

Abstract
Obesity is a complex metabolic disorder that is more prevalent among women. Until now, the only relevant rodent models of diet-induced obesity were via the use of ovariectomized (“postmenopausal”) females. However, recent reports suggest that the xenobiotic nuclear receptor pregnane X receptor (PXR) may contribute to obesity. Therefore, we compared the roles of mouse and human PXRs in diet-induced obesity between wild type (WT) and PXR-humanized (hPXR) transgenic female mice fed either control or high-fat diets (HFD) for 16 weeks. HFD-fed hPXR mice gained weight more rapidly than controls, exhibited hyperinsulinemia, and impaired glucose tolerance. Fundamental differences were observed between control-fed hPXR and WT females: hPXR mice possessed reduced estrogen receptor α (ERα) but enhanced uncoupling protein 1 (UCP1) protein expression in white adipose tissue (WAT); increased protein expression of the hepatic cytochrome P450 3A11 (CYP3A11) and key gluconeogenic enzymes phosphoenolpyruvate carboxykinase and glucose 6-phosphatase, and increased total cholesterol. Interestingly, HFD ingestion induced both UCP1 and glucokinase protein expression in WT mice, but inhibited these enzymes in hPXR females. Unlike WT mice, CYP3A11 protein, serum 17β-estradiol levels, and WAT ERα expression were unaffected by HFD in hPXR females. Together, these studies indicate that the hPXR gene promotes obesity and metabolic syndrome by dysregulating lipid and glucose homeostasis while inhibiting UCP1 expression. Furthermore, our studies indicate that the human PXR suppresses the protective role of estrogen in metabolic disorders. Finally, these data identify PXR-humanized mice as a promising in vivo research model for studying obesity and diabetes in women.
Keywords: obesity, high-fat diet, pregnane X receptor, nuclear receptors, type 2 diabetes, females
1. Introduction
The prevalence of obesity has increased worldwide at an alarming rate and is now recognized as a serious global health problem [1]. In the US, obesity affects every age-group of the population, with 1 in 3 adults considered to be clinically obese [2]. In the context of this global obesity pandemic, a clear gender disparity exists, with women at greater risk of developing obesity than men [2]. Further, obesity is also a disease of racial disparity, in which African-American women are more frequently afflicted than Caucasian women [3]. The biological mechanisms that contribute to these gender and racial disparities remain known.
Similar to humans, several studies have reported differential susceptibility to obesity upon high-fat diet (HFD) ingestion by some strains of rats and mice [4,5,6,7]. Reports indicate that C57BL/6 (B6) mice will develop severe obesity, hyperglycemia, and insulin resistance if weaned unto a HFD compared to other strains, such as C3H/He, 129/Sv, and A/J mice, which are resistant to obesity and diabetes [7,8,9,10,11,12]. Interestingly, Similar to the B6 mice, AKR/J mice are also sensitive to the development of obesity by a HFD, but are less hyperglycemic [7]. Several factors influence the development of obesity including genetics, sedentary lifestyles, and dietary fat amount and type [13,14,15]. Interestingly, the specific genes that determine sensitivity to dietary obesity remain unknown [13,14].
With respect to gender differences in obesity, both clinical data and animal studies indicate that sex hormones (estrogens in females and androgens in males) are the chief drivers of sexual dimorphisms; gonadal steroids contribute to the regulation of food intake, body weight, and lipid metabolism [16,17]. In females, the loss of estrogen production, either following menopause in humans or ovariectomy in rodents, frequently leads to obesity, suggesting that estrogens appear to play a role in lipid homeostasis to reduce overall body adiposity [18,19].
Consumption of a high caloric diet, rather than genetic makeup, is the most common cause of obesity in humans [20]. Therefore, a clinically relevant animal model of HFD-induced obesity would provide a valuable tool for investigating the molecular basis of obesity in women. Unfortunately, unlike humans, female rodents are more resistant to HFD-induced obesity than their male counterparts, and among female rodents, only ovariectomized animals are capable of significant HFD-induced weight gain [21]. Therefore, female mice are less prone to develop diabetes than humans [22]. Thus, data from both human and animal studies suggest that estrogen may not be the only factor that influences sexual dimorphism in obesity.
Numerous studies have utilized ovariectomy with or without repletion of estrogen and/or compared male and female mice provided a HFD as models to explore the contribution of estrogen to the sexual dimorphisms in obesity [18,19]. Obesity is the product of a chronic positive energy balance, and the health risks associated with obesity vary depending on the amount and location of adipose tissue [23,24,25]. While the ovariectomized rodent model shares similar characteristics with humans, this model requires a high level of surgical skill and does not completely match the physiology of postmenopausal women, most of whom retain their ovaries. For example, both ovariectomy in female rodents and menopause result in weight gain and increased adipose tissue mass, however, ovariectomy preferentially increases subcutaneous fat whereas menopause increases the metabolically more deleterious intraabdominal visceral fat [24,25,26,27]. Furthermore, ovariectomy leads to an immediate cessation of estrogen secretion and gain in lean body mass, whereas menopause occurs after a gradual cessation of estrogen secretion resulting in loss of lean body mass [26,27]. These dissimilar effects of ovariectomy and menopause suggest that the metabolic consequences of ovariectomy differ from that caused by menopause-induced obesity and that ovariectomy may not fully represent a rodent model for female obesity.
Nevertheless, estrogens and estrogen receptors (ERs) have been the primary focus of studies attempting to unravel the molecular basis of sexual dimorphisms in obesity. Estrogen activity occurs via the ER, a ligand-dependent transcription factor that consists of distinct modular domains, each with unique biological functions [28,29]. Importantly, ER-mediated transcriptional activity modulates other nuclear receptors, including the peroxisome proliferator-activated receptor α (PPARα), the constitutive androstane receptor (CAR), and the pregnane X receptor [PXR; NR1I2, its human homologue, steroid and xenobiotic receptor (SXR)] [30,31,32]. While the role of nuclear receptors in obesity is now well established, information is lacking about the contribution of nuclear receptors to the gender disparity of obesity incidence in humans and the functional differences between mouse and human nuclear receptors in diet-induced obesity.
PXR expressed primarily in the liver, intestine and kidney is a xenobiotic sensing nuclear receptor, involved in detoxification of drugs and other foreign chemicals [33]. While previous studies have established functional links between glucose homeostasis and PXR, the in vivo significance of PXR function in obesity and metabolic syndrome has not been fully explored [34,35,36]. The sequences of human and mouse PXRs share nearly 77% amino acid identity across the C-terminal ligand binding domain (LBD), 96% amino acid identity in the N-terminal DNA-binding domain, and the two PXRs display similar tissue- specific expression patterns [33]. However, there are major differences in responses to ligand activation by human and mouse PXR that are likely due to differences in LBD sequence [37]. As a result, some chemicals that activate human PXR usually have little effect on the mouse PXR and vice versa [38,39]. For example, rifampicin, phytoestrogens, and corticosterone potently activate human PXR, but not mouse PXR. In contrast, pregnenolone- 16α-carbonitrile (PCN) and dexamethasone strongly activate mouse, but not human PXR [38].
To overcome the species differences in ligand specificity, PXR-humanized (hPXR) transgenic mice have provided a relevant in vivo model of the human xenobiotic response [40]. The hPXR mice were generated by bacterial artificial chromosome (BAC) transgenesis, in which the transgene contains the complete human PXR gene and the 5′- and 3′-flanking sequences and then bred with PXR-KO mice to produce hPXR mice carrying a C57BL/6 genetic background [40]. hPXR mice selectively express PXR in the liver and intestine in parallel with cytochrome P450 3A4 (CYP3A4), used to detoxify a wide range of substances [40]. Treatment with PXR ligands revealed a clear species difference between WT and hPXR mice in their response to xenobiotics, suggesting that this BAC transgenic hPXR mouse model is useful for investigating human PXR function in vivo [40]. Indeed, recent reports have demonstrated the utility of this whole body hPXR mouse model in exploring the xenobiotic and endobiotic role of PXR [40,41,42,43,44,45]. Our group and others have shown that the male mouse PXR (mPXR) promotes obesity, however, whether PXR regulates obesity with sexual dimorphism remains unknown [45,46]. Moreover, the effect of the human PXR gene on ER- and diet-induced obesity in a mouse model requires investigation. At the same time, gender-based obesity research has lacked a valid animal model that fully recapitulates the human disease.
To resolve discrepancies in the gender-specific molecular pathology of obesity between rodents and humans, female WT and humanized PXR transgenic mice were investigated with HFD treatment. Our results indicate that the human PXR gene when expressed transgenically in female mice produces an animal model that becomes rapidly obese, glucose intolerant, diabetic, and hyperinsulinemic when exposed to a HFD. These results suggest that human PXR promotes obesity in female mice and that PXR-humanized mice will serve as a valuable in vivo model for obesity research in humans.
