

Abstract
Oncogene–induced DNA damage elicits genomic instability in epithelial cancer cells, but apoptosis is blocked through inactivation of the tumor suppressor p53. In hematological cancers, the relevance of ongoing DNA damage and mechanisms by which apoptosis is suppressed are largely unknown. We found pervasive DNA damage in hematologic malignancies including multiple myeloma, lymphoma and leukemia, which leads to activation of a p53–independent, pro-apoptotic network centered on nuclear relocalization of ABL1 kinase. Although nuclear ABL1 triggers cell death through its interaction with the Hippo pathway co–activator YAP1 in normal cells, we show that low YAP1 levels prevent nuclear ABL1–induced apoptosis in these hematologic malignancies. YAP1 is under the control of a serine–threonine kinase, STK4. Importantly, genetic inactivation of STK4 restores YAP1 levels, triggering cell death in vitro and in vivo. Our data therefore identify a novel synthetic–lethal strategy to selectively target cancer cells presenting with endogenous DNA damage and low YAP1 levels.
Keywords: Hippo pathway, YAP1, leukemia, lymphoma, multiple myeloma, ABL1, STK4, synthetic lethality
Introduction
In epithelial cancers, rampant DNA double strand break (DSB) formation followed by activation of DNA damage response (DDR) occurs in both premalignant and malignant conditions. However, in precancerous settings senescence and apoptotic responses, the so-called ‘tumorigenesis barrier’, prevent progression to malignancy1 until the tumor suppressor TP53 (p53) is inactivated, thereby triggering genomic instability and enhancing tumor cell growth.
Variable levels of γ-H2A.X and pATM have been reported in AML, myelodysplastic syndromes2 and in multiple myeloma (MM)3. However, in blood tumors, the presence and the functional role of ongoing DNA damage in carcinogenesis have not been explored in detail. Additionally, inactivation of p53 does not appear to represent a pivotal event in the evolution from premalignancy toward cancer in these diseases, since p53 mutations are relatively rare and appear late during the course of these tumors. For example, p53 mutations and deletions are rare in newly diagnosed MM patients (5–10%), and present in only 10–20% of patients with relapsed and refractory MM4, while losses of ATM and ATR are even rarer5.
An alternative pathway to p53, downstream of ATM/ATR has been described, which is activated after DSB and induces apoptosis6-10. It is centered on the proto-oncogene ABL1, commonly translocated in chronic myeloid leukemia. Treatment of tumor cell lines with DNA damaging agents results in ABL1 relocalization from the cytoplasm to the nucleus, where it elicits apoptosis. Importantly, the nuclear shuttling of ABL1 has been demonstrated only in vitro in tumor cell lines after drug-induced DNA damage; therefore, its functional and in vivo clinical relevance remains unknown.
Here we elucidate a novel synthetic lethal approach where genetic inhibition of serine– threonine kinase 4 (STK4) reactivates the Hippo mediator YAP1, thereby triggering apoptosis in hematologic malignancies with intrinsic DNA damage. Our data provide the rationale for the development and clinical evaluation of novel STK4 inhibitors.
Results
MM cells evade apoptosis despite pervasive DNA damage
We first explored a panel of MM cell lines and MM cells to confirm the presence of widespread DNA damage3. Eleven of 13 MM cell lines and cells derived from subjects with MM demonstrated increased γ-H2A.X staining (Fig. 1a,b and Supplementary Fig. 1a,b) and an activated DNA damage response (DDR) (Fig. 1c and data not shown). This pattern was not present in normal plasma cells or in peripheral blood mononuclear cells (PBMCs) derived from healthy individuals3 (Fig. 1a–c), mirroring reports in other cellular contexts where the presence of DNA damage discriminates normal tissues from pre–neoplastic and cancerous lesions11,12. Notably, U266 and KMS–34 MM cell lines, which did not show γ-H2A.X foci, were also negative for all markers of DDR activation (Fig. 1c).
Fig. 1. MM cells present ongoing DNA damage, driving ABL1 inside the nucleus.

(a) Immunofluorescence staining of γ-H2A.X in MM cell lines, PBMCs and MM cells. (b) Western blot analysis of γ-H2A.X in MM cell lines, PBMCs and MM cells.
(c) Western blot analysis of p–ATM(Ser1981), p–CHK2(Thr68), p–ATR(Ser428), and p– CHK1(Ser296) and their corresponding un–modified forms in MM cell lines and samples from subjects with MM.
(d) Immunofluorescence staining for γ-H2A.X–Alexa Fluor 568, cleaved caspase 3–Alexa 488, and DAPI in H929 cells. As positive control, MM cell lines and MM cells were treated with 5 nM Bortezomib (Btz) for 24 h.
