

Abstract
Cation-π interactions of methylated ammonium ions play a key role in a broad range of biochemical systems. These include methyl-lysine binding proteins which bind to methylated sites on histone proteins, lysine demethylase enzymes which demethylate these sites, and neurotransmitter receptor complexes which bind choline derived ligands. Recognition in these systems is achieved through an ‘aromatic cage’ motif in the binding site. Here we use high level quantum mechanical calculations to address how cation-π interactions of methylated ammonium ions are modulated by a change in methylation state and interaction geometry. We survey methyl-lysine and choline derived complexes in the Protein Databank to validate our results against available structural data. A quantitative description of cation-π interactions of methylated ammonium systems is critical to structure-based efforts to target methyl-lysine binding proteins and demethylase enzymes in the treatment of unregulated transcriptional control, and neurotransmitter receptors in the treatment of neurological disease. It is our hope that our work will serve as a benchmark for the development of physical chemistry based force fields that can accurately model the contribution of cation-π interactions to binding and specificity in these systems.
Keywords: counterpoise calculation, histone binding proteins, lysine methylation, lysine demethylase, choline
Introduction
Cation-π interactions involving methylated ammonium ions play a significant role in molecular recognition and protein-protein interactions.1-3 Early studies showed that ammonium and methyl-ammonium ions form strong complexes with benzene.4 Subsequent studies showed that water soluble quaternary ammonium compounds could form host guest complexes with natural and synthetic aryl receptors such as cyclophane.5-7 Choline, and choline derived ligands such as acetylcholine and phosphocholine, bind to their receptors via interactions between a trimethyl-ammonium head group and an aromatic cage of two to four Trp, Tyr, or Phe residues.8-10 The same aromatic cage motif is utilized by methyl-lysine binding proteins in the recognition of lysine methylated sites on the tails of histone proteins.11-17
Lysine methylation is a post-translational modification in which one, two, or three methyl groups are added to the side chain nitrogen atom of a lysine residue. In histone proteins, this modification plays a key role in epigenetic control of transcriptional regulation which for the most part is effected indirectly through recruitment of “reader” proteins which bind to the methylated lysines.18-19 Methyl-lysine binding proteins are specific for particular methylation states, for example the Chp1 chromodomain20 and the PHD finger of the ING4 and ING2 tumor suppressors16,21-22 have the highest affinity for trimethyl-lysine, while the Tudor domains, found in the DNA repair protein 53BP1 and its homolog Crb2, preferentially bind to dimethyl-lysine.12,23 It has further been shown that methylation of a single site can result in different functional consequences depending on the number of methyl groups that have been added.25
Over the past ten years it has become apparent that lysine methylation is a dynamic process in which methyl groups can be removed from histone proteins by the action of lysine demethylase enzymes.26 Like methyl-lysine binding proteins, these enzymes are characterized by aromatic binding sites and are specific for methylation state; for example lysine-specific demethylase 1(LSD1) does not demethylate trimethyl-lysine while JMJD2A is specific to the trimethylated state.27,28 Deregulation of either methylation or demethylation results in malfunctioning transcriptional control and is associated with cancer development. Thus, both histone methyl transferases and histone lysine demethylases are considered to be potential targets for inhibition in the treatment of cancer.26,29
Several noncovalent binding forces come into play in determining methyl-lysine binding specificity. In lower methylation states hydrogen bonding occurs between the side chain amino proton and a nearby acidic residue,30,31 and smaller size cavities present steric hindrance against higher methylation states. Hydrophobic desolvation, which refers to the reduction in the free energy cost of desolvating a methylated lysine in place of an unmethylated one, favors binding of higher methylation states. The effect of hydrophobic desolvation has been observed in a range of systems including host systems that prefer methylated over unmethylated guests,32,33 beta hairpin peptides whose stability is enhanced by the interaction between one or more Trp residues and a methylated Lys residue,34-36 and the factor Xa protein which shows higher affinities to more methylated ligands.37 Cation-π interactions represent another noncovalent binding force; these interactions occur between the charged methyl-lysine residue and surrounding aromatic residues. The primarily electrostatic nature of this interaction dictates that the larger size and more dispersed positive charge of the more methylated ion results in weaker interactions for higher methylation states.1 An understanding of methyl-lysine binding specificity depends on precise models which can quantify the competing noncovalent forces which come into play within a particular binding site, and is critical for the development of synthetic, and potentially therapeutic, receptors which bind to these sites.38-40
Cation-π interactions are not easily modeled by traditional molecular mechanics force fields due to effects such as induction, polarization, and quadropole moment interactions, which may all contribute to the strength of the interaction.1, 41 Quantum mechanical studies on model systems are the benchmark for the parameterization and formulation of force fields that can be used to quantify the energetics of chemical and biochemical systems.42-44 Recent works that have utilized quantum mechanical calculations on cation-π systems to parameterize polarizable molecular mechanics force fields include Orabi and Lamoureux45 and Ansorg et al.46 Here we conduct high level quantum mechanical calculations to address how cation-π interactions in methylated ammonium systems are affected by changes in methylation state and interaction geometry. We use ammonium ions that are methylated to different degrees as models for methyl-lysine and quaternary ammonium compounds; and benzene as a model for the aromatic amino acids Trp, Tyr, and Phe. While the most preferred geometry for a simple cation interacting with benzene places the cation directly over the center of the benzene ring (axial position),1 it has been shown that almost half of the cation-π interactions between residues in the Protein Databank (PDB)47 are off-axis.48 Since studies that have shown that even interactions placing the cation in the plane of the benzene ring can be attractive,49 our model systems consist of methylated ammonium ions at angles of 0-90° from the axial position. Finally, we compare our theoretical results with experimental complexes of methyl-lysine binding proteins and receptors of choline derived ligands in the PDB to get a sense of how predicted favored geometries of interaction are reflected in actual protein complexes. It is hoped that our work will contribute to the development of improved models to describe specificity, recognition, and binding energies in methylated ammonium systems.
