

Abstract
Growing evidence supports the hypothesis that epigenetics is a key mechanism through which environmental exposures interact with an individual’s genetic constitution to determine risk for depression throughout life.1 Epigenetics, in its broadest meaning, refers to stable changes in gene expression that are mediated via altered chromatin structure without modification of DNA sequence. According to this hypothesis, severe stress triggers changes—in vulnerable individuals—in chromatin structure at particular genomic loci in the brain’s limbic regions, which drive sustained changes in gene expression that contribute to episodes of depression. A corollary of this hypothesis is that such stress-induced epigenetic modifications also occur early in life and help determine an individual’s lifetime vulnerability or resistance to subsequent stressful events.
There are 2 additional ways in which epigenetic mechanisms might influence depression. One involves stochastic events during development. The other concerns true epigenetic inheritance across multiple generations. As will be seen, these forms of epigenetic contributions to depression remain less established.
Mechanisms of Epigenetic Regulation
DNA is wrapped around histone octamers to form nucleosomes—the unit of chromatin (Figure). A gene’s activity is reflected in the surrounding structure of chromatin: genes within relatively spaced nucleosomes are actively transcribed, whereas those in tightly packed nucleosomes are silenced. Such nucleosome spacing is determined by extremely complex processes, which include the posttranslational modification of histones (Figure) and DNA and the recruitment of large numbers of chromatin regulatory proteins that interact with these modifications.
Figure. Chromatin Structure and Histone Modifications at N-Terminal Histone Tails.
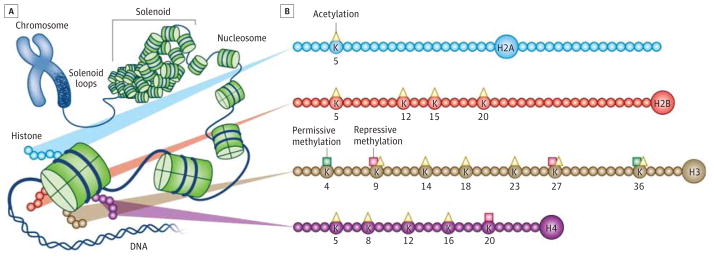
A, The eukaryotic genome is organized by wrapping DNA around histone octamers to form the basic units of chromatin, nucleosomes, which are then further organized and compacted into higher-ordered structures. B, The histone octamer consists of 2 copies each of H2A, H2B, H3, and H4. In addition to globular domains, they each have N-termini tails that protrude from the nucleosome, while H2A also has a C-terminal tail. These tails can be posttranslationally modified, and major forms of mammalian acetylation and methylation modifications on lysine residues on each tail are highlighted. The molecules are drawn approximately proportional to the size of the protein, although the number of residues shown is not meant to reflect the exact size of the N-terminal tails. This image was published in Sun HS, Kennedy PJ, Nestler EJ. Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology Rev. 2013;38:124-137. The author retains the copyright.
Histone acetylation, catalyzed by histone acetyltransferases and reversed by histone deacetylases (HDACs), generally drives a more permissive (open) state of chromatin and increased gene expression. Histone methylation, catalyzed by histone methyltransferases (HMTs) and reversed by histone demethylases (HDMs), can either activate or repress gene transcription depending on the amino acid residue undergoing methylation. DNA methylation, catalyzed by DNA methyltransferases (DNMTs), generally drives repression of gene expression, although variant forms of DNA methylation (hydroxymethylation) may be more related to gene activation. Large numbers of “reader” proteins bind to each of these histone and DNA modifications and mediate changes in chromatin structure and gene transcription. As just one example, chromatin remodeling proteins possessing ATPase activity control nucleosome spacing and move nucleosomes along a DNA during transcription.
Environmental stimuli such as stress regulate these processes in 2 main ways. First, synaptic transmission and neural activity, through intracellular signaling cascades, control the activity and levels of numerous transcription factors—eg, CREB (cAMP [cyclic adenosine monophosphate] response element binding protein) and Fos and Jun family proteins, among many others—which bind to their specific response elements contained within regulatory regions of genes and trigger downstream changes in chromatin structure. Second, the same intracellular signaling pathways directly control the activity of many chromatin regulatory proteins, which then directly drive alterations in gene expression.
Epigenetic Control of Stress Responses
Chronic exposure of rodents to several forms of stress alters chromatin structure in the brain. One type of modification is global: chronic stress alters total cellular levels of acetylated or methylated histones within specific limbic brain regions.2–4 Such global changes presumably reflect a genomewide skew toward a more permissive or restrictive state of chromatin activity. The other type of modification is locus specific, that is, a change in histone acetylation or methylation or in DNA methylation at a particular gene that mediates its altered expression.3,5,6 Interestingly, many of these locus-specific changes defy the coincident global modifications.
