adjacent normal lung tissues. For example, reduced expression of the miRNA let-7 was reported to occur in bronchioloalveolar carcinoma57 and lung adenocarcinoma.58 The miR-146b expression patterns in squamous cell lung cancer were found to predict a poor clinical outcome, and miR-34a expression was found as a biomarker for clinical relapse in surgically resected NSCLC.59 Also, miR-34a was inactivated by CpG methylation and this caused transcriptional silencing in lung cancers.60 The precise mechanisms through which these miRNAs exert their tumor-suppressive effects remain to be determined.
The miR-34 family is reported as a p53-induced tumor-suppressive miRNA family in diverse types of cancers.61–63 As a transcription factor, p53 directly induces miR-34 family transcription. Ectopic miR-34 expression can augment apoptosis, cell-cycle arrest, or senescence. The promoter regions of the miR-34 family are often inactivated by CpG methylation.60 Repression of the miR-34 family was linked to resistance to p53-activating agents that can cause apoptotic response to specific chemotherapy treatments.64
Therefore, specific miRNAs play critical roles in regulating tumorigenicity. It is thus appealing to consider pharmacologic strategies to target specific miRNAs in lung carcinogenesis. These approaches can use miRNA derivatives as anticancer agents. Inhibition of specific oncomirs is achieved by use of optimized antisense derivatives. As an example, locked nucleic acid (LNA) miRNA derivatives represent a class of chemically modified nucleic acids that inhibit specific miRNAs in mouse models65 and in nonhuman primates.66 LNAs and related compounds are designed to be stable in serum and to exert limited off-target toxicities.66 Use of LNAs to target oncomirs in lung cancer is an attractive approach. Other chemically-modified nucleotide compounds were shown to antagonize miRNA activities in in vitro and in vivo models. For example, 2′-O-methyl-modified nucleotide derivatives inhibited miR-122 (cholesterol-conjugated antagomir) in mice, which caused a decrease in cholesterol biosynthesis.67 An analogous approach inhibited miR-10b activity in a mouse mammary tumor model, and this caused a decrease in lung metastases.
In contrast to suppression of oncomirs as a way to confer antitumorigenic effects, restoration of tumor suppressive miRNAs was also examined in preclinical models. A chemically synthesized miR-34a analog was used along with a lipid-based formulation.67 This was delivered to murine models by intravenous delivery, which caused antitumorigenic effects.67 Other examples were that of intratumoral or intranasal delivery of let-7 to established tumors in mouse models of NSCLC, and this conferred antineoplastic effects.68
Agents that affect expression of specific miRNAs were shown to overcome tumor-suppressive or oncogenic miRNA effects. These agents are in the early phases of preclinical testing. Preclinical validation would set the stage for conduct of proof of principle clinical trials targeting desired miRNAs. In this regard, the miRNA expression profile within a patient’s lung cancer might need to be discerned before selection of the miRNA-based therapy that would target aberrant miRNA expression within the tumor.
