

Abstract
Valosin containing protein (VCP) disease (also known as Inclusion Body Myopathy, Paget Disease of Bone and Frontotemporal Dementia [IBMPFD] syndrome) is caused by mutations in the gene encoding VCP classically affecting the muscle, bone and brain. Although the genetic cause has been identified, details regarding the pathogenesis of IBMPFD have not been fully determined. Muscle wasting observed in VCP disease is suggestive of cytokine imbalance. We hypothesized that dysfunctional protein homeostasis caused by VCP mutations leads to cytokine imbalances thereby contributing to the muscle wasting phenotype. Circulating levels of interleukin‐4 (IL‐4), interleukin‐6 (IL‐6), tumor necrosis factor alpha (TNF a) and epidermal growth factor (EGF) were measured in plasma of patients with VCP disease or controls. TNF a and EGF were significantly altered in VCP disease as compared to control. TNF a was up‐regulated, consistent with a cachexia phenotype and EGF levels were increased. No significant differences were observed in IL‐4 and IL‐6. Cytokine imbalances may be associated with VCP disease and may play a contributory role in VCP myopathy. Further understanding of how VCP dysfunction leads to aberrant protein homeostasis and subsequent cytokine imbalances may also aid in the understanding of other proteinopathies and in the development of novel treatments.
Keywords: IBMPFD, cytokines, VCP
Introduction
Muscle wasting is found in a multitude of disease states in reaction to a variety of insults. Muscle atrophy can be secondary to disuse, nutritional deficiencies, neuropathy, vasculopathy, cancer, HIV, and other inflammatory diseases. Inflammatory states occur as a result of cytokine release and subsequent activation of the immune response. If levels of pro‐inflammatory cytokines are elevated for prolonged periods, muscle wasting can result. The importance of both pro‐ and anti‐inflammatory cytokine balances in maintaining skeletal muscle health is increasingly recognized. Cytokine factors such as interleukin‐6 (IL‐6) and tumor necrosis factor alpha (TNFα) have long been implicated in muscle wasting.1, 2 In fact, TNFα was once known as cachectin given its direct effect in muscle atrophy.3 Evidence reported by Horsely et al.,4 suggests interleukin‐4 (IL‐4) plays a role in myoblast fusion and myogenesis and epidermal growth factor (EGF) has been a key component in most commercially available myoblast media since 19925 although its specific role in myogenesis is unclear. Regarding inflammatory pathways, it is especially enticing to consider the role of inflammation in Valosin containing protein (VCP) disease given the progressive muscle cachexia.
VCP disease, also known as Inclusion Body Myopathy, Paget's Disease of Bone and Frontotemporal Dementia (IBMPFD), is a life shortening, crippling disease of muscle, bone and brain.6 It is an autosomal dominant, adult onset disease caused by mutations in the VCP gene.6 VCP is an ATP driven shuttling/chaperone protein that normally participates in protein homeostasis through its interaction with degradative pathways including the ubiquitin‐proteasome and autophagy pathways.7, 8, 9 VCP plays a role in numerous cellular processes such as cell cycle progression, chromatin remodeling, homotypic membrane fusion (in both the endoplasmic reticulum [ER] and golgi apparatus) and mitochondrial turnover.10, 11 Of those affected with VCP disease, 92% have myopathy, 50% have Paget's disease of the bone, and 30% have frontotemporal dementia. Although VCP is expressed in almost every cell, its dysfunction in muscle is the most destructive and life shortening. Myogenic tissues either fail to regenerate after injury or degenerate prematurely causing a muscle wasting phenotype.
