

Abstract
Background
Following peroral Toxoplasma (T.) gondii infection, susceptible mice develop acute ileitis due to a microbiota-dependent Th1 type immunopathology. Toll-like-receptor (TLR)-9 is known to recognize bacterial DNA and mediates intestinal inflammation, but its impact on intestinal microbiota composition and extra-intestinal sequelae following T. gondii infection has not yet been elucidated.
Methods and results
Seven days following peroral infection (p.i.) with 100 cysts of T. gondii ME49 strain, TLR-9-/- and wildtype (WT) mice suffered from comparable ileitis, whereas ileal parasitic loads as well as IFN-γ and nitric oxide levels were higher in TLR-9-/- compared to WT mice. Locally, TLR-9-/- mice exhibited increased ileal CD3+, but not FOXP3+ cell numbers at day 7 p.i.; in mesenteric lymph nodes IFN-γ-producing CD4+ cell numbers and TNF-α and IFN-γ concentrations were also increased in TLR-9-/- compared to WT mice. T. gondii DNA levels, however, did not differ in mice of either genotype. Differences in intestinal microbiota were rather subtle except for bifidobacteria that were virtually absent in both, naïve and T. gondii infected TLR-9-/-, but not WT mice. Extra-intestinally, TLR-9-/- mice displayed less distinct systemic immune responses as indicated by lower serum IL-6, and splenic TNF-α and IFN-γ levels as compared to WT mice despite higher translocation rates of intestinal bacteria to extra-intestinal compartments such as liver, spleen, kidney, and cardiac blood. Most importantly, brains were also affected in this inflammatory scenario as early as day 7 p.i. Remarkably, TLR-9-/- mice exhibited more pronounced inflammatory infiltrates with higher numbers of F4/80+ macrophages and microglia in the cortex and meninges as compared to WT mice, whereas T. gondii DNA levels did not differ.
Conclusion
We here show that TLR-9 is not required for the development of T. gondii induced ileitis but mediates distinct inflammatory changes in intestinal and extra-intestinal compartments including the brain.
Keywords: Toxoplasma gondii, Acute ileitis, TLR-9, Th1-type immunopathology, Extra-intestinal immune responses, Intracerebral inflammation, Pro-inflammatory cytokines, Intestinal microbiota composition, Bacterial translocation, Systemic inflammatory response, Bifidobacteria, FOXP3, Regulatory T cells, Gut-brain-axis
Introduction
Seven days following peroral infection with 100 cysts of Toxoplasma (T.) gondii ME49 strain, susceptible mice develop massive necrosis in the terminal ileum and succumb to the infection [1]. This fatal scenario is due to a classical Th1-type immunopathology orchestrated by intestinal epithelial cells, granulocytes, macrophages, monocytes, dendritic cells, and lymphocytes [2]. Early upon T. gondii infection, parasitic interaction with antigen presenting cells results in activation of CD4+ T cells and over-production of mediators such as IFN-γ, TNF-α, nitric oxide (NO), IL-6, and MCP-1 among others comprising a “pro-inflammatory cytokine storm” [3-7]. Hence, the underlying immunopathogenesis resembles key features of chronic inflammatory bowel diseases (IBD) such as Crohn’s disease in the acute stage [2,8]. Furthermore, we recently showed that lipopolysaccharide (LPS) derived from the intestinal microbiota mediates T. gondii induced immunopathology via Toll-like-receptor (TLR) -4 signaling [9-11].
Bacterial unmethylated CpG DNA is the classical ligand for TLR-9 signalling [12-14]. Upon binding, transcription factors such as NF-κB, IFN regulatory factor-7, and AP-1 among others become activated in a MyD88-dependent fashion leading to Th1 type immune responses [15,16]. Initially, TLR-9 was shown to be crucial for an effective Th1-type immune response following oral T. gondii infection of mice [17]. However, the direct recognition of T. gondii molecules through TLR-9 is controversially discussed [18]. Parasitic DNA and RNA have been shown to activate innate immune responses via TLR-7 and TLR-9, but mice lacking TLR-9 alone were not susceptible to T. gondii infection [19]. In addition, TLR-11 and TLR-12 acting as heterodimers were shown to be essentially required for sensing of Toxoplasma profilin [19].