CSC is the obstacle to chemotherapy
Tumor drug resistance is a major issue in the management of lung cancer patients as almost all lung tumors are either intrinsically resistant or quickly develop acquired resistance to chemotherapeutic drugs. Cancer drug resistance has recently been linked, at least in part, to the existence of CSLCs, a small subpopulation of cells within the tumor that possess stem-like properties.13
The major criteria of stem cells is their ability to self-renew and differentiate into various cellular lineages via asymmetric division. Moreover, stem cells are characterized by slow division kinetics, drug and irradiation resistance, and an increased capacity of invasion, metastasis, tumor formation, and proliferation.69,70 CSCs are cancer cells (found within tumors or hematological cancers) that possess characteristics associated with normal stem cells; specifically, the ability to give rise to all cell types found in a particular cancer sample. CSCs are, therefore, tumorigenic in contrast to other nontumorigenic cancer cells. It is assumed that lung cancer derives from a multipotent stem-like cell that is able to generate these various histological components because lung cancers show frequent intratumoral heterogeneity.71,72
So far, little heed has been taken of the specific drug resistance mechanisms of CSCs in antineoplastic treatment. The ability for asymmetric cell division, the slow division kinetics or even quiescence, and the expression of several drug efflux pumps and DNA repair proteins represent the major escape mechanisms of CSCs.73–75 As most classical cytostatics require cell division in order to be effective, quiescence or slow division kinetics protects tumor cells against lethal DNA damage. Moreover, novel molecular drugs such as Akt-TKI might support rather than suppress CSCs as they favor asymmetric cell division and the generation of slowly-cycling drug-resistant tumor cells.76
Besides division symmetry and slow division kinetics, CSCs show an increased expression of DNA repair proteins and drug efflux transporters. Salnikov et al and Ota et al demonstrated that overexpression of TS and ERCC1 (both are DNA repair proteins), which might predict resistance against pemetrexed and cisplatin, respectively, correlated with the expression of the stem cell antigens cluster of differentiation (CD)133 and BCRP1;77,78 The findings of Ota et al demonstrated a correlation between protein expression of ERCC1 and the stem cell antigen BCRP1 (P=0.012), and the results of Salnikov et al, who demonstrated a correlation between protein expression of TS and the stem cell antigen CD133 (P,0.01), indicate that tumor cells with predominant expression of these enzymes might bear stem cell characteristics.77,78 CSCs possess several mechanisms to overcome irreversible damage by cytostatic drugs. These mechanisms have neither been studied sufficiently nor have they been considered in cancer therapy so far.
As the ability of asymmetric cell division and the generation of slowly cycling or quiescent progenies represent important escape mechanism of CSCs, pivotal molecules and mechanisms involved in the regulation of division symmetry might provide one important target in treating NSCLC. Currently, several Phase I and II studies testing inhibitors of the Wnt, Notch, and Hedgehog pathway as well as the PI3K/Akt/mTOR signal transduction axis are in progress. Eradication of these CSCs might contribute to better response and prolonged survival in lung cancer.79
Summary
Over the last decade, our understanding of human cancer development has greatly increased and much progress has been made in cancer therapy. Nevertheless, our ability to develop clinically effective therapies based on this knowledge has had limited success.80 After an apparently successful initial therapy, many tumors often relapse in a more aggressive form than the original tumor. Lung cancer is the worldwide leading cause of cancer-related deaths, and one of the most incurable cancers due to late presentation, disease relapse, and low rate of curative therapy.81 Indeed, after an apparent good response to initial therapy, lung cancer has a particularly poor prognosis, with 5-year survival lower than 15%.
Drug resistance has been one of the hindrances to the progress of cancer treatment. The irresponsiveness to chemotherapies at a later stage of treatment leads to poor prognosis. To study the methods combating resistance, NSCLC has been used frequently as a model. Looking into the resistance mechanism in the standard and relatively efficient EGFR-TKIs in lung cancer offers the opportunity to develop second-generation TKIs to improve drug concentrations during treatment. Moreover, the emerging evidence of CSCs and miRNA pathways provide other novel molecular targets for postponing resistance onset (Figure 2). Although at preliminary research stage, CSCs and miRNAs are potential powerful targets to efficiently combat resistance in that they are both involved in several aspects of cancer-cell development. Besides the hope to develop novel drugs through these targets, an alternative idea is to develop efficient drug delivery systems, which can both maintain blood drug concentration, especially within tumor cells, and at the same time initiate apoptosis. However, as discussed above, combating drug resistance is not only an issue in cancers. Although the mechanisms have been studied for decades, individual diseases have specific mechanisms to be elucidated. Maintaining drug concentration through either novel targets or novel delivery strategies, and stimulating the internal apoptosis signaling through immunity onset, remain the two most focused aims to delay resistance and prolong therapeutic effects.