Methods
Quantum Mechanical Calculations
Initial coordinates for all geometries were set up in Maestro50 using the default geometries for the ammonium ion and benzene. Following the work of others44,49 geometries for the benzene-ammonium complexes are described based on three parameters as shown in Figure 1: R, the distance between the ammonium nitrogen atom and the center of the benzene ring; theta (θ), the angle between the axial to the ring and the direction of R (such that θ=0° places the cation directly above the center of the ring); and psi (ϕ) for off- axis geometries, referring to whether the cation is aligned on a vertical place with a benzene hydrogen atom (ϕ=0°) or is positioned in between two hydrogen atoms (ϕ=30°). Geometries are labeled θ_ϕ (for example 30_0) in the context of this work. For consistency, interaction geometries for the mono-, di-, tri- and tetra-methylated ammonium ions were generated directly from the benzene-ammonium geometries by adding the appropriate number of methyl groups to the ammonium ion. Methyl groups were added to the position that would place the group farthest from the benzene ring. While this choice is arbitrary, subsequent optimization steps allow for the relaxation of the configuration of the methylated ion.
Figure 1.
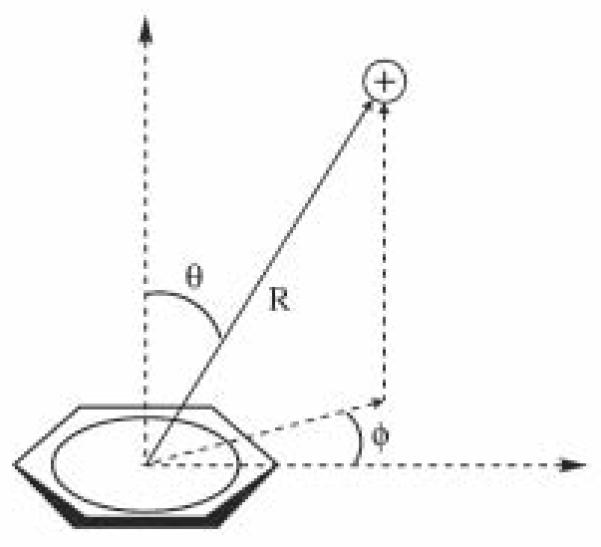
Geometries for Cation-π Systems.49
All calculations were carried out with the Jaguar software package.51 Potential energy surfaces (PES) were generated for each geometry by adjusting the distance R from two to seven Angstroms at one Angstrom intervals, and then at intervals of 0.05 Angstroms around the region of the minimum. The benzene molecule and ammonium cations were held rigid and single point energies were calculated using the M06−2X52 functional with the 6−31++G** basis set. Default grids and SCF convergence criteria as implemented in Jaguar were used. Interaction energies were obtained by subtracting the energies of the monomers from the energy of the complex.
The calculation of the PES surface was followed by a more rigorous quantum mechanical calculation for each complex at its minimum energy distance based on the PES. Counterpoise calculations53 were implemented in Jaguar using the M06−2X52 functional with the aug-cc-pVTZ without f functions basis set. The counterpoise calculation involves geometry optimization of the complex, geometry optimization of each of the monomers in its own basis set, single-point calculations of each of the monomers in its own basis set in the geometries adopted in the complex, and single-point counterpoise calculations on each of the monomers in the geometries adopted in the complex using the basis set of the complex. For the geometry optimization of the complex, constraints were implemented to fix the value of theta at 0°, 30°, 60°, or 90° as it was set in the initial geometry. This was done by positioning a dummy atom in the axial position directly above the center of the ring and constraining the angle between the dummy atom, the centroid of the benzene ring, and the nitrogen atom of the cation. The position of the centroid was also constrained with respect to the ring. For two cases in which the SCF calculation did not converge, the value of R was increased by 0.1 Angstroms above its value from the minimum of the PES, and the calculation was repeated. Interaction energies and basis set superposition errors (BSSE)53 were calculated for each system.