Given that bacterial DNA derived from the commensal microbiota provide pivotal immune-stimulatory molecules for effective host defense against parasitic infection [20], the distinct microbiota composition displays an essential determinant for the immunopathogenesis in murine T. gondii induced acute ileitis. In the present study we performed a comprehensive survey of quantitative and qualitative changes in the intestinal microbiota composition of TLR-9-/- and WT control mice following ileitis induction. Furthermore, we assessed immunopathological sequelae following peroral T. gondii infection in intestinal as well as extra-intestinal compartments such as spleen, liver, kidneys, and brain.
Results
Acute ileal immunopathology in TLR-9-/- mice following peroral T. gondii infection
In order to induce acute ileitis TLR-9-/- and wildtype (WT) mice were subjected to a peroral infection with 100 cysts of T. gondii ME49 strain. Seven days thereafter mice of either genotype were compromised due to wasting disease and suffered from comparable relative body weight losses (Figure 1A). In addition, TLR-9-/- and WT mice displayed similar severity of acute ileitis as indicated by comparable ileal histopathological scores (Figure 1B). Notably, TLR-9-/- mice harbored significantly higher parasitic DNA loads in the small intestines as compared to WT controls at day 7 p.i. (p < 0.05; Figure 1C).
Figure 1.
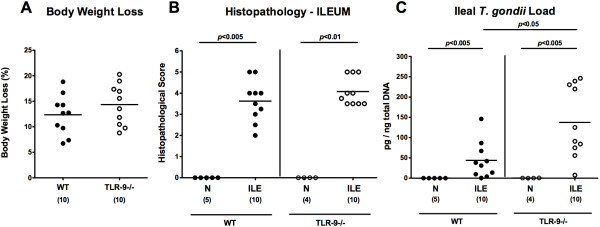
Clinical conditions and small intestinal histopathology following ileitis induction upon peroral T. gondii infection. In order to induce acute ileitis, C57BL/6 wildtype (WT; black circles) and TLR-9 deficient (TLR-9-/-; white circles) mice were perorally infected with 100 cysts of T. gondii ME49 strain on day 0. (A) Relative loss of body weights between day 7 following ileitis induction (ILE) and day 0 (naïve mice; N) were determined (in %). Furthermore, (B) histopathological mucosal changes were assessed in ileal paraffin sections from respective mice seven days following peroral T. gondii infection applying a standardized histopathological score. (C)T. gondii DNA was determined in ex vivo ileal biopsies at day 7 p.i.. Naïve mice served as negative controls (N). Numbers of analyzed mice are given in parentheses. Medians (black bars) and significance levels (P-values) determined by Mann–Whitney-U test are indicated. Data shown are pooled from three independent experiments.
We next quantitatively assessed ileum mucosal apoptotic cells as well as the influx of distinct immune cell populations into the small intestinal mucosa and lamina propria during acute ileitis applying in situ immunohistochemical stainings of ileal paraffin sections. At day 7 p.i., numbers of caspase-3+ apoptotic cells increased to comparable levels in ilea of TLR-9-/- and WT mice (Figure 2A), thus further underlining the clinical and histopathological results. In addition, numbers of T cells, the major driving forces of T. gondii induced acute ileitis, as well as of neutrophilic granulocytes, monocytes, and macrophages exerting oxidative stress to the intestinal epithelium increased comparably in TLR-9-/- and WT mice until day 7 p.i. (Figure 2B-D). Interestingly, FOXP3+ regulatory T cell (Treg) numbers increased in WT, but not TLR-9-/- mice upon ileitis induction (p < 0.001; Figure 2E). Taken together, TLR-9-/- mice were not protected from T. gondii induced acute ileitis and were unable to control replication of the parasite.
Figure 2.