(e) Subcellular fractionation of p53–WT and p53–mutant MM cell lines. Cell lysates from cytoplasmic (C) and nuclear (N) fractions were analyzed by western blot for ABL1 expression. α-tubulin and Histone H3 were used as loading controls for C and N fractions, respectively.
(f) Immunohistochemical ABL1 staining on one representative sample derived from an individual affected by MM (MM–004). Left panel, 400x. Right panel, 1000x magnification.
Surprisingly, despite this pervasive DNA damage and DDR activation, we did not detect any significant cell death under basal conditions (Fig. 1d and Supplementary Fig. 1c,d), implying that MM cells have mechanisms to escape the apoptotic response triggered in normal cells.
ABL1 relocalization in the nucleus of MM cells
As mentioned above, p53 genetic inactivation appears to be less relevant in hematopoietic neoplasms including MM than in epithelial cancers4. Indeed, modifications associated with DNA damage were present at a comparable level in both p53–wild type and – mutated MM cell lines (Fig. 1c and data not shown). A second pathway involved in the apoptotic response after DSBs involves nuclear re–localization of ABL1 upon DNA damage7-10. We therefore asked whether ABL1 is localized in the nucleus in MM. Strikingly, ABL1 demonstrated a prominent and preferential localization inside the nucleus in most MM cells, regardless of their p53 mutational status (Fig. 1e), in contrast to HeLa cells, in which ABL1 shuttles inside the nucleus only after doxorubicin treatment (Supplementary Fig.2a). Immunohistochemical staining also confirmed prominent nuclear ABL1 localization in cells derived from individuals with MM (Fig. 1f and Supplementary Fig. 2b).
Following DNA damage, ATM is phosphorylated13. As a result, activated JNK phosphorylates 14–3–3 proteins leading to ABL1 release, which in turn can shuttle inside the nucleus14. Indeed treatment of MM cell lines with an ATM inhibitor, Ku5593315 or JNK1 inhibitor, SP60012516 reduced nuclear and increased cytoplasmic ABL1 (Fig. 2a,b and Supplementary Fig. 3a,b) in both p53–WT and p53–mutant MM cell lines. Taken together, these results suggest that ongoing DNA damage in MM activates ATM and JNK, leading to nuclear relocalization of ABL1.
Fig. 2. pATM and pJNK modulate ABL1 nuclear re–localization.

(a) MM cell lines were treated with 2–10 μM of ATM kinase inhibitor Ku55933 for 1–2 h in MM.1S and for 2 h in UTMC–2, JJN–3 and KMS–20 cells. Lysates were blotted for ABL1, α- tubulin (C loading control), and p84 (N loading control).
(b) MM cell lines were treated with 10 μM of JNK1 inhibitor SP600125 for 2 h. Lysates were blotted for ABL1, α-tubulin (C loading control), and Histone H3 (N loading control).
(c) Left panel: Western blot analysis for γ-H2A.X, p–ATM(Ser1981) and p–JNK (Thr183/Tyr185) on total lysates in U266 MM cell line after Dmso or doxorubicin (Doxo; 40 nM) treatment for 72 h. Right panel: Western blot analysis for ABL1, Histone H3, and α- tubulin in U266 cells after Dmso or Doxo (40 nM) treatment for 72 h.
(d) Annexin V–FITC/PI staining (left panel) and MTT absorbance assay (right panel) in U266 cells after 48 h of Dmso, Doxo (40 nM) or Doxo plus imatinib (10 μM) treatment. Data are mean values ± SD of triplicates, Student's t test.
To determine whether nuclear ABL1 is able to induce apoptosis in MM cells, we used the U266 MM cell line, which does not show γ-H2A.X foci, pATM or nuclear ABL1 under basal conditions. Treatment with DNA–damaging agent doxorubicin induced multiple γ-H2A.X foci and strong ATM and JNK phosphorylation (Fig. 2c–left panel). Importantly, ABL1 moved into the nuclear compartment (Fig. 2c–right panel), and a marked increase in apoptotic cells was evident (Fig. 2d). Treatment with imatinib significantly increased viability, suggesting a prominent role for nuclear ABL1 in inducing cell death in MM. An increase in γ-H2AX, pATM, and pJNK after exposure to doxorubicin was also evident in MM.1S cell line that presents endogenous DNA damage and DSBs, associated with a major increase in nuclear ABL1 and apoptosis (Supplementary Fig. 3c–e). However, in the presence of imatinib, cell viability significantly increased. In contrast, PBMCs lacked ongoing DNA damage (Fig. 1b) and did not demonstrate nuclear ABL1 relocalization or apoptosis after doxorubicin–induced DNA damage (Supplementary Fig. 4a–d).
We hence propose a model whereby MM cells live in a delicate equilibrium, withstanding high levels of persistent DNA damage that, through pATM and pJNK, leads to ABL1 nuclear relocalization, that should lead to cell death. Notwithstanding, no significant apoptosis was evident in MM cells, suggesting that additional mechanisms are engaged in these cells to prevent their own demise.