Methylated Lysine Residues in the Protein Databank
We searched the Protein Databank (PDB)47 for all entries containing a methyl-lysine residue using the three letter codes MLZ, MLY, and M3L to search for mono-, di- and tri-methyl lysine respectively. In all, we identified four, nine, and 25 entries containing mono-, di- and tri- methyl lysine respectively. Choline and choline derived ligands such as phosphocholine and acetylcholine are also characterized by the ‘aromatic cage’ recognition mode; as such, our study includes twenty PDB entries containing choline and choline derived ligands. These structures were drawn from a table compiled by Cheng et al. of all choline ligand containing entries as of August 2012;54 our set includes the structure with the highest resolution from each bolded entry (indicating the presence of cation-π interactions) on that list. For each methyl-lysine residue or choline ligand, we identified all interactions with aromatic residues using the literature reference included in the PDB entry; in all, 11, 30, 74, and 46 interactions were identified for mono-, di-, tri-methyl lysine residues, and choline ligands, respectively. NMR entries, or entries that did not have an associated literature reference, were excluded from our study. For each interaction, we calculated R, the distance between the ammonium nitrogen atom and the center of the aromatic ring of the interacting Trp, Tyr, or Phe residue; and theta (θ), the angle between the axial to the ring and the direction of R.
Results
Quantum Mechanical Calculations
The Potential energy surfaces (PES) and the counterpoise calculations show that for all methylation states, the most favored geometry places the cation directly above the center of the benzene ring (Table I and Figs. 2 and 3). Methylation results in less favorable interaction energies and a slight shift to the right in the minimum energy distance (R) which becomes more notable in the transition from partial (mono, di, tri) to full methylation (tetra) (Fig. 2 and Table II). All off-axis geometries, i.e. θ=30°, 60° and 90°, show attractive interaction energies.
Table I.
Counterpoise energies and basis set superposition errors for methylammonium ions interacting with benzene, after constrained optimization using the aug-cc-pVTZ without f functions basis set and M06−2X functional.
| Initial Geometry | Counterpoise Energies (kcal/mol) | ||||
|---|---|---|---|---|---|
| NH4+ | CH3NH3+ | (CH3)2NH2+ | (CH3)3NH+ | (CH3)4N+ | |
| 0_0 | −19.0±0.1 | −18.1±0.6 | −17.0±0.7 | −16.7±1.1 | −8.8±0.7 |
| 30_0 | −14.6±0.2 | −14.0±0.1 | −12.9±0.5 | −12.7±0.7 | −8.2±0.7 |
| 30_30 | −15.1±0.0 | −13.0±0.2 | −12.9±0.6 | −12.8±0.6 | −8.1±0.7 |
| 60_0 | −9.0±0.1 | −8.5±0.2 | −7.8±0.6 | −7.5±0.7 | −3.9±0.7 |
| 60_30 | −9.0±0.1 | −8.7±0.2 | −7.9±0.6 | −7.6±0.7 | −3.9±0.7 |
| 90_0 | −4.7±0.1 | −4.1±0.2 | −4.0±0.6 | −3.9±0.6 | 0.5±0.1 |
| 90_30 | −4.7±0.1 | −4.3±0.2 | −4.3±0.6 | −3.8±0.5 | −0.5±0.2 |
Figure 2.
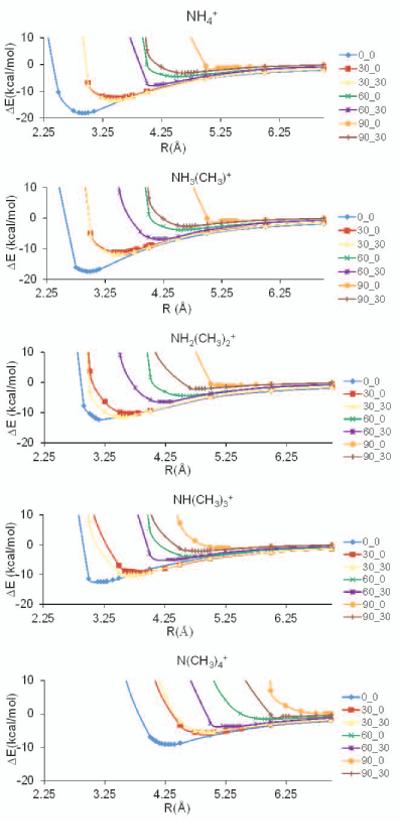
Potential energy surfaces for ammonium, mono-, di-, tri-, and tetra-methyl ammonium ions interacting with benzene, generated with 631**++ basis set and M06−2X functional.
Figure 3.