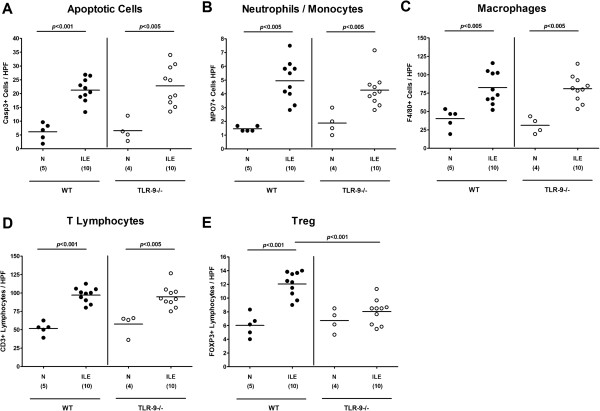
Small intestinal pro-inflammatory immune cell responses following ileitis induction. Pro-inflammatory immune cell responses were assessed in ileal paraffin sections derived from C57BL/6 wildtype (WT; black circles) and TLR-9 deficient (TLR-9-/-; white circles) mice seven days following ileitis induction (ILE) applying in situ immunohistochemistry. The average numbers of ileal (A) apoptotic cells (positive for caspase-3; Casp3), (B) neutrophilic granulocytes and monocytes (positive for MPO-7), (C) macrophages (positive for F4/80), (D) T lymphocytes (positive for CD3), and (E) regulatory T cells (Treg, positive for FOXP3) were determined in six high power fields (HPF, 400 × magnification) per animal by light microscopy. Naïve mice served as negative controls (N). Numbers of analyzed mice (in parentheses), means (black bars) and levels of significance (P-values) as compared to the respective groups (determined by Mann–Whitney-U test) are indicated. Data shown were pooled from three independent experiments.
We next determined pro-inflammatory cytokine levels in ex vivo ileal biopsies taken before and seven days after ileitis induction. Until day 7 p.i., intestinal IFN-γ, TNF-α, nitric oxide (NO), and IL-6 levels had increased in mice of either genotype (p < 0.05-0.001; Figure 3). TLR-9-/- mice, however, displayed even higher ileal IFN-γ and NO concentrations as compared to WT controls at day 7 p.i. (p < 0.05; Figure 3A,C). Notably, small intestinal concentrations of the anti-inflammatory cytokine IL-10 essentially involved in counteracting T. gondii induced immunopathology [21] increased to comparable levels in mice of either genotype until day 7 p.i. (Figure 3E). In the following we assessed pro-inflammatory cytokine responses in mesenteric lymph nodes (MLNs) draining the small intestinal tract. Again, IFN-γ, TNF-α, and NO protein levels increased multifold upon T. gondii infection (p < 0.005-0.001; Figure 4A-C), but the former two cytokines were significantly higher in TLR-9-/- mice as compared to WT controls (p < 0.005 and p < 0.05, respectively; Figure 4A,B). Furthermore, flow cytometry analysis of lymphocytes isolated from MLNs revealed a higher abundance of IFN-γ producing CD4+ cells in TLR-9-/- versus WT mice at day 7 p.i. (2.84% versus 0.88%; Figure 5), whereas naïve mice of either genotype exhibited similar frequencies of CD4+ IFN-γ + cells. Furthermore, relative frequencies of CD3+ CD4+ cells were comparable in MLNs of naïve and T. gondii infected mice of either genotype, whereas percentages of activated T lymphocytes (i.e. CD69+ CD4+ cells) increased comparably upon T. gondii infection in both, TLR-9-/- and WT animals (Figure 5).
Figure 3.
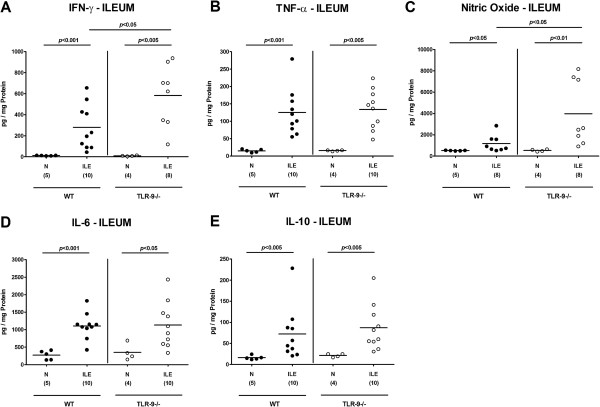
Ileal cytokine secretion following ileitis induction. (A) IFN-γ, (B) TNF-α, (C) nitric oxide, (D) IL-6, and (E) IL-10 protein levels were determined in ex vivo ileal biopsies derived from C57BL/6 wildtype (WT; black circles) and TLR-9 deficient (TLR-9-/-; white circles) mice seven days following ileitis induction (ILE) as described in methods. Naïve mice served as negative controls (N). Numbers of analyzed mice (in parentheses), means (black bars) and levels of significance (P-values) as compared to the respective groups (determined by Mann–Whitney-U test) are indicated. Data shown were pooled from three independent experiments.