2. Materials and Methods
2.1. Animals
Breeding-pairs of adult male and female C57BL/6 (WT) mice were purchased from Jackson Laboratories (Bar Harbor, ME). All mice were housed in polycarbonate cages on racks directly vented via the facility's exhaust system at 22°C with a 12/12-h light/dark cycle at the Animal Resources Complex at North Carolina Central University. Breeding pairs of hPXR mice on a C57BL/6 background were transferred from the colony housed at the National Cancer Institute (NIH, Bethesda, Maryland) [40]. Age-matched female WT and hPXR mice, were each randomly separated at weaning (∼3 weeks old) into two groups (n = 7-11 for each group) and fed with either a control diet (12% of calories as fat; Research Diet catalogue #: D03032702); or a high-fat diet (HFD, 45% of calories from fat; Research Diets catalogue #: D03032705) for 16 weeks. The gross energy density in the control diet and the HFD were calculated to be 3.8 and 4.7 Kcal/g, respectively. Food and water were provided ad libitum throughout the entire feeding period. However, we observed that both the control-and HFD-fed mice were most active and fed more at night. Throughout the experimental period, the mice were housed in groups of 3-5 mice/cage. All diets were prepared by Research Diets Inc. (New Brunswick, NJ) and the detailed diet composition was previously described except that HFD in this study provided 45% of calories from lard and soybean oil, instead of 43% [47]. Furthermore, the HFD and the control chow diets were matched in the type of fat and other nutrients. The food consumption and the change in body weight were monitored weekly. All procedures conducted in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals were approved by the North Carolina Central University Institutional Animal Care and Use Committee. After 16 weeks on the diet, mice were anesthetized with isoflurane and sacrificed in the morning (09:00–11:00 hours) without starvation. Mice were not starved overnight before sacrifice because previous reports indicate that fasting induces PXR expression in the liver [48,49]. Furthermore, a detrimental role of PXR activation by both the human and rodent agonists on glucose tolerance has been reported [50]. After sacrifice, sections of liver, brown adipose tissue (BAT, removed from the back), and WAT were rapidly dissected, weighed, snap-frozen in liquid nitrogen and kept at –80°C. The WAT comprised parametrial, perirenal, and mesenteric fat. Blood samples collected by cardiac puncture were centrifuged at 3000 rpm for 15 min to collect serum to determine liver enzymes, triglycerides and cholesterol, glucose, 17β-estradiol, leptin, and insulin concentrations.
2.2. Serum alanine aminotransferase (ALT), cholesterol, triglyceride, glucose, insulin, leptin, and 17β-estradiol measurements
Blood was collected by cardiac puncture and serum processed and stored at -80°C. Serum ALT activity and glucose levels were determined with the ALT/AST (parts # 12-788) and Lipid profile-GLU (parts # 10-991) Test Cassettes, respectively, using the Cholestech LDX analyzer (Cholestech Corporation, Hayward, CA) [51]. Serum leptin (catalog # 90030) and insulin (catalog # 90080) levels were determined using enzyme-linked immunosorbent assay (ELISA) kits according to protocols provided by the kit manufacturers (Crystal Chem Inc., Dowers Grove, IL). Serum triglyceride and cholesterol levels were quantified using a Triglyceride (catalog #s, 461-08992, 461-09092, and 461-01601) and Cholesterol (catalog # 439-17501) test kits (Wako pure Chemical Industries, Richmond,VA). Serum levels of 17β-estradiol were determined using an ELISA kit (catalog # 582251, Cayman Chemical, Ann Arbor, MI).
2.3. Hepatic triglyceride, cholesterol, and nonesterified free fatty acid (NEFA) levels
Total liver lipids were extracted from 100 mg of liver homogenate using methanol and chloroform as previously described [52]. Hepatic triglyceride, cholesterol, and NEFA levels were quantified using a Triglyceride, Cholesterol, and the HR series NEFA-HR (2) (Catalog #s 999-34691, 995-34791, 991-34891, 993-35191, and 276-76491) test kits (Wako pure Chemical Industries, Richmond, VA).
2.4. Glucose tolerance test (GTT)
GTT were performed on WT and hPXR mice (n = 6/group) on the control diet or HFD for 15 weeks using females. Glucose tolerance tests were conducted in 12-15 hours fasting mice, before a single dose of D-glucose (10% solution in water, 10 mL/kg) was intraperitoneally (i.p.) injected. Glucose levels were measured from blood collected from the tail vein immediately before and 15, 30, 60, 90, and 120 min after the injections using Contour TS blood glucose meter (Bayer HealthCare LLC, Mishawaka, IN), an instrument that reads blood glucose levels.
2.5. Preparation of liver and white adipose tissue (WAT) extracts for Western blot analyses
Frozen livers, were homogenized at 4°C and then subjected to Western blot analysis as described previously [53]. Frozen WAT (500 mg of parametrial fat) were homogenized at 4°C, followed by centrifugation at 14,000 rpm for 20 min as we described previously [45]. The supernatant was collected as the cytosolic fraction. The liver homogenate or cytosolic fraction (30-40 μg/lane), were mixed in Laemmli loading buffer containing β-mercaptoethanol, boiled for 5 min, and were separated by 10 or 15% SDS-PAGE gels, and transferred to polyvinylidene difluoride (PVDF) membrane. The membranes were probed overnight with rabbit polyclonal primary antibodies from Abcam Inc. (Cambridge, MA) at a concentration of 1:1000 for anti-CYP2E1 (ab28146), anti-glucokinase (ab37796), anti-glucose 6-phosphatase (G6Pase, ab83690), anti-UCP1 (ab10983) or anti-PEPCK1 (ab70358). Primary antibodies from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA) were used at a concentration of 1:200 for rabbit polyclonal anti-ERα (Sc-542), rabbit polyclonal anti-ERβ (Sc-8974), or goat polyclonal anti-CYP3A (Sc-30621). Blots were then incubated with the appropriate peroxidase-conjugated anti-rabbit IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) secondary antibodies diluted in TBST plus 1% milk for 60 min at room temperature. Following probing, the blots were stripped and reprobed with anti-α-tubulin antibody (Cell Signaling Technology, Danvers, MA) at a concentration of 1:1000. Proteins were viewed using enhanced chemiluminescence. The intensities of the bands were quantified using ImageJ software (U. S. National Institutes of Health, Bethesda, MD). Protein contents in the liver homogenate and cytosolic fractions prepared from the WAT were determined by the BCA™ protein assay kit (Thermo Scientific, Rockford, IL).
2.6. Statistical Analysis
Data are presented as means ± SEM (n = 7-11). Statistical analysis was performed using one-way ANOVA followed by Bonferroni post-hoc test. A P value of < 0.05 was considered statistically significant. The analyses were performed with the statistical software IBM SPSS Statistics 20 (Armonk, NY).
3. Results
3.1 The hPXR gene predisposes female mice to high fat diet-induced obesity
We characterized the role of the human PXR gene in a widely studied HFD-induced model of obesity using female WT and hPXR mice. While the effect of HFD on weight gain in WT mice was not apparent until after 6 weeks (Fig. 1a), body weight differences between hPXR mice fed control diet and those fed a HFD were statistically significant as early as 2 weeks from the start of their respective diets (Fig. 1a). Within this same timeframe (2 weeks), significant body weight differences were also observed between the hPXR and WT genotypes receiving a HFD; these genotype differences in response to HFD persisted until the end of the study at 16 weeks (Fig. 1a). By the end of the study period, HFD-fed hPXR mice had gained an average of 30.3 g, while HFD-fed WT mice gained only 20.3 g (Fig. 1b). This difference in weight gain between the two genotypes did not occur among animals fed control chow (Fig.1a and b).
Fig. 1.

Body weight, weight gain, and average food intake of mice during a 16 week control diet or a high-fat diet (HFD) regimen. Female wild type (WT) and hPXR mice were fed a control or a HFD for 16 weeks. Body weight was recorded weekly. The gross energy density in the control diet and the HFD were calculated to be 3.8 and 4.7 Kcal/g, respectively. Time-dependent weight gain in mice maintained on control or a HFD (a); total weight gain (b); and average food intake (c). Data represent mean ± SEM (n = 7-11). #P < 0.05 between mice fed control and HFD. †P < 0.05 between WT mice fed a HFD and hPXR mice fed a HFD.
Consonant with trends in weight gain, animals of both genotypes ate more on a HFD than on a normal diet (Fig. 1c). Also consistent with weight gain patterns, there was no difference in daily food intake between genotypes on the control diet. Curiously, however, even though HFD-fed hPXR animals gained more weight than their WT counterparts, this weight gain came without an increase in food intake by hPXR mice (Fig. 1c).
HFD ingestion increased parametrial and perirenal fat pads to the same extent in both WT and hPXR mice (Fig 2a and b). In contrast, the increase in mesenteric fat pads by HFD ingestion was higher in hPXR mice (410%) than in WT mice (230%, Fig. 2c). Furthermore, HFD ingestion increased the weight of the BAT to a greater extent in hPXR mice (312%) than in WT mice (224%, Fig. 2d). HFD ingestion also increased liver weight only in hPXR (180%) mice (Fig 2e). Taken together, these data indicate that the human PXR gene rapidly predisposes female mice to HFD-induced obesity and imply that PXR-dependent weight gain occurs in mesenteric fat, BAT, and liver but does not result from increased food intake.
Fig. 2.