Footnotes
Disclosure
This research is supported by a grant from Tianjin Health Bureau (No. 2011KZ116, No. 2010KZ106), Tianjin Natural Science Foundation (No. 10JCYBJC23700) and the National Natural Science Foundation (No. 81370183). The authors declare no conflicts of interest.
References
- 1.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83:584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Travis WD, Brambilla E, Noguchi M, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6(2):244–285. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stewart DJ, Johnson C, Lopez A, Glisson B, Rhee JM, Bekele BN. Extensive disease small cell lung cancer dose-response relationships: implications for resistance mechanisms. J Thorac Oncol. 2010;5(11):1826–1834. doi: 10.1097/JTO.0b013e3181f387c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almeida GM, Duarte TL, Farmer PB, Steward WP, Jones GD. Multiple end-point analysis reveals cisplatin damage tolerance to be a chemoresistance mechanism in a NSCLC model: implications for predictive testing. Int J Cancer. 2008;122(8):1810–1819. doi: 10.1002/ijc.23188. [DOI] [PubMed] [Google Scholar]
- 5.Donev IS, Wang W, Yamada T, et al. Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clin Cancer Res. 2011;17(8):2260–2269. doi: 10.1158/1078-0432.CCR-10-1993. [DOI] [PubMed] [Google Scholar]
- 6.Garofalo M, Romano G, Di Leva G, et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med. 2012;18(1):74–82. doi: 10.1038/nm.2577. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Catuogno S, Cerchia L, Romano G, Pognonec P, Condorelli G, de Franciscis V. miR-34c may protect lung cancer cells from paclitaxel-induced apoptosis. Oncogene. 2013;32(3):341–351. doi: 10.1038/onc.2012.51. [DOI] [PubMed] [Google Scholar]
- 8.Romano G, Acunzo M, Garofalo M, et al. MiR-494 is regulated by ERK1/2 and modulates TRAIL-induced apoptosis in non-small-cell lung cancer through BIM down-regulation. Proc Natl Acad Sci U S A. 2012;109(41):16570–16575. doi: 10.1073/pnas.1207917109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrison R, Schleicher SM, Sun Y, et al. Targeting the mechanisms of resistance to chemotherapy and radiotherapy with the cancer stem cell hypothesis. J Oncol. 2011;2011:941876. doi: 10.1155/2011/941876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23(25):5900–5909. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 11.Milella M, Nuzzo C, Bria E, et al. EGFR molecular profiling in advanced NSCLC: a prospective phase II study in molecularly/clinically selected patients pretreated with chemotherapy. J Thorac Oncol. 2012;7(4):672–680. doi: 10.1097/JTO.0b013e31824a8bde. [DOI] [PubMed] [Google Scholar]
- 12.Yu S, Wang Y, Li J, et al. Gefitinib versus erlotinib as salvage treatment for lung adenocarcinoma patients who benefited from the initial gefitinib: A retrospective study. Thoracic Cancer. 2013;4(2):109–116. doi: 10.1111/j.1759-7714.2012.00152.x. [DOI] [PubMed] [Google Scholar]
- 13.Freitas DP, Teixeira CA, Santos-Silva F, Vasconcelos MH, Almeida GM. Therapy-induced enrichment of putative lung cancer stem-like cells. Int J Cancer. 2014;134(6):1270–1278. doi: 10.1002/ijc.28478. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Qu L, Wei X, et al. Clinical observation of EGFR-TKI as a first-line therapy on advanced non-small cell lung cancer. Zhongguo Fei Ai Za Zhi. 2012;15(5):299–304. doi: 10.3779/j.issn.1009-3419.2012.05.09. Chinese. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Z, Wang Z, Bai H, et al. The detection of EGFR mutation status in plasma is reproducible and can dynamically predict the efficacy of EGFR-TKI. Thoracic Cancer. 2012;3(4):334–340. doi: 10.1111/j.1759-7714.2012.00133.x. [DOI] [PubMed] [Google Scholar]
- 16.Li R, Lou Y, Zhang Y, et al. Clinical analysis of Gefitinib in the treatment of stage IV lung adenocarcinoma with unknown EGFR gene mutations. Thoracic Cancer. 2013;4(4):433–439. doi: 10.1111/1759-7714.12044. [DOI] [PubMed] [Google Scholar]