Figure 4.
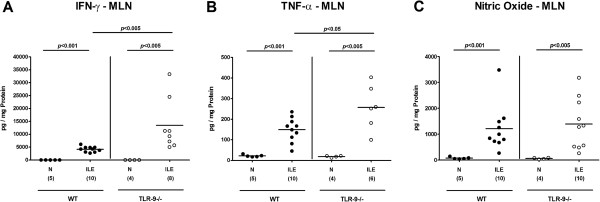
Pro-inflammatory cytokine responses in mesenteric lymph nodes following ileitis induction. (A) IFN-γ, B) TNF-α, and (C) nitric oxide protein concentrations were determined in ex vivo biopsies of mesenteric lymph nodes (MLN) derived from C57BL/6 wildtype (WT; black circles) and TLR-9 deficient (TLR-9-/-; white circles) mice seven days following ileitis induction (ILE). Naïve mice served as negative controls (N). Numbers of analyzed mice (in parentheses), means (black bars) and levels of significance (P-values) as compared to the respective groups (determined by Mann–Whitney-U test) are indicated. Data shown were pooled from three independent experiments.
Figure 5.
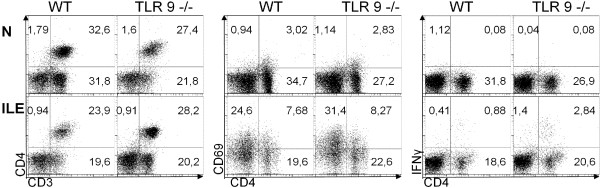
Flow cytometry analysis of lymphocyte populations in mesenteric lymph nodes following ileitis induction. Lymphocyte populations were isolated from mesenteric lymph nodes (MLN) derived from C57BL/6 wildtype (WT) and TLR-9 deficient (TLR-9-/-) mice before (naïve) and seven days following ileitis induction (ILE) and analyzed by flow cytometry. Frequencies of CD4+ cells producing IFN-γ (right panel), CD4+ CD3+ cells (left panel), as well as CD69+ CD4+ cells (middle panel), are indicated in representative FACS plots (in %). Data shown are representative for three independent experiments.
Intestinal microbiota changes and bacterial translocation to extra-intestinal compartments in TLR-9-/- mice with acute ileitis
Given that the commensal intestinal microbiota composition mediates induction and perpetuation of intestinal immunopathogenesis [2,9,10,22], we next performed a comprehensive quantitative molecular survey of the main fecal bacterial groups derived from TLR-9-/- and WT mice, before and seven days after ileitis induction. Until day 7 p.i., the total eubacterial load increased slightly, but only reached statistical significance in TLR-9-/- mice (p < 0.001; Figure 6A). Higher total bacterial loads at day 7 as compared to day 0 were due to increases in enterobacteria, enterococci, Bacteroides/Prevotella spp. loads in both, WT and TLR-9-/- mice (Figure 6B,C,F), and of Mouse Intestinal Bacteroides in the latter (Figure 6I). Notably, naive TLR-9-/- mice harbored slightly lower fecal enterobacteria and Mouse Intestinal Bacteroides DNA, whereas Clostridium leptum loads were higher compared to naïve WT mice (Figure 6B, I, G), Remarkably, bifidobacteria were virtually absent in TLR-9-/-, but not WT mice at either time point (Figure 6E).
Figure 6.