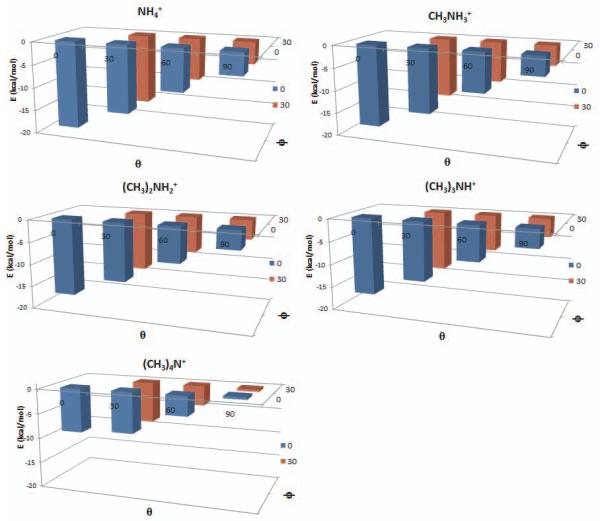
Counterpoise energies for ammonium and methylammonium ions interacting with benzene, after constrained optimization using the aug-cc-pVTZ without f functions basis set and M06−2X functional. Axes indicate theta and psi angles of starting geometries, and energies of the optimized systems.
Table II.
Interaction geometries for methylammonium ions interacting with benzene, after constrained optimization using the aug-cc-pVTZ without f functions basis set and M06−2X functional. R refers to the distance between the ammonium nitrogen and the center of the benzene ring, θ refers to the angle between the ammonium nitrogen and the axial to the ring, and ϕ refers to where the cation is positioned with respect to the benzene hydrogen atoms.
| Initial Geometry θ_ϕ |
NH4+ | CH3NH3+ | (CH3)2NH2+ | (CH3)3NH+ | (CH3)4N+ | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| R(Å) | ϕ | R(Å) | ϕ | R(Å) | ϕ | R(Å) | ϕ | R(Å) | ϕ | |
| 0_0 | 2.9 | - | 3.0 | - | 3.0 | - | 3.0 | - | 4.2 | - |
| 30_0 | 3.4 | 5-10 | 3.5 | 20-25 | 3.5 | 25-30 | 3.5 | 25-30 | 4.3 | 0-5 |
| 30_30 | 3.5 | 10-15 | 3.4 | 5-10 | 3.5 | 5-10 | 3.6 | 25-30 | 4.4 | 0-5 |
| 60_0 | 4.1 | 25-30 | 4.2 | 25-30 | 4.1 | 25-30 | 4.2 | 25-30 | 5.1 | 25-30 |
| 60_30 | 4.1 | 25-30 | 4.2 | 25-30 | 4.2 | 25-30 | 4.2 | 25-30 | 5.2 | 25-30 |
| 90_0 | 4.4 | 25-30 | 4.4 | 25-30 | 4.6 | 25-30 | 4.6 | 25-30 | 8.0 | 15-20 |
| 90_30 | 4.4 | 25-30 | 4.4 | 25-30 | 4.6 | 25-30 | 4.6 | 25-30 | 6.4 | 25-30 |
Figure 4 shows the optimized geometries produced by the counterpoise calculations for the 0_0, 30_0, 60_0, 90_0 geometries; these were generated by superimposing the benzene molecules of each of the optimized systems. Results show that the cations adopt a similar configuration in the off-axis θ=0° and θ=30° geometries, which changes as the cation moves further away from the axial position to θ=60° and θ=90°.
Figure 4.

Optimized geometries of methylammonium ions interacting with benzene, after constrained optimization using the aug-cc-pVTZ without f functions basis set and M06−2X functional.
For the off axis-geometries (θ=30°, 60°, 90°) the PES show a preference, in the form of more favorable interaction energies, for the cation being positioned in between two hydrogen atoms (ϕ=30°) instead of directly above a hydrogen atom (ϕ=0°) (Fig. 2). Figure 5 shows the optimized 60_0 geometry of tetramethyl-ammonium as an example. This preference is particularly notable in the counterpoise calculations (Table II). Independent of the starting geometry (i.e. for both ϕ=0° and ϕ=30°), the optimized systems show the cation at or near the ϕ=30° position. The angle ϕ was measured by positioning the benzene ring in the plane of the Maestro visualization window, drawing a bond between the nitrogen atom of the cation and the ring centroid, and inspecting the position of the bond with respect to the two closest carbon atoms.
Figure 5.
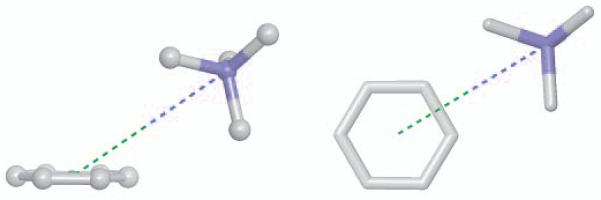
Two views of optimized geometry for tetramethyl-ammonium interacting with benzene highlighting θ=60° (left) and ϕ=30° (right).
Cation-π Interactions in Methylated Protein Structures
In all, 161 cation-π interactions between methyl-lysine or choline groups, and aromatic residues, were identified among experimental structures in the PDB. These include 11 mono-, 30 di-, 74 tri-methyllysine, and 46 choline ligands; interacting with 73 Trp, 60 Tyr, and 28 Phe residues. Notably, only one of the 46 choline interactions is with a Phe residue. Figure 6 shows interaction geometries for these cation-π interactions. Geometries are characterized by R, the distance between the ammonium nitrogen atom and the center of the aromatic ring of the interacting Trp, Tyr, or Phe residue; and theta (θ), the angle between the axial to the ring and the direction of R.