YAP1 is deleted or consistently down–regulated in MM cells
ABL1 forms a complex with the tumor suppressor p73, a p53 homologue17, and the Hippo pathway effector YAP1 (Yes-associated protein)18,19. YAP1 and TAZ are the main transcriptional co-activators downstream of the Hippo pathway, controlling organ size and regulating stem and progenitor cell proliferation. In response to DNA damage, ABL1 induces apoptosis through YAP1phosphorylation, which in turn stabilizes p73 and co–activates p73 pro– apoptotic target genes19,20. Therefore we sought to determine whether endogenous nuclear relocalization of ABL1 in MM is unable to induce apoptosis due to disruption of the ABL1/YAP1/p73 axis.
YAP1 behaves as an oncogene in several epithelial cancers (see discussion). However, a detailed analysis of published gene expression profiling datasets revealed a noteworthy pattern: YAP1 was consistently up–regulated in tumor cell lines of epithelial origin, but profoundly downregulated in hematologic malignancies including lymphomas, leukemias, and MM (Fig. 3a). Human YAP1 maps to chromosome 11 at the 11q22.1 locus, which is a site of focal homozygous deletions in 5 to 13% MM samples21-24. The genes implicated as targets of this deletion are BIRC2 and BIRC3, which control the pro-oncogenic NF-κB pathway21,22. Reassessing previously published data by others and us21,23,24, we noticed that the deletion in this locus consistently involves YAP1 in addition to BIRC2 and BIRC3 in all MM cell lines and most MM samples (Fig. 3b). At the gene expression level, probe sets reporting for YAP1 reflected low values overall, including in normal hematopoietic tissues. Importantly, however, when MM samples were subdivided in two groups based on YAP1 expression, low–expressors had a significantly shorter survival than high–expressors (Fig. 3c). Moreover, in various datasets there was a consistent, significant reduction in YAP1 expression levels, progressing from normal plasma cells to monoclonal gammopathy of undetermined significance (MGUS) to MM (Fig. 3d and Supplementary 5a–c). Among MM cell lines, there are subsets presenting YAP1 homozygous deletions (KMS–18, KMS–20, and KMS–28PE); others with no detectable YAP1 at the mRNA and protein level despite no genomic losses at chromosome 11; and finally, cell lines with robust expression of the gene (Fig. 3e,f and Supplementary Fig. 5d,e).
Fig. 3. YAP1 deletions and expression in MM cell lines and samples from subjects with MM.
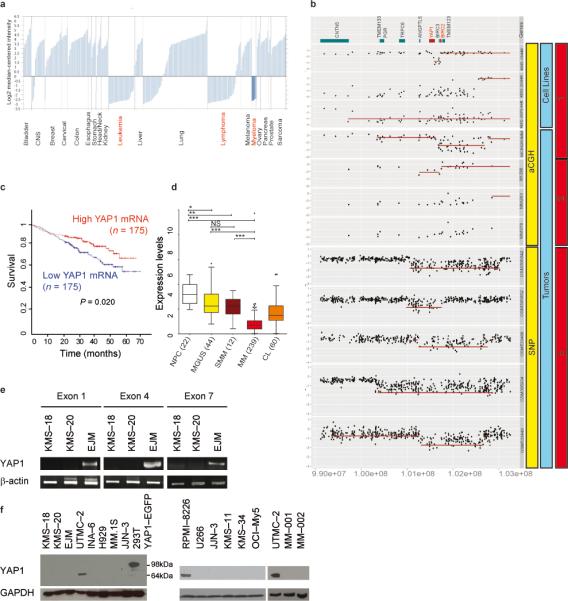
(a) YAP1 mRNA expression in solid tumors and hematological cancers (Oncomine at www.oncomine.org; Wooster Cell Line dataset).
(b) Gene dosage comparison across cell lines and representative tumors demonstrating homozygous deletions at the YAP1 locus. Number 1 corresponds to data derived from 21, 2 from 23, and 3 from 24.
(c) Survival curve relative to YAP1 expression in individuals affected by MM, obtained from www.canevolve.org, and based on GSE2658.
(d) Expression data comparing plasma cells from healthy subjects, MGUS, MM, and cell lines (CL), combining data from GSE5900, GSE2658, and from the MMRC collection (http://www.broadinstitute.org/mmgp), probe set 224895_at. *: P < 0.05; **: P < 0.01; ***: P < 0.001, one-way Anova, Dunn's Multiple Comparison Test.
(e) Non–quantitative PCR on genomic DNA from YAP1–deleted MM cell lines KMS–18 and KMS–20 and control cell line (EJM).