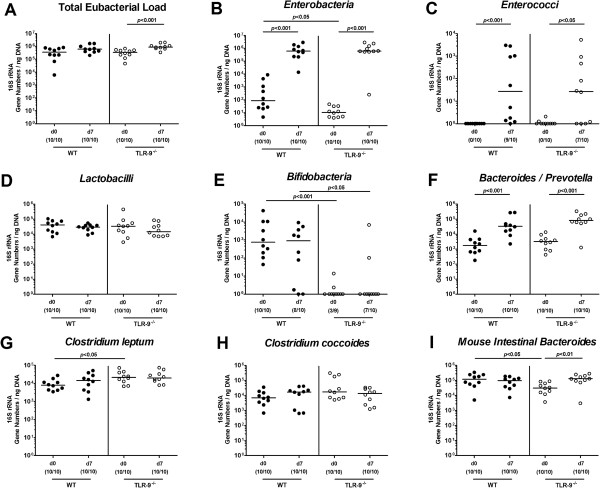
Intestinal microbiota composition of mice following ileitis induction. Main intestinal bacterial groups were quantified by molecular analysis of fecal samples derived from C57BL/6 wildtype (WT; black circles) and TLR-9 deficient (TLR-9-/-; white circles) mice before (N, naïve) and seven days after ileitis induction by peroral T. gondii infection (ILE). Quantitative real time-PCR analyses amplified bacterial 16S rRNA variable regions and 16S rRNA gene numbers/ng DNA from the following bacterial groups: (A) Total eubacterial load, (B)Enterobacteria, (C)Enterococci, (D)Lactobacilli, (E)Bifidobacteria, (F)Bacteroides/Prevotella spp, (G)Clostridium leptum group, (H)Clostridium coccoides group, and (I)Mouse intestinal Bacteroides. Numbers of mice harboring the respective bacterial 16S rRNA out of the total number of analyzed animals are given in parentheses. Medians and significance levels (P-values) determined by Mann–Whitney-U test are indicated. Data shown were pooled from three independent experiments.
Given that a compromised epithelial barrier facilitates bacterial translocation from the intestinal lumen to sub-epithelial and further extra-intestinal sites, we next assessed translocation rates of viable bacteria. Of interest, whereas commensal intestinal bacteria could be cultured from MLNs of TLR-9-/- and WT mice at identical frequencies (80%), mean translocation rates to spleen (20% vs 10%, respectively), liver (40% vs 20%, respectively), and kidneys (20% vs 10%, respectively) were higher in TLR-9-/- as compared to WT mice (Figure 7). Furthermore, even 10% of cardiac blood samples derived from TLR-9-/- mice were culture-positive whereas all blood cultures were negative in WT animals at day 7 p.i. (Figure 7). Cultured translocated bacteria comprised commensal intestinal species such as E. coli, Enterococcus spp. and Lactobacillus spp., but no obligate anaerobic bacteria (not shown). Notably, respective translocation frequencies were identical in independent experiments. Taken together, these data point towards a more severe damage to the ileal epithelial barrier in diseased TLR-9-/- mice as compared to WT controls, thus facilitating translocation of live commensal bacteria originating from the intestinal lumen to extra-intestinal compartments.
Figure 7.
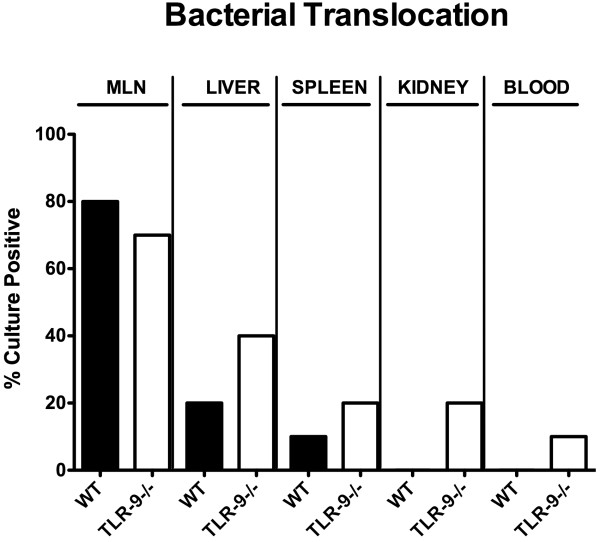
Bacterial translocation following ileitis induction. Relative translocation frequencies (%) of live bacteria were determined in ex vivo biopsies of mesenteric lymph nodes (MLN), liver, spleen, kidneys, and cardiac blood derived from C57BL/6 wildtype (WT; black bars; n = 10) and TLR-9 deficient (TLR-9-/-; white bars; n = 10) mice by culture in enrichment broths seven days after ileitis induction. Data shown were pooled from three independent experiments.