Adipose tissue and liver weights in female wild type (WT) and PXR-humanized (hPXR) transgenic mice fed control or a high-fat diet (HFD) for 16 weeks. After 16 weeks on the diet, mice were sacrificed without starvation and sections of white adipose tissue (WAT) comprising [parametrial fat (a), perirenal fat (b), and mesenteric fat (c)], brown adipose tissue (BAT, removed from the back) (d), and liver (e) were rapidly dissected and weighed. Data represent mean ± SEM (n = 7-11). #P < 0.05 between mice fed control and HFD. †P < 0.05 between WT mice fed a HFD and hPXR mice fed a HFD.
3.2 Basal uncoupling protein 1 (UCP1) protein expression is elevated, but suppressed by HFD in WAT of hPXR mice
To attempt to define the cause of severe obesity in hPXR mice upon HFD ingestion, we examined the expression of the BAT specific-protein, UCP1. Previous reports indicate that induction of UCP1 in WAT is effective at reducing adiposity, insulin resistance, and obesity [54,55,56]. Western blot analysis indicated that the UCP1 protein was barely detectable in WAT of WT mice fed the control diet (Fig 3a). However, unexpectedly, a strong up-regulation (5.4-fold) of the UCP1 protein in WAT of control chow-fed hPXR mice compared to WT mice fed the same diet was observed (Fig 3a). Most interestingly, while UCP1 protein was induced in WT mice (2.5-fold), the UCP1 protein was almost completely inhibited in hPXR mice, when both WT and hPXR mice were placed on a HFD (Fig. 3a). We also explored the association between mouse vs. human PXRs and WAT ERα levels using Western blot analysis. Among control diet-fed animals, WAT ERα expression was markedly decreased (15% of WT mice on control diet) in the hPXR mice (Fig. 3b). Further, HFD feeding significantly decreased ERα protein levels in WT mice (24% of chow-fed animals) (Fig. 3b). Somewhat surprisingly, HFD ERα protein levels in hPXR mice were unresponsive to HFD feeding (Fig. 3b). We also examined ERβ protein levels in WAT. Unexpectedly, ERβ protein levels did not differ significantly among control-fed or HFD-fed mice of either genotype (Fig. 3c). Although hPXR animals may have had slightly reduced ERβ expression levels, these differences were not statistically significant.
Fig. 3.

Immunoblot analysis of white adipose tissue (WAT) uncoupling protein 1 (UCP1), estrogen receptor α (ERα), and (ERβ) in female wild type (WT) and PXR-humanized (hPXR) transgenic mice fed control diet or a HFD for 16 weeks. Cytosolic fractions prepared from frozen WAT (500 mg of parametrial fat) (30 μg/lane) of mice fed a control diet or a HFD were electrophoresed on 10 or 15% SDS-polyacrylamide, transferred to nitrocellulose membranes, and incubated with anti-UCP1 (a), anti-ERα (b), and anti-ERβ (c) antibody as indicated in representative blots. Quantitative analysis of UCP1, ERα, and ERβ levels plotted after normalizing with α-tubulin in WT and hPXR mice fed control diet (□) or a HFD (■) for 16 weeks. The intensities of the bands were quantified using ImageJ software (U. S. National Institutes of Health, Bethesda, MD, USA). Data represent mean ± SEM of 2 independent experiments from 3-4 mice/group. *P < 0.05 between WT mice fed control diet and hPXR mice fed the control diet. #P < 0.05 between mice fed control and HFD.
3.3 The hPXR mice exhibit hyperinsulinemia under basal conditions
Levels of serum 17β-estradiol tended to be lower in control chow-fed hPXR mice (68%), compared with WT controls; but the difference failed to reach statistical significance (Fig. 4a). However, the levels of serum 17β-estradiol were significantly reduced only in HFD-fed WT mice (30% of control-fed WT mice) (Fig. 4a). While serum insulin levels remained unchanged in HFD-fed WT mice, the levels were increased 4.7-fold in hPXR mice fed a HFD (Fig. 4b). Serum insulin levels in control-fed hPXR mice were elevated (2.0-fold) (Fig. 4b). Furthermore, HFD feeding elevated fasting blood glucose levels in both WT and hPXR mice (data not shown). Serum levels of alanine aminotransferase (ALT, a marker of liver injury) increased after HFD ingestion by 1.5- and 1.6-fold in WT and hPXR mice, respectively (Fig. 4c). Basal serum cholesterol levels were elevated (21%) in hPXR mice fed the control chow diet, compared to control chow-fed WT mice (Fig. 4d). As expected, HFD feeding increased serum cholesterol levels in both mice lines where the increase in serum cholesterol levels by HFD in hPXR mice (44%) was significantly higher than that in HFD-fed WT mice (32%) (Fig. 4d). In contrast, HFD increased serum triglycerides by 36% in hPXR mice alone (Fig. 4e). HFD-feeding increased leptin levels in WT (4.0-fold) and hPXR (3.0-fold) mice above the levels found in control-fed mice (Fig. 4f). We also noted that basal (control diet) serum leptin levels were higher in hPXR mice (1.5-fold) than in WT mice, but these differences were not statistically significant (Fig. 4f).
Fig. 4.

Serum 17β-estradiol, insulin, alanine aminotransferase (ALT), cholesterol, and triglyceride, and leptin levels in mice fed control diet or high-fat diet (HFD). Female wild type (WT) and PXR-humanized (hPXR) transgenic mice were fed control diet (□) or a HFD (■) for 16 weeks. 17β-estradiol (a), insulin (b), ALT (c), cholesterol (d), triglyceride (e), and leptin (f) levels in serum were determined as described in Materials and Methods. Data represents means ± SEM (n = 5-6). *P < 0.05 between WT mice fed control diet and hPXR mice fed the control diet. #P < 0.05 between mice fed control and HFD. †P < 0.05 between WT mice fed a HFD and hPXR mice fed a HFD.
3.4 The basal hepatic protein levels of CYP3A11 are up-regulated while ERα are suppressed in hPXR mice
In this study, HFD ingestion decreased serum 17β-estradiol levels in WT mice (Fig. 4a). Among the cytochrome P450 enzyme system, the current study focused only on CYP3A11 and CYP2E1 based on previous reports indicating that CYP3A, a PXR target gene, can metabolize steroid hormones including estrogen, while CYP2E1 participates in oxidation of a variety of xenobiotic compounds [57,58]. Furthermore, the biological functions of estrogen are mediated by binding to its receptors including ERα [59]. Western blot analyses were used to examine hepatic CYP3A11, CYP2E1 and ERα expression. Basal hepatic CYP3A11 protein levels were increased in hPXR (1.8-fold) mice (Fig. 5a). HFD ingestion did not increase CYP3A11 protein levels in hPXR mice, however, it increased the levels in WT (1.5-fold) mice (Fig 5a). CYP2E1 expression in hPXR animals on either diet was not different compared to that in control-fed WT mice. HFD feeding increased CYP2E1 protein levels in WT (2.6-fold) mice and the increase was significantly higher than in HFD-fed hPXR mice (Fig. 5b). The basal hepatic ERα protein levels were decreased by 57% in hPXR mice, compared to control chow-fed WT mice (Fig. 5c). HFD did not have any significant effect on hepatic ERα expression in both genotypes (Fig. 5c).
Fig. 5.

Immunoblot analysis of hepatic cytochrome P450 (CYP) 3A11, CYP2E1, and estrogen receptor α (ERα) in female wild type (WT) and PXR-humanized (hPXR) transgenic mice fed control diet or a HFD for 16 weeks. Liver homogenate (40 μg/lane) prepared from mice fed control diet or a HFD were electrophoresed on 10% SDS-polyacrylamide, transferred to nitrocellulose membranes, and incubated with anti-CYP3A11 (a), anti-CYP2E1 (b) or anti-ERα (c), as indicated in representative blots. Quantitative analysis of CYP3A11, CYP2E1, and ERα protein levels plotted after normalizing with α-tubulin in WT and hPXR mice fed control diet (□) or a HFD (■) for 16 weeks. The intensities of the bands were quantified using ImageJ software (U. S. National Institutes of Health, Bethesda, MD, USA). Data represent mean ± SEM of 2 independent experiments from 3-4 mice/group. *P < 0.05 between WT mice fed control diet and hPXR mice fed the control diet. #P < 0.05 between mice fed control and HFD. †P < 0.05 between WT mice fed a HFD and hPXR mice fed a HFD.
3.5 The human PXR gene disrupts hepatic cholesterol homeostasis
Previous reports have shown that elevated levels of NEFA in the liver is a stimulus for gluconeogenesis [60]. It is interesting to note that both hepatic cholesterol and NEFA levels were elevated in hPXR mice on either diet (Fig. 6a and c), in contrast to the increase in liver weight seen only in HFD-fed hPXR females (Fig. 2e). Both genotypes had increases in hepatic triglyceride levels on a HFD, which were more pronounced in hPXR mice (Fig. 6b).
Fig. 6.