- 17.Antonicelli A, Cafarotti S, Indini A, et al. EGFR-targeted therapy for non-small cell lung cancer: focus on EGFR oncogenic mutation. Int J Med Sci. 2013;10(3):320–330. doi: 10.7150/ijms.4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 19.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 20.Thongprasert S, Duffield E, Saijo N, et al. Health-related quality-of-life in a randomized phase III first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients from Asia with advanced NSCLC (IPASS) J Thorac Oncol. 2011;6(11):1872–1880. doi: 10.1097/JTO.0b013e31822adaf7. [DOI] [PubMed] [Google Scholar]
- 21.Yoshimura N, Okishio K, Mitsuoka S, et al. Prospective assessment of continuation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of pemetrexed. J Thorac Oncol. 2013;8(1):96–101. doi: 10.1097/JTO.0b013e3182762bfb. [DOI] [PubMed] [Google Scholar]
- 22.Johnson ML, Riely GJ, Rizvi NA, et al. Phase II trial of dasatinib for patients with acquired resistance to treatment with the epidermal growth factor receptor tyrosine kinase inhibitors erlotinib or gefitinib. J Thorac Oncol. 2011;6(6):1128–1131. doi: 10.1097/JTO.0b013e3182161508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadgeel SM, Ruckdeschel JC, Heath EI, Heilbrun LK, Venkatramanamoorthy R, Wozniak A. Phase II study of gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), and celecoxib, a cyclooxygenase-2 (COX-2) inhibitor, in patients with platinum refractory non-small cell lung cancer (NSCLC) J Thorac Oncol. 2007;2(4):299–305. doi: 10.1097/01.JTO.0000263712.61697.69. [DOI] [PubMed] [Google Scholar]
- 24.Reckamp KL, Krysan K, Morrow JD, et al. A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clin Cancer Res. 2006;12(11 Pt 1):3381–3388. doi: 10.1158/1078-0432.CCR-06-0112. [DOI] [PubMed] [Google Scholar]
- 25.Bivona TG, Hieronymus H, Parker J, et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471(7339):523–526. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadara H, Shen L, Fujimoto J, et al. Characterizing the molecular spatial and temporal field of injury in early-stage smoker non-small cell lung cancer patients after definitive surgery by expression profiling. Cancer Prev Res (Phila) 2013;6(1):8–17. doi: 10.1158/1940-6207.CAPR-12-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarhini A, Kotsakis A, Gooding W, et al. Phase II study of everolimus (RAD001) in previously treated small cell lung cancer. Clin Cancer Res. 2010;16(23):5900–5907. doi: 10.1158/1078-0432.CCR-10-0802. [DOI] [PubMed] [Google Scholar]
- 28.Nurwidya F, Takahashi F, Takahashi K. Meeting report: Current cancer perspectives from the 9th Annual Meeting of the Japanese Society of Medical Oncology. Thoracic Cancer. 2012;3(1):94–97. doi: 10.1111/j.1759-7714.2011.00076.x. [DOI] [PubMed] [Google Scholar]
- 29.Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17(17):5530–5537. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13(5):528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 32.Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67(24):11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 33.Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2(11):e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerecitano J, Gounder S, Teruya-Feldstein J, et al. Tissue microarray analysis reveals protein expression patterns and potential biomarkers of clinical benefit to bortezomib in relapsed/refractory non-Hodgkin lymphoma. Br J Haematol. 2012;158(2):290–292. doi: 10.1111/j.1365-2141.2012.09137.x. [DOI] [PubMed] [Google Scholar]