(f) Western blot analysis of YAP1 in MM cell lines and cells derived from subjects with MM. As positive control, lysates from 293T cells transfected with YAP1–EGFP vector were used (band at 98 kDa).
YAP1 expression in MM cells induces ABL1-mediated apoptosis
We next explored the functional role of YAP1 in MM. In gain–of–function experiments, reintroduction of YAP1–EGFP25 in deleted cell lines (KMS–18 and KMS–20) significantly reduced cell number and increased apoptosis (Fig. 4a,b and Supplementary Fig. 6a,b). Conversely, downregulation of YAP1 with specific shRNAs in MM cell lines expressing YAP1 induced a significant increase in proliferation and survival proportional to the reduction in YAP1 levels, while YAP1 overexpression did not affect cell count or apoptosis (Fig 4c and Supplementary Fig. 6c-f).
Fig. 4. YAP1 re–expression leads to ABL1- dependent reduced proliferation and cell death.

(a) YAP1 re–expression in KMS–20 (left) and KMS–18 (right) cells. Western blot analysis at 72h after pLENTI4–YAP1–EGFP or pLENTI4–LACZ transfection. Cell number is evaluated by cell counting with trypan blue exclusion. Data are representative of three independent experiments and mean values ± SD of triplicates, Student's t test.
(b) Annexin V–PE/7–AAD staining.
(c) YAP1 silencing in UTMC–2 cells using a lentiviral delivery system. Left panel: western blot analysis in pLKO.1–infected cells after 48h 2μg mL−1 puromycin selection. Right panel: Cell number and apoptosis as in (a) and (b) respectively. Data are representative of two independent experiments.
(d) Effects of YAP1 re–expression at 72 h after transfection. Western blotting, cell counting and Annexin V–PE/7–AAD staining as in (a) and (b), respectively.
(e) YAP1 re–expression in combination with imatinib treatment. Left panel: Cell number evaluated as in (a), in the presence of imatinib (10 μM) or Dmso (from day 0 of transfection).
Right panel: Western blot analysis after 48h incubation with Dmso or 10 μM imatinib. Data are mean values ± SD of triplicates, Student's t test.
(f) Western blot analysis in KMS–20 transfected cells, at 72 h.
(g) mRNA levels of p73–target genes in the same cells.
(h) dWW–YAP1 re–expression. Left panel: Western blot analysis for YAP1, at 48h. Middle panel: Cell number as in (a). Right panel: Apoptosis as in (b). Data are mean values ± SD of triplicates, Student's t test.
As mentioned above, YAP1 is not expressed in a consistent number of MM cell lines, in the absence of deletions at chromosome 11. We therefore assessed whether reintroduction of YAP1 was able to affect cell proliferation and apoptosis in this specific MM subset as well. YAP1 over-expression in MM.1S cell line dramatically reduced proliferation and increased apoptosis to levels comparable to YAP1–deleted cells (Fig. 4d and Supplementary Fig. 6g). These results suggest that re–expression of YAP1 might induce apoptosis and reduce proliferation not only in MM cells where YAP1 is deleted, but also in the larger population of subjects with MM where YAP1 is not expressed despite normal copy number.
YAP1–induced apoptosis was mediated by the aberrant presence of ABL1 in the nucleus, since treatment with imatinib significantly reduced the apoptotic response suggesting that YAP1 phosphorylation by ABL1 is required for the apoptotic response, as previously described19
(Figure 4e–left panel and Supplementary Fig. 7a). Imatinib treatment also specifically reduced phospho–Y357 YAP1, a crucial step for activation of proapoptotic genes mediated by YAP119 (Figure 4e–right panel). Similar effects were obtained in the subset of MM cell lines with low YAP1 levels (Supplementary Fig. 7a and data not shown). These results indicate that apoptosis induced by the nuclear relocalization of ABL1 in MM cells is prevented, at least in part, by low YAP1 levels.
Due to the functional interaction between YAP1 and p7319,20,26,27, we next explored the relationship between YAP1 and p73 upon DNA damage in MM. Re–expression of YAP1 in the deleted MM cell lines remarkably increased p73 protein levels with moderate effects on p73 mRNA levels, while p53 and TP63 (p63) protein levels were not altered (Fig. 4f and Supplementary Fig. 7b–d). Accordingly, levels of transcriptional p73 targets such as BAX, PUMA, and CDKN1A (p21), significantly increased at both the mRNA and protein levels (Fig. 4f,g), whereas p53/p73 target NOXA did not vary.