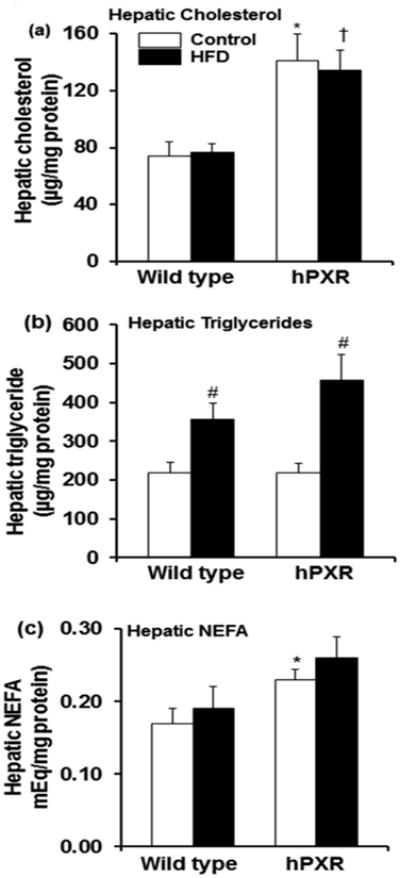
Hepatic cholesterol, triglyceride and nonesterified fatty acid (NEFA) levels in female wild type (WT) and PXR-humanized (hPXR) transgenic mice fed control or a high-fat diet (HFD) diet for 16 weeks. Hepatic cholesterol levels (a), hepatic triglyceride levels (b), and hepatic NEFA levels (c), from different treatment groups were performed as described in Materials and Methods. Data represents means ± SEM (n = 5-6). *P < 0.05 between WT mice fed control diet and hPXR mice fed the control diet. #P < 0.05 between mice fed control and HFD. †P < 0.05 between WT mice fed a HFD and hPXR mice fed a HFD.
3.6 Glucose tolerance is impaired in both HFD-fed WT and hPXR mice
To establish whether glucose clearance varies between females that possess mouse vs. human PXRs, we performed intraperitoneal glucose tolerance tests (IPGTT) on the control- and HFD-fed PXR genotypes. Serum glucose levels peaked at 15 minutes after glucose challenge in both WT and hPXR mice fed the control diet (Fig. 7a). WT mice fed a HFD cleared systemic glucose spikes more efficiently and more rapidly than their hPXR counterparts (Fig. 7a and b) did. Glucose elimination was severely impaired in HFD-fed hPXR mice and the total systemic glucose [represented by the area under the curve (AUC)] significantly increased by 121%, as the result of excess dietary lipids, indicative of glucose intolerance (Fig. 7a and b). It was also noted that the AUC in HFD-fed WT animals significantly increased by 67% compared to their control-fed counterparts (Fig. 7a and b).
Fig. 7.

Blood glucose levels during glucose tolerance test (GTT) in mice fed control or HFD. Female wild type (WT) and PXR-humanized (hPXR) transgenic mice were fed control diet or a HFD for 16 weeks. GTT was performed after mice (WT and hPXR) had been on the control diet or HFD for 15 weeks (n = 6/group). Intraperitoneal glucose tolerance test (IPGTT) was conducted in overnight feed-deprived conditions (12-15 hours of starvation) and mice were intraperitoneally (i.p.) injected a single dose of D-glucose (10% solution in water, 10 mL/kg). Glucose levels were measured from blood collected from the tail vein immediately before and after 15, 30, 60, 90, and 120 minutes using Contour TS blood glucose meter (Bayer HealthCare LLC, Mishawaka, IN), an instrument that reads blood glucose levels (a). Area under the curve (AUC) for the GTT were calculated using Sigma Plot 12.0 (Systat Software Inc, San Jose, CA), to estimate glucose tolerance in corresponding mice (b). #P < 0.05 between mice fed control and HFD. †P < 0.05 between WT mice fed a HFD and hPXR mice fed a HFD.
3.7 Gluconeogenic enzymes PEPCK1 and G6Pase protein are constitutively activated in hPXR mice
Western blot analyses were employed to establish whether the hyperglycemia and severely impaired glucose tolerance observed in HFD-fed hPXR mice resulted from altered expression of liver gluconeogenic enzymes such as PEPCK1 and G6Pase and glucokinase involved in glycolysis [61]. Hepatic PEPCK1 protein levels in hPXR mice fed the control diet were increased 5.0 fold compared with WT mice on the same diet (Fig. 8a). However, HFD feeding did not, further increase the high basal PEPCK1 protein levels in hPXR mice, but did enhance PEPCK1 protein expression in WT mice (3.1-fold), but significantly lower than the level found in HFD-fed hPXR mice (Fig. 8a). The basal protein levels of G6Pase, were increased in control chow-fed hPXR (2.9-fold) mice (Fig. 8b). HFD suppressed G6Pase protein levels in WT mice by 47%, but not in HFD-fed hPXR mice and G6Pase levels remained significantly higher than in HFD-fed WT mice (Fig. 8b). HFD ingestion increased the glucokinase protein in WT (2.1-fold) mice, however, it inhibited the glucokinase protein by 64% in hPXR mice (Fig. 8c).
Fig. 8.

Immunoblot analysis of hepatic PEPCK1, glucose 6-phosphatase (G6Pase), and glucokinase levels in female wild type (WT) and PXR-humanized (hPXR) transgenic mice fed control diet (□) or a HFD (■) for 16 weeks. Liver homogenate (40 μg/lane) from mice fed control diet or a HFD were electrophoresed on 10% SDS-polyacrylamide, transferred to nitrocellulose membranes, and incubated with anti-PEPCK1, anti-G6Pase, or anti-glucokinase antibody as indicated in representative blots (a, b and c). Quantitative analysis of PEPCK1 protein after normalizing with α-tubulin (a), G6Pase protein after normalizing with α-tubulin (b) and glucokinase protein after normalizing with α-tubulin (c) in WT and hPXR mice fed control diet or a HFD for 16 weeks. The intensities of the bands were quantified using ImageJ software (U. S. National Institutes of Health, Bethesda, MD). Data represent mean ± SEM of 2 independent experiments from 3-4 mice/group. *P < 0.05 between WT mice fed control diet and hPXR mice fed the control diet. #P < 0.05 between mice fed control and HFD. †P < 0.05 between WT mice fed a HFD and hPXR mice fed a HFD.
Discussion
The major finding of this study is that the human, but not mouse, PXR gene significantly potentiated HFD-induced obesity, glucose intolerance, and insulin resistance. Our data also indicate that the increased sensitivity of female hPXR mice to HFD-induced obesity was not the result of increased food intake relative to their HFD-fed WT counterparts, but must be due to the function of the PXR gene alone and in combination with exposure to HFD. Specifically, while both HFD-fed WT and hPXR mice were hyperphagic in comparison with control chow-fed mice, there was no difference in food intake between the two HFD-fed genotypes. An obesity metabolic phenotype occurs in HFD-fed hPXR mice, which was associated with decreased basal protein levels of WAT ERα, but increased expression of the hepatic gluconeogenic enzymes PEPCK1 and G6Pase, compared to levels of these enzymes in WT mice fed the control diet. Interestingly, two of these phenomena, reduced WAT ERα protein expression and enhanced hepatic PEPCK1 protein expression were also present in the HFD-induced obesity and a type 2 diabetic profile in our female WT mice. Unexpectedly, it was observed that hPXR mice possess increased basal expression of the UCP1 in WAT compared to WT mice. The most important difference between these two genetic strains, after HFD ingestion, is inhibition of WAT UCP1 and hepatic glucokinase protein in hPXR mice leading to robust obesity and severe glucose intolerance, in contrast to induction of both UCP1 and glucokinase protein in WT mice, probably leading to increased energy expenditure and some protection against diet-induced obesity in female rodents.
The increased health risks associated with obesity vary depending on the amount and location of adipose tissue [23,24,25]. Adipose tissue exists in two forms, WAT and BAT. WAT stores excess energy in the form of triglycerides and releases molecular signals such as leptin to inform the body of its overall energy state, while the main function of BAT is to regulate thermogenesis, producing heat instead of ATP [62,63,64]. WAT distributed in the abdominal viscera is associated with a greater risk of metabolic disorders [23,24,25]. Thus, the size of the different intra-abdominal WAT depots and BAT in both control chow-fed and HFD-fed WT and hPXR mice was investigated in this study. As expected, HFD promoted an increase in weight of fat pads in both WT and hPXR mice, resulting in a significant increase in body weight compared with mice fed the control diet. However, compared with WT mice fed a HFD, fat accumulation in WAT, especially mesenteric WAT and BAT, were significantly higher in HFD-fed hPXR mice, and not an excess buildup in parametrial and perirenal fat. Interestingly, the selective deposition of fat in the mesentery in the hPXR mice models a common form of human obesity, since majority of the increased fat mass in diabetic, obese humans is found in the mesentery [24,65]. Liver weight was also higher in HFD-hPXR mice compared with WT mice fed the same diet. Obesity is also prevalent in children, with more than 17 percent of United States children and adolescents classified as obese [2]. Obesity among children is a significant risk factor for type 2 diabetes and insulin resistance [66]. The importance of developmental origins of adult diseases as a contributor to the epidemic of obesity has been recognized [67]. Interestingly, reports indicate that offspring exposed to maternal obesity were at increased risk of developing components of metabolic syndrome (obesity, dyslipidemia, insulin resistance and hypertension) supporting the concept of fetal programming [67,68]. Thus, it will be of interest to examine the development of metabolic syndrome in the offspring of hPXR dams that were fed a HFD during pregnancy compared with dams that received control chow diets. Overall, our data indicate that increased fat mass together with enlarged organs may contribute to the development of increased body weight in hPXR mice.