- 35.Oxnard GR, Miller VA, Robson ME, et al. Screening for germline EGFR T790M mutations through lung cancer genotyping. J Thorac Oncol. 2012;7(6):1049–1052. doi: 10.1097/JTO.0b013e318250ed9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sasaki H, Endo K, Takada M, et al. EGFR exon 20 insertion mutation in Japanese lung cancer. Lung Cancer. 2007;58(3):324–328. doi: 10.1016/j.lungcan.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 37.Su Z, Dias-Santagata D, Duke M, et al. A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. J Mol Diagn. 2011;13(1):74–84. doi: 10.1016/j.jmoldx.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109(31):E2127–E2133. doi: 10.1073/pnas.1203530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’Angelo SP, Janjigian YY, Ahye N, et al. Distinct clinical course of EGFR-mutant resected lung cancers: results of testing of 1118 surgical specimens and effects of adjuvant gefitinib and erlotinib. J Thorac Oncol. 2012;7(12):1815–1822. doi: 10.1097/JTO.0b013e31826bb7b2. [DOI] [PubMed] [Google Scholar]
- 40.Reungwetwattana T, Dy GK. Targeted therapies in development for non-small cell lung cancer. J Carcinog. 2013;12:22. doi: 10.4103/1477-3163.123972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14(10A):1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar R, Xi Y. MicroRNA, epigenetic machinery and lung cancer. Thoracic Cancer. 2011;2(2):35–44. doi: 10.1111/j.1759-7714.2011.00043.x. [DOI] [PubMed] [Google Scholar]
- 43.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 44.Bernstein E, Kim SY, Carmell MA, et al. Dicer is essential for mouse development. Nat Genet. 2003;35(3):215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 45.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39(5):673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 46.Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101(9):2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galluzzi L, Kepp O, Kroemer G. A new role for cytoplasmic p53: binding and destroying double–stranded RNA. Cell Cycle. 2010;9(13):2491–2492. doi: 10.4161/cc.9.13.12191. [DOI] [PubMed] [Google Scholar]
- 48.Ranade AR, Cherba D, Sridhar S, et al. MicroRNA 92a-2*: a biomarker predictive for chemoresistance and prognostic for survival in patients with small cell lung cancer. J Thorac Oncol. 2010;5(8):1273–1278. doi: 10.1097/JTO.0b013e3181dea6be. [DOI] [PubMed] [Google Scholar]
- 49.Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9(3):189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 50.Silva J, García V, Zaballos Á, et al. Vesicle-related microRNAs in plasma of nonsmall cell lung cancer patients and correlation with survival. Eur Respir J. 2011;37(3):617–623. doi: 10.1183/09031936.00029610. [DOI] [PubMed] [Google Scholar]
- 51.Corsten MF, Miranda R, Kasmieh R, Krichevsky AM, Weissleder R, Shah K. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007;67(19):8994–9000. doi: 10.1158/0008-5472.CAN-07-1045. [DOI] [PubMed] [Google Scholar]
- 52.Asangani IA, Rasheed SA, Nikolova DA, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27(15):2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 53.Ribas J, Ni X, Haffner M, et al. miR-21: an androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res. 2009;69(18):7165–7169. doi: 10.1158/0008-5472.CAN-09-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seike M, Goto A, Okano T, et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc Natl Acad Sci U S A. 2009;106(29):12085–12090. doi: 10.1073/pnas.0905234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Sempere LF, Ouyang H, et al. MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J Clin Invest. 2010;120(4):1298–1309. doi: 10.1172/JCI39566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma Y, Fiering S, Black C, et al. Transgenic cyclin E triggers dysplasia and multiple pulmonary adenocarcinomas. Proc Natl Acad Sci U S A. 2007;104(10):4089–4094. doi: 10.1073/pnas.0606537104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inamura K, Togashi Y, Nomura K, et al. let-7 microRNA expression is reduced in bronchioloalveolar carcinoma, a non-invasive carcinoma, and is not correlated with prognosis. Lung Cancer. 2007;58(3):392–396. doi: 10.1016/j.lungcan.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 58.Takamizawa J, Konishi H, Yanagisawa K, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64(11):3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 59.Raponi M, Dossey L, Jatkoe T, et al. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009;69(14):5776–5783. doi: 10.1158/0008-5472.CAN-09-0587. [DOI] [PubMed] [Google Scholar]