ABL1–mediated phosphorylation of YAP1 at Y357 enhances its affinity toward p73 binding28. Indeed, imatinib treatment reduced the interaction of p73 with YAP1 (Supplementary Fig. 7e). To confirm the role of p73 in driving YAP1–mediated apoptosis, we transfected KMS– 20 with a YAP1 mutant construct that lacks the WW domain necessary to interact with p7328. This mutant, unlike wild type YAP1, was unable to trigger apoptosis and inhibit proliferation (Fig. 4h). Taken together, these results suggest that apoptosis in MM induced by DNA damage and YAP1 restoration is mediated by stabilization of p73 and increased expression of its downstream pro–apoptotic targets.
Inactivation of kinase STK4 enhances YAP1 and apoptosis
A cytoplasmic serine–threonine kinase, STK4, interacts with LATS1 and significantly reduces YAP1 levels29,30. STK4 downregulation with specific shRNAs leads to a robust increase of YAP1 protein levels, compared to scrambled shRNA (Fig 5a). Notably, YAP1 appeared both in the nucleus and in cytoplasm upon STK4 downregulation (Supplementary Fig. 8a). We further explored whether STK4 downregulation impacted on YAP1 mRNA levels. A moderate increase in YAP1 mRNA levels was evident after STK4 inhibition (Supplementary Fig. 8b). Of note, gene expression profiling data revealed a significant, inverse correlation between STK4 and YAP1 expression levels in MM samples (P < 0.0001, Supplementary Fig. 8c). Additionally, treatment of MM.1S cells with the proteasome inhibitor bortezomib robustly increased YAP1 protein levels (Supplementary Fig. 8d). Taken together, these results indicate that STK4 controls YAP1 both at the mRNA and protein levels.
Fig. 5. STK4 knock–down triggers YAP1 re-expression, reducing MM cell proliferation and increasing apoptosis in vitro and in vivo.

(a) STK4 silencing using lentiviral vector pLKO.1 (after 48h 2μg mL−1 puromycin selection) in MM.1S cells. Western blot for YAP1 and STK4 in STK4–silenced cells (3 representative shRNAs).
(b) Cell number evaluated by cell counting with trypan blue exclusion in MM.1S cells silenced for STK4. Left panel, pLKO.1 lentivectors; right panel, inducible pTRIPZ lentivectors. One representative experiment of three is shown. Data are mean values ± SD of triplicates, Student's t test.
(c) Combined effects of STK4 knock–down associated with treatment with various compounds. Apoptosis evaluated with Annexin V–FITC/PI staining in stably infected cells after 48h incubation with 5 nM bortezomib (Btz) and 40 nM doxorubicin (Doxo).
(d) STK4 silencing by transfection using pLKO.1 scrambled or shRNA #5 vectors. Cell counting as in (b).
(e) MM.1S cells, infected with inducible STK4–shRNAs, after 24 h of 2 μg mL−1 doxycycline were transfected by electroporation with pJ3H–STK4–wild type or pJ3H–STK4–K59R mutant. Left panel: Cell number evaluated as in (b). Right panel: Western blot after 48h transfection.
(f) In vivo evaluation of the effects of STK4 knock–down on MM cells. Left panel: injection scheme and tumor size in three representative mice. Right panel: Growth curve assessing tumor size after injection of MM.1S cells transduced with STK4–specific shRNAs or scrambled–control vectors. Data are mean values ± SD of triplicates, Student's t test.
(g) Proposed model for the ABL1/YAP1/p73 axis and effects of STK4 inhibition on YAP1 levels in MM.
We then assessed whether up–regulation of YAP1 induced by STK4 knockdown was associated with reduced proliferation. Indeed, all shRNAs which effectively downregulated STK4 expression and increased YAP1 levels also significantly inhibited MM cell proliferation (Fig. 5b–left panel) and induced a robust apoptotic response (Fig. 5c and Supplementary Fig. 9a). We further confirmed this phenotype using an independent set of inducible shRNA sequences inserted into another vector or in different MM cell lines (Fig. 5b–right panel and Supplementary Fig. 9b–e). Importantly, treatment with bortezomib or doxorubicin enhanced this effect (Fig. 5c). Additionally, inhibition of STK4 failed to reduce proliferation and increase apoptosis in the YAP1–deleted cell lines KMS–18 and KMS–20 (Fig. 5d and Supplementary Fig. 10a,b). To further confirm that YAP1 mediates the phenotypes induced by STK4 inhibition, the expression of STK4 and YAP1 was concomitantly reduced in MM.1S cells with the respective shRNAs, rescuing the phenotype (Supplementary Fig. 10c). These data demonstrate that the effects of STK4 inhibition in MM cells are mediated by restoration of YAP1.
Re–expression of a STK4 mutant devoid of kinase activity, K59R31, in MM.1S and H929 MM cells down–regulated for STK4, failed to repress YAP1 levels, rescue proliferation, or prevent apoptosis, suggesting that STK4 kinase activity is required to suppress YAP1 thereby preventing apoptosis (Fig. 5e and Supplementary Fig. 11a,b).