BAT is a major site involved in energy dissipation and the protein responsible for BAT thermogenesis is UCP1 [64]. UCP1 expression does not normally occur in muscle or white fat, however, previous reports indicate that ectopic UCP1 expression in muscle or WAT causes an increase in energy expenditure and prevents obesity [54,55,56]. Previous reports have shown that overexpression of UCP1 reduces weight gain in obesity prone-mice [56,69,70]. Moreover, the ability of the A/J mice to resist diet-induced obesity compared to the obesity-prone C57BL/6J mice is associated with a strain-specific increase in UCP1 expression in adipose tissue [70]. Unexpectedly, we observed that UCP1 protein was strongly induced in parametrial WAT of control chow-fed hPXR mice, but barely detectable in WT mice. Interestingly, while HFD ingestion induced UCP1 protein expression in WT mice, in contrast, it completely obliterates UCP1 protein expression in hPXR mice. Thus, the failure to upregulate or maintain WAT UCP1 expression to promote thermogenesis and increase in energy dissipation, may contribute to lipid accumulation and obesity in HFD-fed hPXR mice. Conversely, induction of WAT UCP1 protein expression in WT mice will increase thermogenesis capacity in WAT, promote energy dissipation and suppress obesity in WT mice fed the HFD diet as seen in the current study. Although, the reason for the unexpected UCP1 expression in WAT in hPXR mice requires further investigation, it is well established that leptin regulates UCP1 gene [71]. Furthermore, the PPARγ agonist pioglitazone, essential fatty acids and retinoids have been reported to activate UCP1 gene expression [72,73,74,75]. We have recently shown that the basal serum leptin levels are higher in both male PXR-KO and hPXR mice than in WT mice [45]. The current study also observed a slightly higher, though not significantly, basal serum leptin levels in the female hPXR mice than in WT mice. Hence, it is plausible to suggest that the hPXR gene may encode for brown fat cell properties in white fat cells by indirectly increasing leptin secretion.
Evidence indicates functional interactions between sex steroids and insulin in several tissues [76]. Previous reports have shown that the deficiency of estrogens or estrogen receptors results in visceral fat accumulation, impairment of insulin sensitivity and lipid metabolism, hypercholesterolemia, and cardiovascular disease risk [77,78]. Furthermore, estrogen withdrawal causes obesity and increased appetite in rodents [79]. In this study, both hPXR mice and WT mice fed a HFD were hyperphagic and obese, compared to their respective chow-fed littermates. The hPXR mice on the control chow diet also exhibited hypercholesterolemia, an important risk factor associated with estrogen deficiency [77,80,81]. In mice, the estrous cycle, an indicator of reproductive function, is 4-5 days with four stages, (proestrus, estrus, diestrus and metaestrus) and is associated with significant variation in the serum estrogen concentrations [82,83]. Interestingly, disruption of the estrous cycle occurs in female obese rodents [84,85]. Reports indicate that total energy intake of female rodents decrease when 17β-estradiol levels are high (in estrus) and increases when estradiol levels are low (in diestrus) [86,87]. In this study, serum 17β-estradiol levels were also measured, however, the mice we were not sacrificed based on the stage of the estrous cycle. While 17β-estradiol concentrations in hPXR mice fed control diet tended to be lower, significantly reduced levels of 17β-estradiol were observed only in HFD-fed WT mice. Thus, although our mice may be at different stages of estrous at sacrifice, it is possible to suggest that the phenotype observed in both female WT and hPXR mice fed a HFD may be due to alterations in the estrogen/ER signaling.
Two major ERs, alpha and beta (ERα and ERβ), mediate the physiological signaling of estrogens (17β-estradiol) [59]. We therefore examined ERα and/or ERβ expression in WAT and liver of both hPXR and WT mice. Unexpectedly, the basal ERα expression in WAT and liver were markedly lower in hPXR mice fed the control chow diet compared with WT mice fed this diet. ERα-knockout mice and aromatase-knockout mice, which are unable to synthesize estrogen, both exhibit adipocyte hyperplasia and obesity [77,88]. Interestingly, like the ERα-knockout mice, hPXR mice on a control chow diet possessed elevated fasting blood glucose (data not shown), serum insulin and hepatic cholesterol levels [77,80]. In contrast, ERβ-knockout mice have normal fasting blood glucose, glucose tolerance, and cholesterol levels, suggesting that ERα, but not ERβ, has a protective role in metabolic disorders [89]. Intriguingly, hPXR animals expressed only low WAT ERα levels but had normal ERβ levels. These findings strongly suggest that the increase in WAT accumulation and obesity in HFD-fed hPXR females might result from decreased WAT ERα expression. Along these same lines, the moderate obesity observed in HFD-fed WT mice also corresponded with decreased WAT ERα protein levels, suggesting that ERα signaling is a major factor involved in HFD-induced obesity and diabetes in female mice of both genotypes. Together, an intriguing finding here is that resistance to diet-induced obesity in female non-ovariectomized rodents may not be solely due to the ERα, but instead may be due to induction of WAT UCP1 protein by HFD.
Our results resemble the metabolic status of humans who lack ERα and are insulin resistant [90]. In premenopausal females, ovaries are the principal site for the biosynthesis of estrogen and the major contributors to systemic estrogen levels [91]. However, extragonadal tissues, including adipose tissue, become important as the organism ages for the maintenance of adequate local estrogen levels [91]. Since hPXR female mice are fertile (and therefore possess functional ovaries), it is likely that the suppressive effects of hPXR on estrogen/ER signaling observed in these mice involves only peripheral tissues capable of estrogen biosynthesis such as WAT.
PXR mediates the genomic effects of several steroid hormones including estrogen, progesterone, pregnenolone, and glucocorticoids in the mouse, rat, and human [92,93]. Besides liver and intestine, PXR expression occurs in reproductive organs including ovary, uterus, and placenta, and PXR mRNA levels are significantly increased during pregnancy in these organs [58,94]. Moreover, PXR ligands such as endogenous steroids including estrogen, progesterone, and pregnenolone activate PXR-mediated transcriptional activity. Once activated, PXR binds to specific DNA sequences as a heterodimer with the retinoid X receptor (RXR) to promote xenobiotic metabolism and elimination [37,93,94]. In contrast, ligand-bound ERα either binds to DNA directly as a homodimer or interacts indirectly with DNA by tethering to other transcriptional factors in estrogen-responsive target genes [95]. Interestingly, ligand binding induces conformational changes in both hPXR and ERα, leading to the recruitment of co-activators or co-repressors that ultimately determine biological activity [96]. Previous reports indicate that xenobiotic nuclear receptors such as hPXR share a similar pool of co-activators with the ERs, providing a platform for inhibitory “cross-talk” between nuclear receptors [97,98]. In this respect, recent studies have demonstrated that ER-mediated transcriptional activity influences other nuclear receptors [30,31,32]. In this study hepatic protein expression of CYP3A, a PXR target gene was elevated in control chow-fed hPXR mice, suggesting that PXR is activated in these mice [58]. Whether the decreased WAT and hepatic ERα expression observed in hPXR mice is due to suppression of or competition with ERα by overexpressed and activated hPXR or the involvement of a distinct nuclear receptor co-activator/co-repressor of an hPXR-ERα interaction remains to be determined. In this study, the basal hepatic ERα protein levels were decreased in the livers of hPXR mice, and since adipose tissue does not express hPXR, it is possible that the decreased WAT ERα is secondary to the liver effect. Interestingly, CAR, a sister xenobiotic hormone receptor of PXR which is not expressed in adipose tissue, is also able to transduce its metabolic effects in both WAT and BAT [99]. Thus, both hPXR-induced and HFD-induced suppression of ERα protein expression in WAT and liver cause obesity and glucose intolerance in both hPXR and WT mice fed a HFD.
Excessive glucose production by the liver contributes significantly to diabetic hyperglycemia. Mounting evidence indicates that PXR is capable of down-regulating the expression of gluconeogenic genes [34,35,36]. However, the present study demonstrates that hPXR impairs glucose tolerance in the presence of high serum lipids, raising doubts about the anti-diabetogenic actions of PXR [35,36,99]. The current data indicate that induction of the key hepatic gluconeogenic enzymes, PEPCK1 and G6Pase, in hPXR mice is independent of their dietary regimen. Furthermore, HFD-fed hPXR mice became more glucose intolerant than HFD-fed WT mice. HFD-fed hPXR mice also exhibited decreased insulin sensitivity. Supportive of these findings is a recent report indicating a detrimental role of PXR activation by both the human and rodent agonists on postprandial glucose tolerance [50]. In the liver, glucokinase determines the rate of glucose utilization and glycogen synthesis by catalyzing the phosphorylation of glucose to glucose-6-phosphate [100]. Moreover, glucokinase-deficient mice die with severe hyperglycemia [101]. In this study, HFD increased glucokinase protein expression while inhibiting that of G6Pase in WT mice. In contrast, HFD-induced increases in glucokinase protein levels were inhibited while maintaining the elevated G6Pase protein levels in hPXR mice. We speculate that the hyperglycemia and impaired glucose tolerance seen in HFD-fed hPXR mice were due to reduced levels of glucokinase and lost capacity to use or store glucose in the livers of these mice. Together, these data suggest that much of the insulin resistance, hyperglycemia, and impaired glucose tolerance in HFD-fed hPXR mice can be accounted for by 1) overexpression of PEPCK1, 2) marked induction of G6Pase protein, 3) downregulation of glucokinase, and 4) defective inhibition of PEPCK1 expression by insulin or hPXR.