Figure 6.
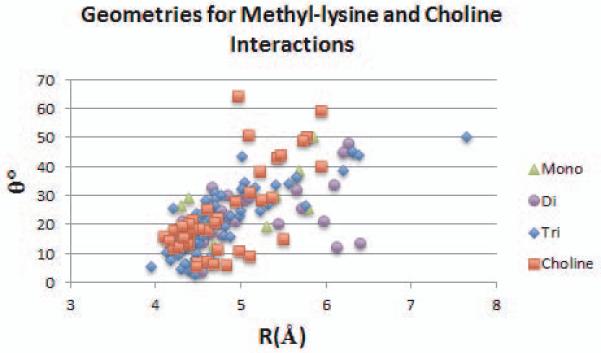
Geometries of interaction for methyl-lysine residues and choline ligands interacting with aromatic residues in 58 protein complexes in the Protein Databank. R refers to the distance between the ammonium nitrogen and the center of the aromatic ring, and θ refers to the angle between the axial to the center of the ring and the direction of R.
For all systems, ammonium-aromatic interactions show a clustering at R=4.0-4.5 Å and θ=0-30°. For the combined set of trimethyl-lysine and choline structures, average values are R=4.8±0.6 Å and θ=22.1±11.9°. These averages are consistent across residue types, for Tyr, Trp, and Phe residues in this set, average R values are 4.8±0.6 Å, 4.7±0.5 Å, and 5.2±0.8 Å, and average theta values are 20.0±11.5°, 23.0 ±10.5°, and 25.8±15.0° respectively. For interactions between partially methylated lysine residues and Tyr, there is potential for hydrogen bonding between the amide hydrogen and the hydroxyl of Tyr. To address whether hydrogen bonding, rather than cation-π interactions, accounts for these interactions, we measured the N-O distance between the nitrogen of the mono- or di-methyllysine amide and the oxygen of the Tyr hydroxyl group. For these seventeen cases, the N-O distance ranges between 4.5 and 7.5 Å (hydrogen bond cutoffs are typically ~4 Å) and as such hydrogen bonding is not likely to play a role.
Discussion
Energies calculated in the counterpoise calculations compare well with reported experimental results for ammonium ions interacting with benzene; these values from the literature are −19.3, −18.8, −15.9 and −9.4 kcal/mol for ammonium, mono-, tri- and tetramethylammonium respectively.2,55 Thus our results demonstrate that a counterpoise calculation using the aug-cc-pVTZ without f functions basis set and M06−2X functional is an effective approach to calculating the absolute and relative binding energies of simple cation-π systems. Our findings that off-axis geometries, including orthogonal (θ=90°) interactions, may be attractive, and the preference for the ϕ=30° position in between benzene hydrogen atoms, are consistent with the results of Marshall et al.49 for complexes of Li+, Na+, K+ and NH4+, with benzene. Our results show that methylation of the ammonium ions results in less favorable interaction energies, with the most significant increase in energy occurring in the transition from partial to full methylation. This is to be expected, as the positive charge is increasingly dispersed over the larger methyl groups in place of the smaller hydrogen atoms.
For the unmethylated ammonium ion we compare our results with those of Marshall et al.49 which were performed using the CCSD(T) method, often considered the gold standard for quantum mechanical calculations.43,56 For all systems, our calculations show more favorable interaction energies. This difference results in part from the geometry optimization step of our protocol in which only the value of theta is fixed, while the PES curves presented by Marshall et al. were performed with rigid monomers at fixed values of both theta and psi. For the optimized θ=0° and θ=30° geometries, our reported energies are lower than the corresponding CCSD(T) energies by 1.6-2.6 kcal/mol. For the optimized θ=60° and θ=90° geometries, which are all characterized by ϕ≈30°, our reported energies are within 1.1-1.5 kcal of the corresponding 60_30 and 90_30 CCSD(T) energies. The changes in interaction energy as the cation is incrementally moved from the axial (θ=0°) to orthogonal (θ=90°) position are comparable in both sets of calculations, and both show that in the orthogonal position, the attractive energy retains a significant part of its value at θ=0° (25% for our calculations, 21% for the CCSD(T) calculations). The R values of our optimized geometries are within 0.2 Å of the minimum energy distances of the corresponding CCSD(T) curves which were calculated for a dense set of grid points.49
Our survey of methyl-lysine and choline cation-π interactions in the PDB shows that geometries are clustered at 4.0≤R≤4.5 Å and 0°≤θ≤30° with average values of R=4.8±0.6 Å and θ=22.1±11.9°. The clustering around the axial position can be understood in light of the more favorable interaction energies for the θ=0° and θ=30° geometries as compared to those further away from the axial position. The average R value for all interactions characterized by 0°≤θ≤30° (81% of interactions) is 4.6±0.6 Å, which is consistent with the R values of the optimized tetramethyl-ammonium systems generated by our quantum mechanical calculations. An earlier survey48 of 910 Arg and Lys cation-π interactions in 96 PDB entries showed that 51% of all geometries were characterized by 0°≤θ≤30°, with an average R value for these interactions of 4.1±0.2 Å . The higher average R value for the structures in our study is consistent with results of our quantum mechanical calculations which show that the most favorable distance for ammonium-aromatic interactions increases with increasing methylation of the ammonium ion.