How mutations in VCP lead to poor regeneration/degeneration and the subsequent muscle wasting phenotype is unclear. VCP appears to directly interact with proteins involved in catabolic pathways and thereby affect protein fate. In fact, VCP affects many protein pathways that ultimately influence cell fate and tissue health. The footprint of VCP's effect on cytokine signaling was first described by Asai et al.,12 when VCP expression was shown to affect nuclear factor kappa‐B activity (NFkB, a downstream target of TNFα). In this report, they induced VCP expression and then observed an increase in the activation of NFκB.12
Clinically, VCP myopathy appears similar to inflammatory‐mediated wasting diseases as described in cachexia. Although VCP disease clinically resembles a chronic inflammatory syndrome, there is no inflammatory cell infiltrate and patient plasma shows a normal erythrocyte sedimentation rate (ESR, a global marker of inflammation). It is important to note, however, that ESR is not a 100% sensitive for inflammatory conditions. For example, asthmatic patients can have a waxing and waning chronic inflammation that does not necessarily show an elevated ESR in patient plasma. Because the clinical picture implicating inflammation was so compelling, we decided to investigate the cytokine profile of patient plasma.
Our hypothesis is that the dysfunctional protein homeostasis in VCP disease causes a perturbation of cytokine signals that are classically associated with impaired muscle regeneration and muscle wasting. We predict patient plasma will show a cytokine profile enriched for factors associated with muscle cachexia (TNFα and IL‐6) and poor in factors that promote muscle regeneration (EGF and IL‐4).
Methods
Clinical evaluation and consenting of patients
All subjects were consented and approved for the study in accordance with the institutional review board's consent procedure at the University of California, Irvine. Clinical evaluations were conducted as part of the natural history of IBMPFD. And 16 patients with IBMPFD, 2 presymptomatic mutation carriers and 16 controls participated in this study. Patients with a probable clinical diagnosis of VCP disease had full VCP gene sequencing to confirm the diagnosis. Cascade testing of family members was completed with the specific proband mutation. Controls were age‐matched and gender‐matched, were not taking medications and were free of inflammatory conditions. Patients with a probable clinical diagnosis of VCP disease had sequential testing for the specific VCP familial mutation or gene sequencing for hot spots followed by complete gene sequencing to confirm the diagnosis. Confirmatory CLIA testing of the VCP gene mutations was performed by MitoMed Laboratory (University of California‐Irvine). Thirty‐one different point mutations have been found in patients with VCP disease.13
ELISA cytokine quantification
Blood samples were collected in heparin tubes and spun down at 3,000g × 15 minutes to separate plasma. Plasma was isolated, aliquoted and frozen until the day of the assay. Plasma cytokine levels were evaluated using commercially available Enzyme‐Linked Immunosorbent Assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) and included interleukin‐6 (IL‐6: intra‐assay and interassay CV<10%, sensitivity <7 pg/mL), interleukin‐4 (IL‐4: intra‐assay and interassay CV < 10%, sensitivity < 0.22 pg/mL, tumor necrosis factor a (TNFα: intra‐assay and interassay CV < 10%, sensitivity < 0.19 pg/mL and EGF (intra‐assay and interassay CV < 10% sensitivity < 0.7 pg/mL). Out of the patient samples, three TNFα, two EGF samples and four IL‐4 samples were excluded. Four IL‐4 control samples were excluded. These samples were excluded because they contained an unknown precipitant that interfered with the accuracy of our assays. The precipitant could have been from poor buffy coat separation. Despite several attempts, we were unable to get rid of this unknown precipitant without losing a significant volume of the samples. The “n” used for the statistical analysis for each group is provided in Table 1.
Table 1.
Patient plasma samples analyzed per cytokine versus control groups
| Cytokine | Disease group | Control group |
|---|---|---|
| Interleukin‐6 | 16 | 16 |
| Interleukin‐4 | 11 | 12 |
| TNFα | 13 | 16 |
| EGF | 14 | 16 |
Statistical analysis
Analysis of covariance (ANCOVA) was employed to evaluate the difference between affected and unaffected subjects while adjusting for gender and age. Outliers as determined by each assay were excluded for analysis and log‐transformation was only applied to IL‐6 and EGF in order to satisfy the normality assumption of residuals. Data were presented with mean and standard deviation at nontransformed scale (pg/mL). Significance level was set at 0.05 level and all tests were performed using SAS 9.2 (Cary, NC, USA).