- 60.Lodygin D, Tarasov V, Epanchintsev A, et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7(16):2591–2600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 61.He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435(7043):828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007;67(18):8433–8438. doi: 10.1158/0008-5472.CAN-07-1585. [DOI] [PubMed] [Google Scholar]
- 63.Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26(34):5017–5022. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- 64.Zenz T, Mohr J, Eldering E, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009;113(16):3801–3808. doi: 10.1182/blood-2008-08-172254. [DOI] [PubMed] [Google Scholar]
- 65.Elmén J, Lindow M, Silahtaroglu A, et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36(4):1153–1162. doi: 10.1093/nar/gkm1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elmén J, Lindow M, Schütz S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452(7189):896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 67.Wiggins JF, Ruffino L, Kelnar K, et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010;70(14):5923–5930. doi: 10.1158/0008-5472.CAN-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trang P, Medina PP, Wiggins JF, et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2010;29(11):1580–1587. doi: 10.1038/onc.2009.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 70.Sullivan JP, Minna JD, Shay JW. Evidence for self-renewing lung cancer stem cells and their implications in tumor initiation, progression, and targeted therapy. Cancer Metastasis Rev. 2010;29(1):61–72. doi: 10.1007/s10555-010-9216-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fraire AE, Roggli VL, Vollmer RT, et al. Lung cancer heterogeneity. Prognostic implications. Cancer. 1987;60(3):370–375. doi: 10.1002/1097-0142(19870801)60:3<370::aid-cncr2820600314>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 72.Long H, Zhang S, Liu C, et al. Characterization of a stem cell population in lung cancer cell line Glc-82. Thoracic Cancer. 2012;3(1):8–18. doi: 10.1111/j.1759-7714.2011.00089.x. [DOI] [PubMed] [Google Scholar]
- 73.Tomasetti C, Levy D. Role of symmetric and asymmetric division of stem cells in developing drug resistance. Proc Natl Acad Sci U S A. 2010;107(39):16766–16771. doi: 10.1073/pnas.1007726107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 75.Saini V, Shoemaker RH. Potential for therapeutic targeting of tumor stem cells. Cancer Sci. 2010;101(1):16–21. doi: 10.1111/j.1349-7006.2009.01371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dey-Guha I, Wolfer A, Yeh AC, et al. Asymmetric cancer cell division regulated by AKT. Proc Natl Acad Sci U S A. 2011;108(31):12845–12850. doi: 10.1073/pnas.1109632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ota S, Ishii G, Goto K, et al. Immunohistochemical expression of BCRP and ERCC1 in biopsy specimen predicts survival in advanced non-small-cell lung cancer treated with cisplatin-based chemotherapy. Lung Cancer. 2009;64(1):98–104. doi: 10.1016/j.lungcan.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 78.Salnikov AV, Gladkich J, Moldenhauer G, Volm M, Mattern J, Herr I. CD133 is indicative for a resistance phenotype but does not represent a prognostic marker for survival of non-small cell lung cancer patients. Int J Cancer. 2010;126(4):950–958. doi: 10.1002/ijc.24822. [DOI] [PubMed] [Google Scholar]
- 79.Gottschling S, Schnabel PA, Herth FJ, Herpel E. Are we missing the target? Cancer stem cells and drug resistance in non-small cell lung cancer. Cancer Genomics Proteomics. 2012;9(5):275–286. [PubMed] [Google Scholar]
- 80.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 81.Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]