The current study reveals that female hPXR mice fed a HFD constitute a suitable in vivo rodent model to elucidate the molecular mechanisms linking HFD and impaired insulin signaling in women. Our collective observations suggest that the hPXR gene directly or indirectly promotes weight gain and aggravates diet-induced obesity associated with type 2 diabetes in females. Importantly, these findings are in agreement with a recent report by our group and others indicating that ablation of the PXR gene in male mice protects against obesity [45,46]. Considered together, both our previous and current reports indicate that the hPXR genotype is associated with the development of HFD-induced hyperglycemia in both genders [45].
In summary, this report demonstrates that female hPXR mice have markedly reduced basal WAT and hepatic ERα levels but elevated WAT UCP1, hepatic cholesterol, CYP3A11, PEPCK1, and G6Pase protein levels. When subjected to a HFD, hPXR mice display exacerbated diet-induced obesity, hyperinsulinemia, and severely impaired glucose tolerance compared to WT mice subjected to similar diet regimen. Together, these data provide the first evidence that the hPXR genetically engineered in female mice positively regulates obesity, insulin signaling, and glucose metabolism. The impairment of ERα expression in WAT by hPXR and HFD-induced inhibition of both UCP1 in WAT and hepatic glucokinase induction resulting in obesity makes a significant contribution to our understanding of the discrepancy between diet-induced obesity associated metabolic disease in female rodents and women.
Acknowledgments
The authors are grateful to Drs. Thomas Pazdernik and Susan T. Yeyeodu for critical reading of the manuscript. This work was supported by the National Institutes of Health (NIH) grants U54 AA019765, SC1 HL099139, P20 MD000175, RO1 HL064761, and R25HL059868 and the National Cancer Institute Intramural Research Program, NIH.
Abbreviations
- HFD
high-fat diet
- PXR
pregnane X receptor
- mPXR
mouse PXR
- hPXR
PXR-humanized or human PXR
- WT
wild type
- WAT
white adipose tissue
- BAT
brown adipose tissue
- ALT
alanine transaminase
- PPARs
peroxisome proliferator-activated receptors
- LBD
ligand binding domain
- H&E
hematoxylin and eosin
- GTT
Glucose tolerance test
- IPGTT
intraperitoneal glucose tolerance test
- CYP2E1
cytochrome P450 2E1
- CYP3A11
cytochrome P450 3A11
- AUC
area under the curve
- PEPCK1
phosphoenolpyruvate carboxykinase 1
- PCN
pregnenolone 16α-carbonitrile
- UCP1
uncoupling protein 1
- ERα
estrogen receptor α
- ERβ
estrogen receptor β
- CAR
constitutive androstane receptor
- NEFA
nonesterified free fatty acid
- G6Pase
glucose 6-phosphatase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.James WP. The epidemiology of obesity: the size of the problem. J Intern Med. 2008;263:336–352. doi: 10.1111/j.1365-2796.2008.01922.x. [DOI] [PubMed] [Google Scholar]
- 2.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311:806–814. doi: 10.1001/jama.2014.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumanyika SK. Obesity in minority populations: an epidemiologic assessment. Obes Res. 1994;2:166–182. doi: 10.1002/j.1550-8528.1994.tb00644.x. [DOI] [PubMed] [Google Scholar]
- 4.Bouchard C, Tremblay A, Despres JP, Nadeau A, Lupien PJ, Theriault G, Dussault J, Moorjani S, Pinault S, Fournier G. The response to long-term overfeeding in identical twins. N Engl J Med. 1990;322:1477–1482. doi: 10.1056/NEJM199005243222101. [DOI] [PubMed] [Google Scholar]
- 5.Reid JM, Fullmer SD, Pettigrew KD, Burch TA, Bennett PH, Miller M, Whedon GD. Nutrient intake of Pima Indian women: relationships to diabetes mellitus and gallbladder disease. Am J Clin Nutr. 1971;24:1281–1289. doi: 10.1093/ajcn/24.10.1281. [DOI] [PubMed] [Google Scholar]
- 6.Schemmel R, Mickelsen O, Gill JL. Dietary obesity in rats: Body weight and body fat accretion in seven strains of rats. J Nutr. 1970;100:1041–1048. doi: 10.1093/jn/100.9.1041. [DOI] [PubMed] [Google Scholar]
- 7.West DB, Boozer CN, Moody DL, Atkinson RL. Dietary obesity in nine inbred mouse strains. Am J Physiol. 1992;262:R1025–1032. doi: 10.1152/ajpregu.1992.262.6.R1025. [DOI] [PubMed] [Google Scholar]
- 8.Almind K, Kahn CR. Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice. Diabetes. 2004;53:3274–3285. doi: 10.2337/diabetes.53.12.3274. [DOI] [PubMed] [Google Scholar]
- 9.Bachmanov AA, Reed DR, Tordoff MG, Price RA, Beauchamp GK. Nutrient preference and diet-induced adiposity in C57BL/6ByJ and 129P3/J mice. Physiol Behav. 2001;72:603–613. doi: 10.1016/s0031-9384(01)00412-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehrabian M, Wen PZ, Fisler J, Davis RC, Lusis AJ. Genetic loci controlling body fat, lipoprotein metabolism, and insulin levels in a multifactorial mouse model. J Clin Invest. 1998;101:2485–2496. doi: 10.1172/JCI1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, Kuhn CM, Rebuffe-Scrive M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism. 1995;44:645–651. doi: 10.1016/0026-0495(95)90123-x. [DOI] [PubMed] [Google Scholar]
- 12.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 13.Astrup A, Buemann B, Western P, Toubro S, Raben A, Christensen NJ. Obesity as an adaptation to a high-fat diet: evidence from a cross-sectional study. Am J Clin Nutr. 1994;59:350–355. doi: 10.1093/ajcn/59.2.350. [DOI] [PubMed] [Google Scholar]
- 14.Bell CG, Walley AJ, Froguel P. The genetics of human obesity. Nat Rev Genet. 2005;6:221–234. doi: 10.1038/nrg1556. [DOI] [PubMed] [Google Scholar]
- 15.Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256:51–54. [PubMed] [Google Scholar]
- 16.Longcope C, Baker R, Johnston CC., Jr Androgen and estrogen metabolism: relationship to obesity. Metabolism. 1986;35:235–237. doi: 10.1016/0026-0495(86)90206-4. [DOI] [PubMed] [Google Scholar]
- 17.Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S. Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun. 2003;300:167–171. doi: 10.1016/s0006-291x(02)02774-2. [DOI] [PubMed] [Google Scholar]
- 18.Kamei Y, Suzuki M, Miyazaki H, Tsuboyama-Kasaoka N, Wu J, Ishimi Y, Ezaki O. Ovariectomy in mice decreases lipid metabolism-related gene expression in adipose tissue and skeletal muscle with increased body fat. J Nutr Sci Vitaminol (Tokyo) 2005;51:110–117. doi: 10.3177/jnsv.51.110. [DOI] [PubMed] [Google Scholar]
- 19.Stubbins RE, Holcomb VB, Hong J, Nunez NP. Estrogen modulates abdominal adiposity and protects female mice from obesity and impaired glucose tolerance. Eur J Nutr. 2012;51:861–870. doi: 10.1007/s00394-011-0266-4. [DOI] [PubMed] [Google Scholar]
- 20.Hill JO, Peters JC. Environmental contributions to the obesity epidemic. Science. 1998;280:1371–1374. doi: 10.1126/science.280.5368.1371. [DOI] [PubMed] [Google Scholar]
- 21.Hong J, Stubbins RE, Smith RR, Harvey AE, Nunez NP. Differential susceptibility to obesity between male, female and ovariectomized female mice. Nutr J. 2009;8:11. doi: 10.1186/1475-2891-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wade GN, Gray JM, Bartness TJ. Gonadal influences on adiposity. Int J Obes. 1985;9 Suppl 1:83–92. [PubMed] [Google Scholar]
- 23.Bjorntorp P. Body fat distribution, insulin resistance, and metabolic diseases. Nutrition. 1997;13:795–803. doi: 10.1016/s0899-9007(97)00191-3. [DOI] [PubMed] [Google Scholar]