These results indicate that YAP1 downregulation, seen in MM cells and cell lines in the absence of chromosome 11 deletion, can, at least in part, be due to the inhibitory effects of STK4 upon YAP1 levels.
To demonstrate in vivo the relevance of this synthetically lethal interaction, a genetic approach conditionally knocking down STK4 in MM.1S cells injected subcutaneously in mice was performed. Tumors developed exclusively from MM.1S cells infected with a scrambled shRNA, while no growth was evident in STK4 silenced cells (P < 0.0001; Fig. 5f). Taken together, our results demonstrate that STK4 inhibition upregulates YAP1 levels in MM cells, thereby triggering apoptosis both in vitro and in vivo (Fig. 5g).
DNA damage, ABL1, STK4 and YAP1 in lymphoma and leukemia
We next assessed DNA damage in a panel of lymphoma, lymphoblastic and myeloid leukemias, and Waldenström macroglobulinemia cell lines. Staining with γ-H2A.X revealed robust, ongoing DNA damage in the majority of the cell lines assessed (Fig. 6a,b). Moreover, consistent nuclear localization of ABL1 was evident (Fig. 6c and Supplementary Fig. 12a). YAP1 mRNA and protein levels were low, as in MM (Fig. 6d and Supplementary Fig. 12b). Remarkably as in MM, cells derived from individuals with leukemia showing low YAP1 expression had a significantly worse prognosis (Fig. 6d). The reintroduction of YAP1 in ALL (Jurkat) or AML (OCI/AML3)(Fig. 6f,g) cell lines decreased cell number and was associated with apoptosis and induction of p73–target genes (Fig. 6f–h and Supplementary Fig. 13). As in MM, STK4 reduction through STK4 shRNAs increased YAP1 levels, reduced cell number, and enhanced apoptosis (Fig 6i and Supplementary Fig. 14).
Fig. 6. Lymphoma, leukemia and Waldenström's macroglobulinemia cells present ongoing DNA damage and ABL1 nuclear localization, and undergo apoptosis following STK4-mediated YAP1 increased levels.

(a) γ-H2A.X staining of Waldenström's macroglobulinemia (BCWM.1 and MWCL.1), lymphoma (DOHH–2 and RAMOS), B–cell acute lymphoblastic leukemia–B–ALL (RS4;11, CEMO–1, REH, and NALM–6), acute myeloid leukemia–AML (OCI/AML3, KG–1, and HL–60), and T– cell acute lymphoblastic leukemia–T–ALL (Jurkat) cell lines.
(b) Western blot analysis for γ-H2A.X and p–ATM.
(c) Cell lysates from cytoplasmic (C) and nuclear (N) fractions analyzed by Western blot for ABL1 expression. γ-tubulin and Histone H3 were used as loading controls for C and N fractions, respectively.
(d) YAP1 protein expression in ALL, lymphomas, and AML. As positive control, lysates from 293T cells transfected with YAP1–EGFP vector were used (98 kDa–band).
(e) Survival curve relative to YAP1 expression in subjects with AML, obtained from www.canevolve.org, based on GSE12417.
(f) Western blot analysis at 72 h after transfection. Cell number evaluated with cell counting using trypan blue exclusion. Mean values ± SD of triplicate of two experiments are shown. Data are mean values ± SD of triplicates, Student's t test.
(g) Apoptosis evaluated by Annexin V–PE/7–AAD staining after gating on GFP–positive cells.
(h) mRNA levels of YAP1, p73, and p73–target genes (BAX, PUMA, p21).
(i) STK4 knockdown in OCI/AML3 and Jurkat cell lines. Left panel: Western blot for YAP1 and STK4. Middle panel: Cell number measured as in (f) after transfection with scrambled or pLKO.1 #5 vectors. Right panel: Apoptosis by Annexin V–PE/7–AAD staining.
Discussion
In this study, we have demonstrated that MM, lymphomas, and leukemias present pervasive DNA damage. As a result, the pro–apoptotic tyrosine kinase ABL1 relocalizes into the nucleus, uncovering for the first time an unexpected broad role for this protein in inducing apoptosis during the DDR response. Tumor cells nevertheless escape apoptosis due to genetic inactivation or reduced expression of the Hippo co–transcription factor YAP1. Importantly, we elucidate a novel synthetic lethal approach32 in which inhibition of the kinase STK4 reactivates YAP1 and triggers apoptosis, providing the rationale for developing novel STK4 inhibitors, for clinical evaluation in hematological malignancies.
As in neoplasms of epithelial origin1, activation of DNA damage checkpoint might also represent a barrier against the evolution towards cancer in hematological tissues; however, unlike epithelial cancers, hematological malignancies do not seem to require p53 inactivation. Instead, early inactivation of the ABL1/YAP1/p73 axis may substitute for p53 mutations and/or inactivation in hematological tumors.