Results
The individual observation and box‐and‐whisker plots of cytokine data for both VCP disease and control subjects is shown in Figure 1. Circulating levels of cytokines (TNFα and EGF) in the VCP disease group versus healthy control groups were significantly different. After adjusting for the effect of age and gender, the ANCOVA results revealed that both TNFα (VCP 2.1 ± 0.9 vs. control 1.3 ± 0.7 pg/mL, p = 0.024) and EGF (VCP 31.2 ± 40.1 vs. control 8.4 ± 15.5 pg/mL, p = 0.0065) were significantly elevated in VCP disease as compared to control; and neither IL‐6 (VCP 1.9 ± 1.3 vs. control 1.2 ± 1.0 pg/mL, p = 0.06) nor IL‐4 (VCP 0.25 ± 0.16 vs. control 0.50 ± 0.34 pg/mL, p = 0.06) showed a statistically significant difference (Figure 1). We did not find any significant elevations in the levels in those individuals with myopathy who had Paget disease versus those who did not.
Figure 1.
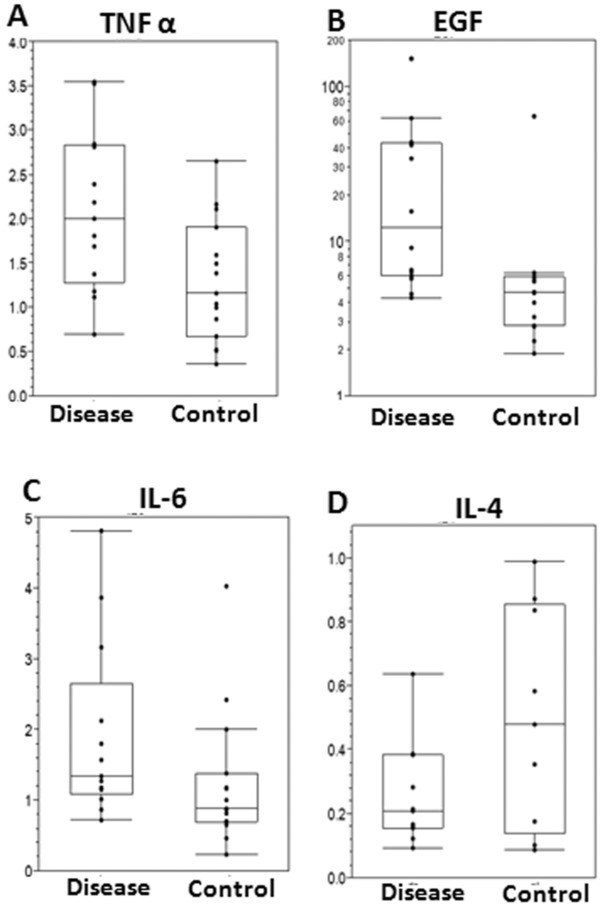
Box‐and‐whisker plot of cytokine data for VCP disease and control subjects with individual data (dot) and box‐and‐whisker plots. The box represents the quartile (Q1), then median and third quartile (Q3), and the whiskers extend to the largest value below Q3 + 1.5*(Q3 − Q1) and to the smallest value above Q1 − 1.5*(Q3 − Q1).
Discussion
The hypothesis that dysfunctional protein homeostasis in VCP disease causes a perturbation of cytokine levels in patient plasma was confirmed. This paper is the first to show a correlation between individuals with VCP mutations and elevated TNFα and EGF in patient plasma. TNFα is a pro‐inflammatory, myogenically important cytokine. Excess TNFα affects myogenesis and can lead to muscle atrophy.14 Elevated TNFα in VCP disease is consistent with the myopathic, muscle wasting clinical phenotype.