Extra-intestinal immunopathology in TLR-9-/- mice with acute ileitis
We next investigated systemic immune responses and potential pro-inflammatory sequelae in extra-intestinal organs following ileitis induction. Irrespective of the genotype of mice, IL-6 and IFN-γ serum levels (Figure 8A,B), splenic TNF-α and IFN-γ (Figure 8C,D) as well as hepatic TNF-α and IL-6 concentrations (Figure 8E,F) increased multifold until day 7 following infection. Remarkably, pro-inflammatory cytokine levels in the respective extra-intestinal compartment were significantly lower in TLR-9-/- as compared to WT mice at day 7 p.i. (p < 0.05-0.005; Figure 8). Furthermore, TLR-9-/- mice exhibited approximately 50% lower IL-6 serum concentrations as compared to WT controls at day 7 p.i. (p < 0.05; Figure 8A). Taken together, the absence of TLR-9 does not prevent the development of T. gondii induced acute ileitis. However, intestinal parasite loads and local immune responses are more distinct in infected TLR-9-/- as compared to WT mice. In contrast, the absence of TLR-9 results in lower levels of extra-intestinal and systemic inflammation.
Figure 8.
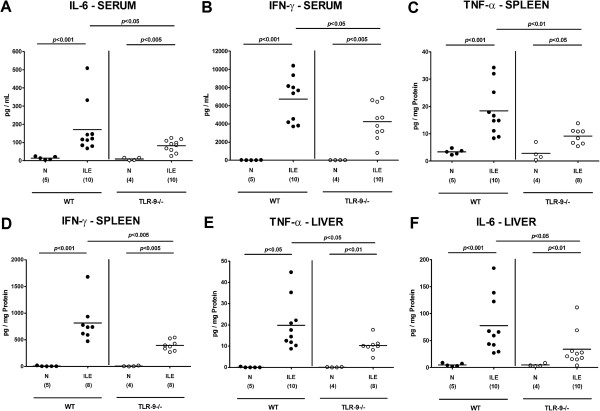
Systemic and extra-intestinal pro-inflammatory cytokine responses following ileitis induction. Serum (A) IL-6 and (B) IFN-γ, splenic (C) TNF-α and (D) IFN-γ as well as hepatic (E) TNF-α and (F) IL-6 protein concentrations were determined in respective ex vivo biopsies derived from C57BL/6 wildtype (WT; black circles) and TLR-9 deficient (TLR-9-/-; white circles) mice seven days following ileitis induction (ILE). Naïve mice served as negative controls (N). Numbers of analyzed mice (in parentheses), means (black bars) and levels of significance (P-values) as compared to the respective groups (determined by Mann–Whitney-U test) are indicated. Data shown were pooled from three independent experiments.
Next we analyzed a potential intracerebral inclusion in T. gondii induced acute small intestinal immunopathology. Most importantly, T. gondii induced acute ileitis and brain inflammation could be observed in mice of either genotype as early as day 7 p.i. (Figure 9); the cortex as well as the meninges of infected, but not naïve animals were affected (Figure 9A). These inflammatory responses, however, were more pronounced in TLR-9-/- as compared to WT mice as indicated by higher histopathological scores (assessing both, cortical and meningeal inflammatory foci) in the former (p < 0.05; Figure 9A). In situ immunohistochemical analyses revealed higher numbers of F4/80+ inflammatory monocytes, macrophages and residential microglia in TLR-9-/- as compared to WT mice (Figure 9B). Interestingly, comparable parasitic DNA loads could be detected in brain tissues derived from mice of either genotype seven days following T. gondii infection (Figure 9C). Cerebral IFN-γ, TNF-α, and IL-6 mRNA expression levels increased multifold upon T. gondii infection, but did not differ between TLR-9-/- and WT mice at day 7 p.i. (Figure 10A-C). Taken together, pro-inflammatory immune responses develop not only in intestinal, but also in extra-intestinal compartments including the brain as early as 7 days after peroral infection with T. gondii. In the absence of TLR-9, T. gondii induced small intestinal and intracerebral pro-inflammatory cytokine responses are higher, whereas systemic (i.e. serum and splenic) levels are lower as compared to WT mice at day 7 p.i.
Figure 9.