Methyl-lysine residues and choline molecule head groups are typically surrounded by an aromatic cage of two to four aromatic residues. It is unknown whether the effect of several aromatic donors is additive; future studies, along the lines of a recent study of a cation-π sandwich model,57 will address the effect of multiple aromatic groups on the interactions described here. Also to be addressed is the effect of the identity of the donor aromatic residue on the strength of the cation-π interaction; among the interactions in our dataset, most are with Trp or Tyr (37% and 45% respectively) and a smaller percentage are with Phe (17%).
Conclusions
This work presents state-of-the-art quantum mechanical calculations focused on cation-π interactions of ammonium ions, and the effect of methylation state and geometry on the energetics of these interactions. Understanding the contribution of cation-π interactions to binding and specificity in histone binding proteins and lysine demethylase enzymes is critical to elucidating the mechanisms of transcriptional regulation, and structure-based therapeutic efforts to target these proteins. Our study also has relevance to receptors of choline and choline derived ligands which are recognized by a trimethyl-ammonium headgroup. Acetylcholine, and many acetylcholine esterase inhibitors currently used or being developed for the treatment of Alzheimer’s disease, utilize cation-π interactions between an ammonium cation and aromatic residues in the enzyme binding site.58 Recent studies have further shown that cation-π interactions of phosphotidylcholine are involved in the recognition of target lipids by peripheral membrane proteins.54,59 It is hoped that our work will contribute to the development of physical chemistry based models that can quantify the contribution of cation-π interactions to binding and specificity across a broad range of biochemical systems.
Acknowledgement
This work was supported by National Institute of Health grants 1R15GM098997-01 to C.R.
References
- 1.Ma JC, Dougherty DA. The cation-pi interaction. Chem Rev. 1997;97:1303–24. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 2.Mahadevi AS, Sastry GN. Cation-π Interaction: Its Role and Relevance in Chemistry, Biology, and Material Science. Chem Rev. 2013;113:2100–2138. doi: 10.1021/cr300222d. [DOI] [PubMed] [Google Scholar]
- 3.Dougherty DA. The Cation-π Interaction. Acc Chem Res. 2013;46:885–893. doi: 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deakynet CA, Meot-Ner M. Unconventional Ionic Hydrogen Bonds. 2. NH+ ..π Complexes of Onium Ions with Olefins and Benzene Derivatives. J Am Chem Soc. 1985;107:474–479. [Google Scholar]
- 5.Smithrud DB, Diederich F. Strength of molecular complexation of apolar solutes in water and in organic solvents is predictable by linear free energy relationships: a general model for solvation effects on apolar binding. J Am Chem Soc. 1990;112:339–343. [Google Scholar]
- 6.Kearney PC, Mizoue LS, Kumpf RA, Forman JA, McCurdy A, Dougherty DA. Molecular recognition in aqueous media. New binding studies provide further insights into the cation-pi interaction and related phenomena. J Am Chem Soc. 1993;115:9907–9919. [Google Scholar]
- 7.Schneider HJ. Interactions in Supramolecular Complexes Involving Arenes: Experimental Studies. Acc Chem Res. 2013;46:1010–1019. doi: 10.1021/ar3000579. [DOI] [PubMed] [Google Scholar]
- 8.Dougherty DA, Stauffer DA. Acetylcholine Choline Binding by a Synthetic Receptor:Implications for Biological Recognition. Science. 1990;250:1558–1560. doi: 10.1126/science.2274786. [DOI] [PubMed] [Google Scholar]
- 9.Pittelkow M, Tschapek B, Smits SHJ, Schmitt L, Bremer E. The Crystal Structure of the Substrate-Binding Protein OpuBC from Bacillus subtilis in Complex with Choline. J Mol Biol. 2011;411:53–67. doi: 10.1016/j.jmb.2011.05.037. [DOI] [PubMed] [Google Scholar]
- 10.Cheng J, Goldstein R, Gershenson A, Stec B, Roberts MF. The Cation-π Box Is a Specific Phosphatidylcholine Membrane Targeting Motif. J Biol Chem. 2013;288:14863–14873. doi: 10.1074/jbc.M113.466532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobs SA, Khorasanizadeh S. Structure of HP1 Chromodomain Bound to a Lysine 9-Methylated Histone H3 Tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 12.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim YC, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 14.Grimm C, Matos R, Ly-Hartig N, Steuerwald U, Lindner D, Rybin V, Muller J, Muller CW. Molecular recognition of histone lysine methylation by the polycomb group repressor dSfmbt. Embo J. 2009;28:1965–1977. doi: 10.1038/emboj.2009.