Although downstream targets of TNFα in VCP myopathy were not investigated in this study, it would be expected that TNFα's downstream targets would correlate with changes in VCP function and expression. One downstream target of TNFα, NFκB, has been correlated with changes in VCP function. Additionally, mutations in the intermediate pathway of NFkB have been implicated in Paget disease of bone. Custer et al.,15 reported that NFkB activity is up‐regulated in C2C12 myoblast cell lines transfected with VCP mutations known to cause human disease. Asai et al.,12 showed that increased native VCP expression corresponded to increased activation of NFkB. Additionally in 2012, Liu et al.,2 showed that VCP inhibition leads to a decrease in NFkB activity through increased levels of I‐kappa‐B‐alpha the inhibitor for NFkB.
While a high level of TNFα in patient plasma is associated with VCP mutations, neither the cell source of TNFα nor the stimulus for its production is understood. The source of excess TNFα does not appear to be cell infiltrate. Myopathic tissue from patient's muscle biopsies do not show inflammatory cell infiltrate. Instead, VCP disease pathology appears to be consistent with a type of sterile inflammation. This is not the first time cytokine levels have not correlated with cell infiltrate. A 1996 study16 investigating inflammatory myositis found no correlation between the level of inflammatory infiltrates and TNFα expression. To that point, several investigators have now found convincing evidence that cytokines can be produced directly from myocytes, so called myokines.16 It is, therefore, possible that cytokine changes seen in patient plasma are not from inflammatory cells, but rather expression from diseased muscle tissue.
Regardless of the source of cytokines, the specific role they are playing in VCP disease muscle atrophy remains to be elucidated. Cytokines such as TNFα are known to affect cell fate decisions such as apoptosis, proliferation and differentiation.17 In vitro cell fusion indices suggest impaired differentiation during myogenesis in VCP disease. Although speculative, these cytokine imbalances (increased TNFα and EGF) may alter the process of myoblast proliferation and differentiation.
EGF is expressed in skeletal and smooth muscle tissue and when bound to its receptor, EGFR, is involved in growth, proliferation and differentiation.18 EGF has been found to stimulate proliferation of porcine myogenic satellite cells.19 EGF is beneficial in maintaining cultured human muscle cells.5 A member of the EGF family, HB‐EGF (identical to EGF except for an additional amino terminal extension) has been implicated as a survival factor for differentiating myoblasts.20 While EGF is elevated in patient plasma, EGF‐receptor expression was found to be down regulated on microarray analysis from VCP patient muscle biopsies.21
In general, it is known that growth factor starved myoblasts exit the cell cycle and become terminally differentiated myotubes.6 One possible consequence of increased EGF levels is the maintenance of myoblasts in an undifferentiated state thus impairing differentiation to functional myotubes. This possibility fits with our unpublished in vivo results, but it is difficult to predict what the overall effect of increased plasma EGF is, given EGF‐receptor down regulation in muscle tissue. The role of EGF and its receptor in myogenesis will be one focus of future research.
Studies researching the role of IL‐6 in myogenesis and muscle cachexia have indicated that exposure time is important in determining the ultimate effects on tissue. Acute elevations of IL‐6 are required for proper myogenesis,22 but chronic exposure to IL‐6 leads to muscle atrophy.1 Therefore, the timing of blood collection and additional sampling that takes into account the onset of VCP disease may help to determine if IL‐6 is truly altered in this disease since some statistical trends towards significance were observed when taking into account age and gender.
IL‐4 is secreted by muscle and IL‐4 alpha receptors are also expressed in skeletal muscle tissue. Evidence from myoblast fusion experiments in mice conducted by Horsley et al.,4 suggests that IL‐4 is important in increasing myotube size after the initial myotube is formed via aiding myoblast fusion. Studies in human tissue before and after strength training show increased expression of both IL‐4 and IL‐4 receptor alpha.23 Our results did not show IL‐4 to be clinically significant (p = 0.06), which may mean that it is simply not involved in the pathogenesis, or that our study is underpowered for this particular cytokine.