- 24.Macotela Y, Boucher J, Tran TT, Kahn CR. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes. 2009;58:803–812. doi: 10.2337/db08-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. 2000;21:697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 26.Gloy V, Langhans W, Hillebrand JJ, Geary N, Asarian L. Ovariectomy and overeating palatable, energy-dense food increase subcutaneous adipose tissue more than intra-abdominal adipose tissue in rats. Biol Sex Differ. 2011;2:6. doi: 10.1186/2042-6410-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lutz TA, Woods SC. Overview of animal models of obesity. Curr Protoc Pharmacol. 2012;Chapter 5 doi: 10.1002/0471141755.ph0561s58. Unit5 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J. Sequence and expression of human estrogen receptor complementary DNA. Science. 1986;231:1150–1154. doi: 10.1126/science.3753802. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 30.Min G. Estrogen modulates transactivations of SXR-mediated liver X receptor response element and CAR-mediated phenobarbital response element in HepG2 cells. Exp Mol Med. 42:731–738. doi: 10.3858/emm.2010.42.11.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Min G, Kim H, Bae Y, Petz L, Kemper JK. Inhibitory cross-talk between estrogen receptor (ER) and constitutively activated androstane receptor (CAR). CAR inhibits ER-mediated signaling pathway by squelching p160 coactivators. J Biol Chem. 2002;277:34626–34633. doi: 10.1074/jbc.M205239200. [DOI] [PubMed] [Google Scholar]
- 32.Nunez SB, Medin JA, Braissant O, Kemp L, Wahli W, Ozato K, Segars JH. Retinoid X receptor and peroxisome proliferator-activated receptor activate an estrogen responsive gene independent of the estrogen receptor. Mol Cell Endocrinol. 1997;127:27–40. doi: 10.1016/s0303-7207(96)03980-9. [DOI] [PubMed] [Google Scholar]
- 33.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 34.Gao J, Xie W. Pregnane X receptor and constitutive androstane receptor at the crossroads of drug metabolism and energy metabolism. Drug Metab Dispos. 2010;38:2091–2095. doi: 10.1124/dmd.110.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol. 2004;24:7931–7940. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kodama S, Moore R, Yamamoto Y, Negishi M. Human nuclear pregnane X receptor cross-talk with CREB to repress cAMP activation of the glucose-6-phosphatase gene. Biochem J. 2007;407:373–381. doi: 10.1042/BJ20070481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blumberg B, Sabbagh W, Jr, Juguilon H, Bolado J, Jr, van Meter CM, Ong ES, Evans RM. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998;12:3195–3205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NC, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- 40.Ma X, Shah Y, Cheung C, Guo GL, Feigenbaum L, Krausz KW, Idle JR, Gonzalez FJ. The PREgnane X receptor gene-humanized mouse: a model for investigating drug-drug interactions mediated by cytochromes P450 3A. Drug Metab Dispos. 2007;35:194–200. doi: 10.1124/dmd.106.012831. [DOI] [PubMed] [Google Scholar]
- 41.Cheng J, Krausz KW, Tanaka N, Gonzalez FJ. Chronic exposure to rifaximin causes hepatic steatosis in pregnane X receptor-humanized mice. Toxicol Sci. 2012;129:456–468. doi: 10.1093/toxsci/kfs211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng J, Ma X, Gonzalez FJ. Pregnane X receptor- and CYP3A4-humanized mouse models and their applications. Br J Pharmacol. 2011;163:461–468. doi: 10.1111/j.1476-5381.2010.01129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng J, Shah YM, Ma X, Pang X, Tanaka T, Kodama T, Krausz KW, Gonzalez FJ. Therapeutic role of rifaximin in inflammatory bowel disease: clinical implication of human pregnane X receptor activation. J Pharmacol Exp Ther. 2010;335:32–41. doi: 10.1124/jpet.110.170225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li F, Lu J, Cheng J, Wang L, Matsubara T, Csanaky IL, Klaassen CD, Gonzalez FJ, Ma X. Human PXR modulates hepatotoxicity associated with rifampicin and isoniazid co-therapy. Nat Med. 2013;19:418–420. doi: 10.1038/nm.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spruiell K, Richardson RM, Cullen JM, Awumey EM, Gonzalez FJ, Gyamfi MA. Role of pregnane X receptor in obesity and glucose homeostasis in male mice. J Biol Chem. 2014;289:3244–3261. doi: 10.1074/jbc.M113.494575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He J, Gao J, Xu M, Ren S, Stefanovic-Racic M, O'Doherty RM, Xie W. PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes. 2013;62:1876–1887. doi: 10.2337/db12-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parra P, Bruni G, Palou A, Serra F. Dietary calcium attenuation of body fat gain during high-fat feeding in mice. J Nutr Biochem. 2008;19:109–117. doi: 10.1016/j.jnutbio.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 48.Bauer M, Hamm AC, Bonaus M, Jacob A, Jaekel J, Schorle H, Pankratz MJ, Katzenberger JD. Starvation response in mouse liver shows strong correlation with life-span-prolonging processes. Physiol Genomics. 2004;17:230–244. doi: 10.1152/physiolgenomics.00203.2003. [DOI] [PubMed] [Google Scholar]
- 49.Buler M, Aatsinki SM, Skoumal R, Hakkola J. Energy sensing factors PGC-1alpha and SIRT1 modulate PXR expression and function. Biochem Pharmacol. 2011;82:2008–2015. doi: 10.1016/j.bcp.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 50.Rysa J, Buler M, Savolainen MJ, Ruskoaho H, Hakkola J, Hukkanen J. Pregnane X receptor agonists impair postprandial glucose tolerance. Clin Pharmacol Ther. 2013;93:556–563. doi: 10.1038/clpt.2013.48. [DOI] [PubMed] [Google Scholar]
- 51.Shemesh T, Rowley KG, Shephard M, Piers LS, O'Dea K. Agreement between laboratory results and on-site pathology testing using Bayer DCA2000+ and Cholestech LDX point-of-care methods in remote Australian Aboriginal communities. Clin Chim Acta. 2006;367:69–76. doi: 10.1016/j.cca.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 52.Gyamfi MA, He L, French SW, Damjanov I, Wan YJ. Hepatocyte retinoid X receptor alpha-dependent regulation of lipid homeostasis and inflammatory cytokine expression contributes to alcohol-induced liver injury. J Pharmacol Exp Ther. 2008;324:443–453. doi: 10.1124/jpet.107.132258. [DOI] [PubMed] [Google Scholar]
- 53.Gyamfi MA, Kocsis MG, He L, Dai G, Mendy AJ, Wan YJ. The role of retinoid X receptor alpha in regulating alcohol metabolism. J Pharmacol Exp Ther. 2006;319:360–368. doi: 10.1124/jpet.106.108175. [DOI] [PubMed] [Google Scholar]
- 54.Flachs P, Rossmeisl M, Kuda O, Kopecky J. Stimulation of mitochondrial oxidative capacity in white fat independent of UCP1: A key to lean phenotype. Biochim Biophys Acta. 2013;1831:986–1003. doi: 10.1016/j.bbalip.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 55.Kalaany NY, Gauthier KC, Zavacki AM, Mammen PP, Kitazume T, Peterson JA, Horton JD, Garry DJ, Bianco AC, Mangelsdorf DJ. LXRs regulate the balance between fat storage and oxidation. Cell Metab. 2005;1:231–244. doi: 10.1016/j.cmet.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Kopecky J, Clarke G, Enerback S, Spiegelman B, Kozak LP. Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J Clin Invest. 1995;96:2914–2923. doi: 10.1172/JCI118363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- 58.Masuyama H, Hiramatsu Y, Mizutani Y, Inoshita H, Kudo T. The expression of pregnane X receptor and its target gene, cytochrome P450 3A1, in perinatal mouse. Mol Cell Endocrinol. 2001;172:47–56. doi: 10.1016/s0303-7207(00)00395-6. [DOI] [PubMed] [Google Scholar]
- 59.Nilsson S, Gustafsson JA. Estrogen receptor transcription and transactivation: Basic aspects of estrogen action. Breast Cancer Res. 2000;2:360–366. doi: 10.1186/bcr81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saloranta C, Groop L. Interactions between glucose and FFA metabolism in man. Diabetes Metab Rev. 1996;12:15–36. doi: 10.1002/(SICI)1099-0895(199603)12:1<15::AID-DMR153>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 61.Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol. 1992;54:885–909. doi: 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- 62.Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr. 2006;83:461S–465S. doi: 10.1093/ajcn/83.2.461S. [DOI] [PubMed] [Google Scholar]
- 63.Guerre-Millo M. Adipose tissue and adipokines: for better or worse. Diabetes Metab. 2004;30:13–19. doi: 10.1016/s1262-3636(07)70084-8. [DOI] [PubMed] [Google Scholar]
- 64.Matthias A, Ohlson KB, Fredriksson JM, Jacobsson A, Nedergaard J, Cannon B. Thermogenic responses in brown fat cells are fully UCP1-dependent. UCP2 or UCP3 do not substitute for UCP1 in adrenergically or fatty scid-induced thermogenesis. J Biol Chem. 2000;275:25073–25081. doi: 10.1074/jbc.M000547200. [DOI] [PubMed] [Google Scholar]