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li HT, Ilin S, Wang WK, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palacios A, Munoz IG, Pantoja-Uceda D, Marcaida MJ, Torres D, Martin-Garcia JM, Luque I, Montoya G, Blanco FJ. Molecular basis of histone H3K4me3 recognition by ING4. J Biol Chem. 2008;283:15956–15964. doi: 10.1074/jbc.M710020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santiveri CM, Lechtenberg BC, Allen MD, Sathyamurthy A, Jaulent AM, Freund SMV, Bycroft M. The malignant brain tumor repeats of human SCML2 bind to peptides containing monomethylated lysine. J Mol Biol. 2008;382:1107–1112. doi: 10.1016/j.jmb.2008.07.081. [DOI] [PubMed] [Google Scholar]
- 18.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 19.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schalch T, Job G, Noffsinger VJ, Shanker S, Kuscu C, Joshua-Tor L, Partridge JF. High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol Cell. 2009;34:36–46. doi: 10.1016/j.molcel.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hung T, Binda O, Champagne KS, Kuo AJ, Johnson K, Chang HY, Simon MD, Kutateladze TG, Gozani O. ING4 mediates crosstalk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol Cell. 2009;33:248–256. doi: 10.1016/j.molcel.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pena PV, Davrazou F, Shi XB, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. Embo Reports. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy S, Musselman CA, Kachirskaia I, Hayashi R, Glass KC, Nix JC, Gozani O, Appella E, Kutateladze TG. Structural insight into p53 recognition by the 53BP1 tandem tudor domain. J Mol Biol. 2010;398:489–496. doi: 10.1016/j.jmb.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trojer P, Li G, Sims RJ, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, Wang YH, Reinberg D. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell. 2007;129:915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 26.Pederson MT, Helin K. Histone Demethylases in Development and Disease. Trends Cell Biol. 2010;20:662–671. doi: 10.1016/j.tcb.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, Cole PA, Yu H. Structural Basis for the Inhibition of the LSD1 Histone Demethylase by the Antidepressant trans-2-Phenylcyclopropylamine. Biochemistry. 2007;46:8058–8065. doi: 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- 28.Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat Struct Mol Biol. 2007;14:689–695. doi: 10.1038/nsmb1273. [DOI] [PubMed] [Google Scholar]
- 29.Albert M, Helin K. Histone methyltransferases in cancer. Semin Cell Dev Biol. 2010;21:209–220. doi: 10.1016/j.semcdb.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Fischle W, Wang W, Duncan EM, Liang L, Murakami-Ishibe S, Allis CD, Patel DJ. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol Cell. 2007;28:677–691. doi: 10.1016/j.molcel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grimm C, Matos R, Ly-Hartig N, Steuerwald U, Lindner D, Rybin V, Muller J, Muller CW. Molecular recognition of histone lysine methylation by the polycomb group repressor dSfmbt. Embo J. 2009;28:1965–1977. doi: 10.1038/emboj.2009.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCurdy A, Jimenez L, Stauffer DA, Dougherty SA. Biomimetic Catalysis of SN2 Reactions through Cation-π Interactions. The Role of Polarizability in Catalysis. J Am Chem Soc. 1992;114:10314–10321. [Google Scholar]
- 33.Whiting AL, Neufiled NM, Hof F. A tryptophan-analog host whose interactions with ammonium ions in water are dominated by the hydrophobic effect. Tetrahedron Lett. 2009;50:7035–7037. [Google Scholar]
- 34.Hughes RM, Waters ML. Influence of N-Methylation on Cation-π Interaction Produces a Remarkably Stable β-Hairpin Peptide. J Am Chem Soc. 2005;127:6518–6519. doi: 10.1021/ja0507259. [DOI] [PubMed] [Google Scholar]
- 35.Reimen AJ, Waters ML. Design of Highly Stabilized β-Hairpin Peptides through Cation-π Interactions of Lysine and N-methyllysine with an Aromatic Pocket. Biochemistry. 2009;48:1525–1531. doi: 10.1021/bi801706k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hughes RM, Benshoff ML, Waters ML. Effects of Chain Length and N-methylation on a Cation-π Interaction in a β-Hairpin Peptides. Chem Eur J. 2007;13:5753–5764. doi: 10.1002/chem.200601753. [DOI] [PubMed] [Google Scholar]
- 37.Salonen LM, Bucher C, Banner DW, Haap W, Mary J, Benz J, Kuster O, Seiler P, Schweizer WB, Diederich F. Cation–π Interactions at the Active Site of Factor Xa: Dramatic Enhancement upon Stepwise N-Alkylation of Ammonium Ions. Ang Chem Int Ed. 2009;48:811–814. doi: 10.1002/anie.200804695. [DOI] [PubMed] [Google Scholar]