VCP disease is a rare disease and getting enough clinical samples remains a challenge. Additional study limitations include coordination of clinical samples with the wide range of severity and onset of disease. Some samples were collected before severe symptoms had begun, while others were collected at the most severe end point of disease. Correlations between cytokine levels and specific points in the pathogenesis of this dynamic, progressive disease process are difficult and therefore, only observations of general trends in cytokine levels during disease pathogenesis were possible. Follow‐up studies will require more long‐term sample collections and storage to more precisely compare specific points in disease pathogenesis. Future directions will investigate cytokine altering therapies and their effects on VCP disease myogenesis using validated cell models. Lastly, because VCP gene mutations have recently been associated with other more common proteinopathies such as Amyotrophic Lateral Sclerosis (ALS) and Parkinson's disease,24, 25 it was not surprising to find that altered cytokines were found in these disorders, suggesting that similar pathological mechanisms may be contributing to the degenerative course in these disorders.21, 26, 27
Conclusion
VCP disease is associated with perturbed TNFα and EGF cytokine levels. Although IL‐4 and IL‐6 trended toward significance, no significant differences were observed in these two cytokines. Further research is required to better understand the role of cytokines in muscle regeneration/degeneration and the specific molecular pathophysiology of VCP mutations in altering cytokine levels.
Conflict of Interest
The authors have no personal financial or institutional interest or conflicts in any of the outcomes or materials described in this paper.
Acknowledgments
We thank the families and their health care providers for their enthusiastic participation and contribution in our research studies. Funding of this study was from the California Institute for Regenerative Medicine at the University of California, Irvine (TG2‐01152). NIAMS, National Institutes of Health (R03 AR 46869, RO1 AR050236), Muscular Dystrophy Association, Paget Foundation and the Institute for Clinical and Translational Science (ICTS), University of California, Irvine.
References
- 1. Haddad F, Zaldivar F, Cooper DM, Adams GR. IL‐6‐induced skeletal muscle atrophy. J Appl Physiol. 2005; 98: 911–917. [DOI] [PubMed] [Google Scholar]
- 2. Liu Y, Hei Y, Shu Q, Dong J, Gao Y, Fu H, Zheng X, Yang G. VCP/p97, down‐regulated by microRNA‐129–5p, could regulate the progression of hepatocellular carcinoma. PloS One. 2012; 7(4): e35800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kawakami M, Pekala PH, Lane MD, Cerami A. Lipoprotein lipase suppression in 3T3‐L1 cells by an endotoxin‐induced mediator from exudate cells. Proc Natl Acad Sci USA. 1982; 79: 912–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Horsley V, Jansen KM, Mills ST, Pavlath GK. IL‐4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell. 2003; 113: 483–494. [DOI] [PubMed] [Google Scholar]
- 5. St Clair JA, Meyer‐Demarest SD, Ham RG. Improved medium with EGF and BSA for differentiated human skeletal muscle cells. Muscle Nerve. 1992; 15: 774–749. [DOI] [PubMed] [Google Scholar]
- 6. Vesa J, Su H, Watts GD, Krause S, Walter MC, Martin B, Smith C, Wallace DC, Kimonis VE. Valosin containing protein associated inclusion body myopathy: abnormal vacuolization, autophagy and cell fusion in myoblasts. Neuromuscul Disord. 2009; 19: 766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ju JS, Weihl CC. p97/VCP at the intersection of the autophagy and the ubiquitin proteasome system. Autophagy. 2010; 6: 283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Isakov E, Stanhill A. Stalled proteasomes are directly relieved by P97 recruitment. J Biol Chem. 2011; 286: 30274–30283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ikeda Y, Demartino GN, Brown MS, Lee JN, Goldstein JL, Ye J. Regulated endoplasmic reticulum‐associated degradation of a polytopic protein: p97 recruits proteasomes to Insig‐1 before extraction from membranes. J Biol Chem. 2009; 284: 34889–34900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA‐ATPase in the ubiquitin system. Nat Cell Biol. 2012; 14: 117–123. [DOI] [PubMed] [Google Scholar]