YAP1 is focally amplified in a vast array of solid tumors including brain, colon and hepatocellular carcinomas, and has been consistently reported as an oncogene in epithelial cancers33. Our data support a role for YAP1 as a tumor suppressor gene in hematological cancers. A possible explanation for this differential function may relate to YAP1 forming complexes with various partners with distinct functional sequelae, depending on the cellular context34. For example, in the absence of DNA damage YAP1 preferentially interacts with oncogenic transcription modulator RUNX, leading to increased degradation of p7320. Thus, transcription modulators can shift YAP1 away from p73 towards other partners, endowing YAP1 with proliferative and anti-apoptotic features.
The heart of the Hippo pathway includes a core of two serine–threonine kinase pairs, STK4 (MST1) and STK3 (MST2), which activate two other kinases, LATS1 and LATS2. In turn, LATS1 and LATS2 regulate YAP1 and TAZ35. The concomitant loss of both STK4 and STK3 is required to foster the development of hepatocellular carcinoma in vivo29. In our study, the used STK4–specific shRNAs did not affect STK3 mRNA levels (Supplementary Fig. 15), suggesting that STK4 exerts a prominent, specific role in controlling YAP1 levels in MM. As for other Hippo pathway members, recent intriguing findings have implicated LATS2 in DNA damage and cell cycle control36,37, suggesting that the Hippo pathway exerts a tight control on DNA damage and reinforcing the notion of a tumor suppressive role for the this pathway in specific contexts such as hematological cancers. Ultimately, YAP1 presents a high degree of homology with TAZ. Although they often present overlapping functions, the two proteins seem to exert independent roles, exemplified by the phenotypes of the mouse knockouts and the specific functions of TAZ in stem cell maintenance and WNT regulation38. Unlike YAP1, we did not detect focal deletions affecting the TAZ locus in MM cell lines and cells derived from individuals with MM, suggesting that tumors of hematological lineage preferentially inactivate YAP1.
Kinases are valuable targets for cancer therapies, as already proven by imatinib, erlotinib, and gefitinib. Therefore, STK4 provides a promising therapeutic target in hematological cancers displaying low YAP1 levels and concomitant DNA damage. Importantly, STK4 knockdown induced apoptosis in both p53–WT and mutant cells, suggesting that restoring YAP1 levels could represent a novel treatment strategy even in tumors with p53 inactivation and poor prognosis.
Experimental Procedures
Materials, Reagents and cell lines
All antibodies and assay reagents were obtained from commercial sources as described in Supplemental Experimental Procedures. All MM cell lines were purchased from American Type Culture Collection (ATCC), established in our laboratory or kindly provided by our collaborators.
Primary cells
Blood samples from healthy volunteers and MM bone marrow samples were processed as described in Supplemental Experimental Procedures.
Cell Culture and Molecular Methods
Methods for cell culture, DNA and RNA extraction, mRNA quantification, subcellular fractioning, immunoblotting, immunofluorescence, as well as assays for viability, cellular growth, and apoptosis are described in Supplemental Experimental Procedures.
Lentiviral mediated gene transfer and transient transfection of MM cell lines.
MM cells (3,000,000) were transiently transfected by nucleoporation (LONZA, Cell Line Nucleofector Kit V, Amaxa Biosystems). Lentiviral infection, selection and induction were performed following standard procedures (see also Supplemental Experimental procedures).
Supplementary Material
Acknowledgments
We thank M. Sudol (Mount Sinai School of Medicine), for the EGFP–YAP1 construct and W. Hahn (Dana-Farber Cancer Institute) for pLKO.1 shRNA lentiviral vectors. We also wish to thank E. Di Cairano and L. Spagnuolo for immunohistochemical stains, F.Ghini and A.M. Gasparri for technical help, the DFCI Flow cytometry facility, D. Kufe and F. Bernassola for insightful suggestions, C.Brennan for the bioinformatics analysis and members of the Anderson and Tonon lab for sharing reagents and critical reading of the manuscript. M.W.K. is supported by the Intramural Research Program of the US National Institutes of Health, National Cancer Institute, Center for Cancer Research. This work was supported by Fondazione CARIPLO, Marie Curie International Reintegration Grant, Associazione Italiana per la Ricerca sul Cancro (AIRC; Investigator Grants and Special Program Molecular Clinical Oncology, 5 per mille no. 9965) (G.T.); AIRC (A.N.); K.C.A. is an American Cancer Society Clinical Research Professor and is supported by NIH grants NIH SPORE P50 100707, PO-1 78378, RO-1 50947.