- 38.Daze KD, Hof F. The Cation-π Interaction at Protein-Protein Interaction Interfaces: Developing and Learning from Synthetic Mimics of Proteins that Bind Methylated Lysines. Acc Chem Res. 2013;46:937–945. doi: 10.1021/ar300072g. [DOI] [PubMed] [Google Scholar]
- 39.Beshara CS, Jones CE, Daze KD, Lilgert BJ, Hof F. A Simple Calixarene Recognizes Post-translationally Methylated Lysine. ChemBioChem. 2010;11:63–66. doi: 10.1002/cbic.200900633. [DOI] [PubMed] [Google Scholar]
- 40.Ingerman LA, Cuellar ME, Waters ML. A small molecule receptor that selectively recognizes trimethyl lysine in a histone peptide with native protein-like affinity. Chem Comm. 2010;46:1839–1841. doi: 10.1039/c000255k. [DOI] [PubMed] [Google Scholar]
- 41.Caldwell JW, Kollman PA. Cation-n Interactions: Nonadditive Effects Are Critical in Their Accurate Representation. J Am Chem Soc. 1995;117:4177–4178. [Google Scholar]
- 42.Grauffel C, Stote RH, Dejaegere A. Force field parameters for the simulation of modified histone tails. J Comput Chem. 2010;31:2434–2451. doi: 10.1002/jcc.21536. [DOI] [PubMed] [Google Scholar]
- 43.Schneebeli ST, Bochevarov AD, Arteum D, Friesner RA. Parameterization of a B3LYP Specific Correction for Noncovalent Interactions and Basis Set Superposition Error on a Gigantic Data Set of CCSD(T) Quality Noncovalent Interaction Energies. J Chem Theory Comput. 2011;7:658–668. doi: 10.1021/ct100651f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du QS, Ling SY, Meng JZ, Huang RB. Empirical Formulation and Parameterization of Cation-pi Interactions for Protein Modeling. J Comp Chem. 2011;33:153–162. doi: 10.1002/jcc.21951. [DOI] [PubMed] [Google Scholar]
- 45.Orabi EA, Lamoureux G. Cation-π and π-π Interactions in Aqueous Solution Studied Using Polarizable Potential Models. J Chem Theory Comput. 2012;8:182–193. doi: 10.1021/ct200569x. [DOI] [PubMed] [Google Scholar]
- 46.Ansorg K, Tafipolsky M, Engels B. Cation-π Interactions: Accurate Intermolecular Potential from Symmetry-Adapted Perturbation Theory. J Phys Chem B. 2013;117:10093–10102. doi: 10.1021/jp403578r. [DOI] [PubMed] [Google Scholar]
- 47.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Databank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crowley PB, Golovin A. Cation–π Interactions in Protein–Protein Interfaces. Structure, Function, and Bioinformatics. 2005;59:231–239. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- 49.Marshall MS, Steele RP, Thanthiriwatte KS, Sherrill CD. Potential energy curves for cation-pi interactions: Off-axis configurations are also attractive. J Phys Chem A. 2009;113:13628–13632. doi: 10.1021/jp906086x. [DOI] [PubMed] [Google Scholar]
- 50.Maestro. version 9.2 Schrödinger, LLC, New York, NY: 2011. [Google Scholar]
- 51.Jaguar . version 9.2 Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 52.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc. 2008;120:215–241. [Google Scholar]
- 53.Boys SF, Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys. 1970;19:553–566. [Google Scholar]
- 54.Cheng J, Goldstein R, Gershenson A, Stec B, Roberts MF. The Cation-π Box Is a Specific Phosphatidylcholine Membrane Targeting Motif. J Biol Chem. 2013;288:14863–14873. doi: 10.1074/jbc.M113.466532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meot-Ner M, Deakyne CA. Unconventional ionic hydrogen bonds. 1. CHδ+ …delta Complexes of quaternary ions with n- and π-donors. J Am Chem Soc. 1985;107:469–474. [Google Scholar]
- 56.Rezac J, Hozba P. Describing Noncovalent Interactions beyond the Common Approximations: How Accurate Is the “Gold Standard,” CCSD(T) at the Complete Basis Set Limit? J Chem Theory Comput. 2013;9:2151–2155. doi: 10.1021/ct400057w. [DOI] [PubMed] [Google Scholar]
- 57.Wireduaah S, Parker TM, Lewis M. Effects of the aromatic substitution pattern in cation-π sandwich complexes. J Phys Chem. 2013;117:2598–2604. doi: 10.1021/jp309740r. [DOI] [PubMed] [Google Scholar]
- 58.Berg L, Andersson CD, Artursson E, Hornberg A, Tunemalm AK, Linusson A, Ekstrom F. Targeting Acetylcholinesterase: Identification of Chemical Leads by High Throughput Screening, Structure Determination and Molecular Modeling. PLoS ONE. 2011;6:e26039. doi: 10.1371/journal.pone.0026039. doi:10.1371/journal.pone.0026039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grauffel C, Yang B, He T, Roberts MF, Gershenson A, Reuter N. Cation–π Interactions As Lipid-Specific Anchors for Phosphatidylinositol-Specific Phospholipase C. J Am Chem Soc. 2013;135:5740–5750. doi: 10.1021/ja312656v. [DOI] [PMC free article] [PubMed] [Google Scholar]