- 11. Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, Milligan RA, Wilson‐Kubalek EM. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat Struct Biol. 2002; 9: 950–957. [DOI] [PubMed] [Google Scholar]
- 12. Asai T, Tomita Y, Nakatsuka S, Hoshida Y, Myoui A, Yoshikawa H, Aozasa K. VCP (p97) regulates NFkappaB signaling pathway, which is important for metastasis of osteosarcoma cell line. Jpn J Cancer Res: Gann. 2002; 93: 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mehta S, Khare M, Ramani R, Watts GD, Simon M, Osann KE, Donkervoort S, Dec E, Nalbandian A, Platt J, et al. Genotype‐phenotype studies of VCP‐associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin Genet. 2013; 83:422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miller SC, Ito H, Blau HM, Torti FM. Tumor necrosis factor inhibits human myogenesis in vitro. Mol Cell Biol. 1988; 8: 2295–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Custer SK, Neumann M, Lu H, Wright AC, Taylor JP. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet. 2010; 19: 1741–1755. [DOI] [PubMed] [Google Scholar]
- 16. Tews DS, Goebel HH. Cytokine expression profile in idiopathic inflammatory myopathies. J Neuropathol Exp Neurol. 1996; 55: 342–347. [DOI] [PubMed] [Google Scholar]
- 17. Jonuleit H, Knop J, Enk AH. Cytokines and their effects on maturation, differentiation and migration of dendritic cells. Arch Dermatol Res. 1996; 289: 1–8. [DOI] [PubMed] [Google Scholar]
- 18. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol, Biol, Phys. 2004; 59: 21–26. [DOI] [PubMed] [Google Scholar]
- 19. Doumit ME, Cook DR, Merkel RA. Fibroblast growth factor, epidermal growth factor, insulin‐like growth factors, and platelet‐derived growth factor‐BB stimulate proliferation of clonally derived porcine myogenic satellite cells. J Cell Physiol. 1993; 157: 326–332. [DOI] [PubMed] [Google Scholar]
- 20. Horikawa M, Higashiyama S, Nomura S, Kitamura Y, Ishikawa M, Taniguchi N. Upregulation of endogenous heparin‐binding EGF‐like growth factor and its role as a survival factor in skeletal myotubes. FEBS Lett. 1999; 459: 100–104. [DOI] [PubMed] [Google Scholar]
- 21. Nalbandian A, Ghimbovschi S, Radom‐Aizik S, Dec E, Vesa J, Martin B, Knoblach S, Smith C, Hoffman E, Kimonis VE. Global gene profiling of VCP‐associated inclusion body myopathy. Clin Transl Sci. 2012; 5: 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baeza‐Raja B, Munoz‐Canoves P. p38 MAPK‐induced nuclear factor‐kappaB activity is required for skeletal muscle differentiation: role of interleukin‐6. Mol Biol Cell 2004; 15: 2013–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prokopchuk O, Liu Y, Wang L, Wirth K, Schmidtbleicher D, Steinacker JM. Skeletal muscle IL‐4, IL‐4Ralpha, IL‐13 and IL‐13Ralpha1 expression and response to strength training. Exerc Immunol Rev. 2007; 13: 67–75. [PubMed] [Google Scholar]
- 24. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010; 68: 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chan N, Le C, Shieh P, Mozaffar T, Khare M, Bronstein J, Kimonis V. Valosin‐containing protein mutation and Parkinson's disease. Parkinsonism Relat Disord. 2012; 18: 107–109. [DOI] [PubMed] [Google Scholar]
- 26. Yin HZ, Hsu CI, Yu S, Rao SD, Sorkin LS, Weiss JH. TNF‐alpha triggers rapid membrane insertion of Ca(2+) permeable AMPA receptors into adult motor neurons and enhances their susceptibility to slow excitotoxic injury. Exp Neurol. 2012; 238: 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor‐alpha (TNF‐alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994; 165: 208–210. [DOI] [PubMed] [Google Scholar]