- 65.Collins S, Martin TL, Surwit RS, Robidoux J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: physiological and molecular characteristics. Physiol Behav. 2004;81:243–248. doi: 10.1016/j.physbeh.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 66.Kaufman FR. Type 2 diabetes mellitus in children and youth: a new epidemic. J Pediatr Endocrinol Metab. 2002;15 Suppl 2:737–744. doi: 10.1515/JPEM.2002.15.s2.737. [DOI] [PubMed] [Google Scholar]
- 67.Breier BH, Vickers MH, Ikenasio BA, Chan KY, Wong WP. Fetal programming of appetite and obesity. Mol Cell Endocrinol. 2001;185:73–79. doi: 10.1016/s0303-7207(01)00634-7. [DOI] [PubMed] [Google Scholar]
- 68.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290–296. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- 69.Prpic V, Watson PM, Frampton IC, Sabol MA, Jezek GE, Gettys TW. Adaptive changes in adipocyte gene expression differ in AKR/J and SWR/J mice during diet-induced obesity. J Nutr. 2002;132:3325–3332. doi: 10.1093/jn/132.11.3325. [DOI] [PubMed] [Google Scholar]
- 70.Watson PM, Commins SP, Beiler RJ, Hatcher HC, Gettys TW. Differential regulation of leptin expression and function in A/J vs. C57BL/6J mice during diet-induced obesity. Am J Physiol Endocrinol Metab. 2000;279:E356–365. doi: 10.1152/ajpendo.2000.279.2.E356. [DOI] [PubMed] [Google Scholar]
- 71.Commins SP, Watson PM, Padgett MA, Dudley A, Argyropoulos G, Gettys TW. Induction of uncoupling protein expression in brown and white adipose tissue by leptin. Endocrinology. 1999;140:292–300. doi: 10.1210/endo.140.1.6399. [DOI] [PubMed] [Google Scholar]
- 72.Alvarez R, Checa M, Brun S, Vinas O, Mampel T, Iglesias R, Giralt M, Villarroya F. Both retinoic-acid-receptor- and retinoid-X-receptor-dependent signalling pathways mediate the induction of the brown-adipose-tissue-uncoupling-protein-1 gene by retinoids. Biochem J. 2000;345 Pt 1:91–97. [PMC free article] [PubMed] [Google Scholar]
- 73.Alvarez R, de Andres J, Yubero P, Vinas O, Mampel T, Iglesias R, Giralt M, Villarroya F. A novel regulatory pathway of brown fat thermogenesis. Retinoic acid is a transcriptional activator of the mitochondrial uncoupling protein gene. J Biol Chem. 1995;270:5666–5673. doi: 10.1074/jbc.270.10.5666. [DOI] [PubMed] [Google Scholar]
- 74.Foellmi-Adams LA, Wyse BM, Herron D, Nedergaard J, Kletzien RF. Induction of uncoupling protein in brown adipose tissue. Synergy between norepinephrine and pioglitazone, an insulin-sensitizing agent. Biochem Pharmacol. 1996;52:693–701. doi: 10.1016/0006-2952(96)00345-0. [DOI] [PubMed] [Google Scholar]
- 75.Sadurskis A, Dicker A, Cannon B, Nedergaard J. Polyunsaturated fatty acids recruit brown adipose tissue: increased UCP content and NST capacity. Am J Physiol. 1995;269:E351–360. doi: 10.1152/ajpendo.1995.269.2.E351. [DOI] [PubMed] [Google Scholar]
- 76.Livingstone C, Collison M. Sex steroids and insulin resistance. Clin Sci (Lond) 2002;102:151–166. doi: 10.1042/cs1020151. [DOI] [PubMed] [Google Scholar]
- 77.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shi H, Seeley RJ, Clegg DJ. Sexual differences in the control of energy homeostasis. Front Neuroendocrinol. 2009;30:396–404. doi: 10.1016/j.yfrne.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shimizu H, Shimomura Y, Nakanishi Y, Futawatari T, Ohtani K, Sato N, Mori M. Estrogen increases in vivo leptin production in rats and human subjects. J Endocrinol. 1997;154:285–292. doi: 10.1677/joe.0.1540285. [DOI] [PubMed] [Google Scholar]
- 80.Hewitt KN, Boon WC, Murata Y, Jones ME, Simpson ER. The aromatase knockout mouse presents with a sexually dimorphic disruption to cholesterol homeostasis. Endocrinology. 2003;144:3895–3903. doi: 10.1210/en.2003-0244. [DOI] [PubMed] [Google Scholar]
- 81.Lu H, Higashikata T, Inazu A, Nohara A, Yu W, Shimizu M, Mabuchi H. Association of estrogen receptor-alpha gene polymorphisms with coronary artery disease in patients with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2002;22:817–823. doi: 10.1161/01.atv.0000014424.18209.21. [DOI] [PubMed] [Google Scholar]
- 82.Nelson JF, Felicio LS, Randall PK, Sims C, Finch CE. A longitudinal study of estrous cyclicity in aging C57BL/6J mice: I. Cycle frequency, length and vaginal cytology. Biol Reprod. 1982;27:327–339. doi: 10.1095/biolreprod27.2.327. [DOI] [PubMed] [Google Scholar]
- 83.Ng KY, Yong J, Chakraborty TR. Estrous cycle in ob/ob and ovariectomized female mice and its relation with estrogen and leptin. Physiol Behav. 2010;99:125–130. doi: 10.1016/j.physbeh.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 84.Akamine EH, Marcal AC, Camporez JP, Hoshida MS, Caperuto LC, Bevilacqua E, Carvalho CR. Obesity induced by high-fat diet promotes insulin resistance in the ovary. J Endocrinol. 2010;206:65–74. doi: 10.1677/JOE-09-0461. [DOI] [PubMed] [Google Scholar]
- 85.Brothers KJ, Wu S, DiVall SA, Messmer MR, Kahn CR, Miller RS, Radovick S, Wondisford FE, Wolfe A. Rescue of obesity-induced infertility in female mice due to a pituitary-specific knockout of the insulin receptor. Cell Metab. 2010;12:295–305. doi: 10.1016/j.cmet.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eckel LA. Ingestive behaviour in female rats: influence of the ovarian cycle. Appetite. 1999;32:274. doi: 10.1006/appe.1999.0225. [DOI] [PubMed] [Google Scholar]
- 87.Huang YS, Doi R, Chowdhury P, Pasley JN, Nishikawa M, Huang TJ, Rayford PL. Effect of cholecystokinin on food intake at different stages of the estrous cycle in female rats. J Assoc Acad Minor Phys. 1993;4:56–58. [PubMed] [Google Scholar]
- 88.Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S A. 2000;97:12735–12740. doi: 10.1073/pnas.97.23.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, Dahlman-Wright K, Nilsson S, Gustafsson JA, Efendic S, Khan A. Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia. 2006;49:588–597. doi: 10.1007/s00125-005-0105-3. [DOI] [PubMed] [Google Scholar]
- 90.Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- 91.Simpson E, Rubin G, Clyne C, Robertson K, O'Donnell L, Davis S, Jones M. Local estrogen biosynthesis in males and females. Endocr Relat Cancer. 1999;6:131–137. doi: 10.1677/erc.0.0060131. [DOI] [PubMed] [Google Scholar]
- 92.Blumberg B, Evans RM. Orphan nuclear receptors--new ligands and new possibilities. Genes Dev. 1998;12:3149–3155. doi: 10.1101/gad.12.20.3149. [DOI] [PubMed] [Google Scholar]
- 93.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 94.Tulchinsky D, Hobel CJ, Yeager E, Marshall JR. Plasma estrone, estradiol, estriol, progesterone, and 17-hydroxyprogesterone in human pregnancy. I. Normal pregnancy. Am J Obstet Gynecol. 1972;112:1095–1100. doi: 10.1016/0002-9378(72)90185-8. [DOI] [PubMed] [Google Scholar]
- 95.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 96.Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor transcription and transactivation: Estrogen receptor alpha and estrogen receptor beta: regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res. 2000;2:335–344. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Glass CK. Going nuclear in metabolic and cardiovascular disease. J Clin Invest. 2006;116:556–560. doi: 10.1172/JCI27913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 99.Gao J, He J, Zhai Y, Wada T, Xie W. The constitutive androstane receptor is an antiobesity nuclear receptor that improves insulin sensitivity. J Biol Chem. 2009;284:25984–25992. doi: 10.1074/jbc.M109.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Iynedjian PB, Pilot PR, Nouspikel T, Milburn JL, Quaade C, Hughes S, Ucla C, Newgard CB. Differential expression and regulation of the glucokinase gene in liver and islets of Langerhans. Proc Natl Acad Sci U S A. 1989;86:7838–7842. doi: 10.1073/pnas.86.20.7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grupe A, Hultgren B, Ryan A, Ma YH, Bauer M, Stewart TA. Transgenic knockouts reveal a critical requirement for pancreatic beta cell glucokinase in maintaining glucose homeostasis. Cell. 1995;83:69–78. doi: 10.1016/0092-8674(95)90235-x. [DOI] [PubMed] [Google Scholar]