F.C. designed research, performed experiments, analyzed data, and wrote the manuscript; T.H. designed research and analyzed data; C.X., E.T.H., S.B., A.M.G. performed mouse experiments, M.S., A.K., K.K.W., G.B., and F.C.C. contributed to designing experiments, M.D., L.A. and A.N. analyzed genomic and microarray data, M.P. and R.C. analyzed immunohistochemistry staining, M.M. and P.G.R. provided patient samples, W.M.K. analyzed data and wrote the manuscript; K.C.A. analyzed data and wrote the manuscript, and G.T. designed research, analyzed data and wrote the manuscript.
References
- 1.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 2.Boehrer S, et al. Suppression of the DNA damage response in acute myeloid leukemia versus myelodysplastic syndrome. Oncogene. 2009;28:2205–2218. doi: 10.1038/onc.2009.69. [DOI] [PubMed] [Google Scholar]
- 3.Walters DK, et al. Evidence for ongoing DNA damage in multiple myeloma cells as revealed by constitutive phosphorylation of H2AX. Leukemia. 2011;25:1344–1353. doi: 10.1038/leu.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu-Monette ZY, et al. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood. 2012;119:3668–3683. doi: 10.1182/blood-2011-11-366062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baskaran R, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387:516–519. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- 7.Kharbanda S, et al. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature. 1995;376:785–788. doi: 10.1038/376785a0. [DOI] [PubMed] [Google Scholar]
- 8.Shafman T, et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387:520–523. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- 9.Yuan ZM, et al. Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1437–1440. doi: 10.1073/pnas.94.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan ZM, et al. Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature. 1996;382:272–274. doi: 10.1038/382272a0. [DOI] [PubMed] [Google Scholar]
- 11.Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 12.Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 13.Brown L, McCarthy N. DNA repair. A sense-abl response? Nature. 1997;387:450–451. doi: 10.1038/387450a0. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nature cell biology. 2005;7:278–285. doi: 10.1038/ncb1228. [DOI] [PubMed] [Google Scholar]
- 15.Hickson I, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer research. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 16.Bennett BL, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White E, Prives C. DNA damage enables p73. Nature. 1999;399:734–735. 737. doi: 10.1038/21539. [DOI] [PubMed] [Google Scholar]
- 18.Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 1994;9:2145–2152. [PubMed] [Google Scholar]
- 19.Levy D, Adamovich Y, Reuven N, Shaul Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Molecular cell. 2008;29:350–361. doi: 10.1016/j.molcel.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 20.Levy D, Adamovich Y, Reuven N, Shaul Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell death and differentiation. 2007;14:743–751. doi: 10.1038/sj.cdd.4402063. [DOI] [PubMed] [Google Scholar]
- 21.Keats JJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Annunziata CM, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrasco DR, et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer cell. 2006;9:313–325. doi: 10.1016/j.ccr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 24.Walker BA, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010;116:e56–65. doi: 10.1182/blood-2010-04-279596. [DOI] [PubMed] [Google Scholar]
- 25.Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Molecular cell. 2003;11:11–23. doi: 10.1016/s1097-2765(02)00776-1. [DOI] [PubMed] [Google Scholar]
- 26.Strano S, et al. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. The Journal of biological chemistry. 2001;276:15164–15173. doi: 10.1074/jbc.M010484200. [DOI] [PubMed] [Google Scholar]
- 27.Strano S, et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Molecular cell. 2005;18:447–459. doi: 10.1016/j.molcel.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 28.Lapi E, et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Molecular cell. 2008;32:803–814. doi: 10.1016/j.molcel.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 29.Zhou D, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou D, et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E1312–1320. doi: 10.1073/pnas.1110428108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Creasy CL, Ambrose DM, Chernoff J. The Ste20-like protein kinase, Mst1, dimerizes and contains an inhibitory domain. J Biol Chem. 1996;271:21049–21053. doi: 10.1074/jbc.271.35.21049. [DOI] [PubMed] [Google Scholar]
- 32.Kaelin WG., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nature reviews. Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 33.Pan D. The hippo signaling pathway in development and cancer. Developmental cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertini E, Oka T, Sudol M, Strano S, Blandino G. YAP: at the crossroad between transformation and tumor suppression. Cell cycle. 2009;8:49–57. doi: 10.4161/cc.8.1.7259. [DOI] [PubMed] [Google Scholar]
- 35.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 36.Reuven N, Adler J, Meltser V, Shaul Y. The Hippo pathway kinase Lats2 prevents DNA damage-induced apoptosis through inhibition of the tyrosine kinase c-Abl. Cell Death Differ. 2013;20:1330–1340. doi: 10.1038/cdd.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tschop K, et al. A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev. 2011;25:814–830. doi: 10.1101/gad.2000211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piccolo S, Cordenonsi M, Dupont S. Molecular pathways: YAP and TAZ take center stage in organ growth and tumorigenesis. Clin Cancer Res. 2013;19:4925–4930. doi: 10.1158/1078-0432.CCR-12-3172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.