Such epigenetic regulation of stress responses is illustrated by our work in the chronic social defeat model of depression. Under chronic social stress, susceptible mice manifest a depressionlike syndrome, while resilient ones avoid most of these deleterious symptoms despite being subjected to the same level of stress.1 We have shown that such susceptibility vs resilience is controlled in part by changes in histone acetylation and a repressive form of histone methylation—dimethylation of Lys9 of histone H3 (H3K9me2)—in nucleus accumbens.2,3 Global levels of histone acetylation are increased, and this adaptation promotes a resilient outcome because HDAC inhibition specifically in this brain region opposes susceptibility and induces antidepressant-like responses.2 In contrast, global increases in H3K9me2 are pathological: local knockout or inhibition of the HMT that mediates this mark promotes susceptibility, whereas its activation promotes resilience.3 We have used genomewide measures to identify numerous genes that display alterations in these histone modifications, which provide unprecedented insight into the molecular basis of susceptibility vs resilience.6
There is also strong support for the importance of epigenetic modifications occurring early in life and driving lifelong vulnerability to stress. In landmark experiments in rats by Weaver and colleagues,7 offspring of mothers that received low levels of maternal care/grooming (LG) showed increased stress reactivity and anxiety-related behavior in adulthood compared with those that received high levels of maternal care/grooming (HG). LG rats show decreased hippocampal glucocorticoid receptor (GR) mRNA expression and corresponding decreased H3K9 acetylation and increased DNA methylation around the GR gene promoter compared with HG animals. Such differences in GR expression help determine the maternal care delivered by these animals, thereby perpetuating patterns of LG vs HG behavior from generation to generation.7 It is likely that epigenetic regulation of many additional genes is also involved. This form of trans-generational transmission of stress responsiveness is thus behaviorally transmitted and does not represent true epigenetic inheritance.
Studies of epigenetic mechanisms in stress models throughout life are revealing fundamentally new insight into the range of genes and biochemical pathways that are altered within specific brain regions to govern risk for depression and the reversal of symptoms under antidepressant treatment. Such knowledge can now drive efforts to identify novel antidepressant medications by moving well beyond the current focus on monoamine transporters and receptors.
A related question is whether epigenetic proteins themselves might be suitable targets for such treatments. As noted here in, HDAC inhibitors exert antidepressantlike actions in several rodent models,2,5 and there is evidence to suggest that drugs that control H3K9 methylation or even DNA methylation may be similarly effective.1–5,7,8 However, given the ubiquity of HDACs, HMTs, HDMs, and DNMTs, it is likely that such drugs will cause too many adverse effects for utility in depression. It remains to be seen whether there are other means of influencing such processes by targeting brain-enriched regulatory proteins, which can be exploited in antidepressant drug discovery.
Other Epigenetic Contributions to Depression
As the brain develops, generating 100 billion neurons and 100 trillion synapses, innumerable stochastic events occur that generate diversity despite constant genetics and environment. The highly divergent fingerprints or patterns of cerebral gyri exhibited by identical twins provide examples of such phenomena. While epigenetics is a likely contributor, it is difficult to obtain experimental proof. For example, in our social defeat model, we can demonstrate that genetically identical animals with nearly uniform environmental exposures diverge strikingly into susceptible vs resilient outcomes after social stress.1,6 However, it is not currently possible to define epigenetic contributions to such phenotypic outcomes because this would require tools to characterize the epigenetic state of genes within animals prior to stress exposure. Nevertheless, stochastic, epigenetic differences in brain function could be one reason why it has been difficult to identify genes that confer risk for depression.
The term epigenetics is used by some to denote transgenerational transmission of traits without a change in DNA sequence. Indeed, stressful life events have been shown to alter stress susceptibility in subsequent generations. Male mouse pups subjected to maternal separation display lifelong increases in stress susceptibility and generate offspring that display similarly enhanced stress susceptibility over several generations.9 Likewise, male mice subjected to chronic social defeat stress as adults generate offspring that are more vulnerable to social stress than the offspring of control mice.10 However, the mechanisms underlying this transgenerational transmission of stress vulnerability remain controversial. Stress produces epigenetic modifications at particular genes in the sperm of stressed mice,9 yet it is far from certain whether such modifications contribute to the differences seen in stress vulnerability. Studies of in vitro fertilization suggest that, while epigenetic changes in sperm might be a small factor in transgenerational transmission of stress vulnerability, much of the observed transmission may be behavioral, with females altering their maternal care based on their procreation with previously stressed fathers.10 Further work is therefore needed to understand whether and to what extent true epigenetic inheritance of stress vulnerability adds to the well-established and powerful influence of genetics and environmental exposures in determining an individual’s susceptibility vs resilience to stress throughout life.
Acknowledgments
Funding/Support: Preparation of this review was supported by grants from the National Institute of Mental Health.
Role of the Sponsor: The National Institute of Mental Health had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Conflict of Interest Disclosures: None reported.
References
- 1.Vialou V, Feng J, Robison AJ, Nestler EJ. Epigenetic mechanisms of depression and antidepressant action. Annu Rev Pharmacol Toxicol. 2013;53:59–87. doi: 10.1146/annurev-pharmtox-010611-134540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Covington HE, III, Maze I, LaPlant QC, et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29(37):11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Covington HE, III, Maze I, Sun H, et al. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011;71(4):656–670. doi: 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunter RG, McCarthy KJ, Milne TA, Pfaff DW, McEwen BS. Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc Natl Acad Sci U S A. 2009;106(49):20912–20917. doi: 10.1073/pnas.0911143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9(4):519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson MB, Xiao GH, Kumar A, et al. Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J Neurosci. 2009;29(24):7820–7832. doi: 10.1523/JNEUROSCI.0932-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weaver IC, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7(8):847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 8.Jiang Y, Jakovcevski M, Bharadwaj R, et al. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J Neurosci. 2010;30(21):7152–7167. doi: 10.1523/JNEUROSCI.1314-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franklin TB, Russig H, Weiss IC, et al. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry. 2010;68(5):408–415. doi: 10.1016/j.biopsych.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 10.Dietz DM, Laplant Q, Watts EL, et al. Paternal transmission of stress-induced pathologies. Biol Psychiatry. 2011;70(5):408–414. doi: 10.1016/j.biopsych.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]