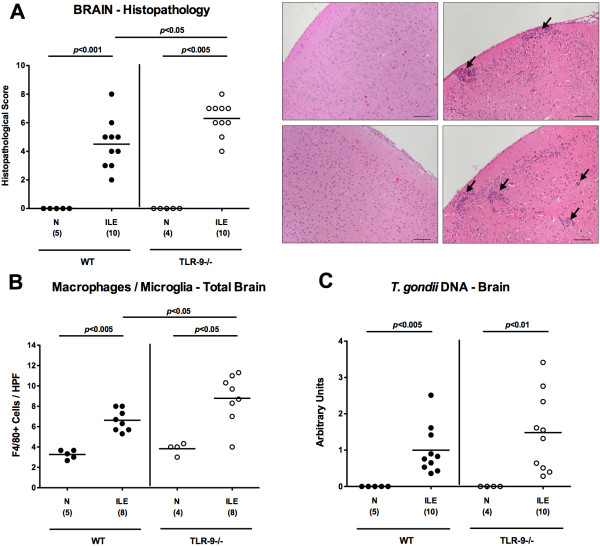
Cerebral histopathological changes following acute ileitis induction. C57BL/6 wildtype (WT; black circles) and TLR-9 deficient (TLR-9-/-; white circles) mice were perorally infected with 100 cysts of T. gondii ME49 strain on day 0 to induce acute ileitis. Naïve mice served as negative controls (N). (A) Cerebral histopathological changes (meninges and cortex) were assessed in H&E-stained paraffin sections of brains seven days following ileitis induction (ILE) applying a standardized histopathological score (as described in methods) and illustrated by representative photomicrographs (WT: upper panel, TLR-9-/-: lower panel; N: left panel, ILE: right panel). Arrows indicate inflammatory foci (100 × magnification, scale bar 100 μm). (B) Brain paraffin sections were stained for F4/80 by in situ immunohistochemistry and the average numbers of intracerebral macrophages and microglial cells were determined in six high power fields (HPF, 400 × magnification) per animal by light microscopy. (C)T. gondii DNA loads were determined in brain homogenates by semi-quantitative RT-PCR and normalized relative to the M. musculus ASL gene (Arbitrary Units). Numbers of analyzed mice (in parentheses), means (black bars) and levels of significance (P-values) as compared to the respective groups (determined by Mann-Whitney-U test) are indicated. Data shown were pooled from three independent experiments.
Figure 10.
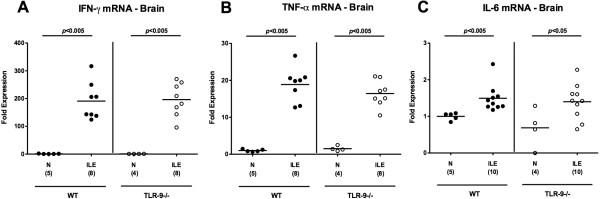
Cerebral histopathological changes following acute ileitis induction. C57BL/6 wildtype (WT; black circles) and TLR-9 deficient (TLR-9-/-; white circles) mice were perorally infected with 100 cysts of T. gondii ME49 strain on day 0 to induce acute ileitis. Naïve mice served as negative controls (N). Intracerebral (A) IFN-γ, (B) TNF-α, and (C) IL-6 mRNA expression levels were measured in ex vivo brain biopsies applying quantitative RT-PCR. Numbers of analyzed mice (in parentheses), means (black bars), and levels of significance (P-values) determined by Mann–Whitney-U test are indicated. Data shown were pooled from three independent experiments.
Discussion
In the present study we show that the absence of TLR-9 does not protect mice from the development of acute ileitis following peroral infection with T. gondii. TLR-9-/- mice succumbed to intestinal immunopathology as did WT mice. Despite a comparable clinical and intestinal (e.g. histopathological) outcome of infection we observed several TLR-9-mediated effects. For instance, TLR-9-/- mice displayed higher parasitic loads in the small intestines as compared to WT mice, which was accompanied by higher levels of pro-inflammatory cytokine such as IFN-γ and NO in ilea and IFN-γ and TNF-α in MLNs in the absence of TLR-9. The T. gondii induced Th1 type immune response in TLR-9-/- mice was further supported by a higher abundance of IFN-γ producing T cells in MLNs derived from TLR-9-/- as compared to WT mice at day 7 p.i. Our results are in contrast to a previous study by Minns and colleagues demonstrating that T. gondii infected TLR-9-/- mice were protected from disease due to a diminished Th1-type immune response [17]. TLR-9 deficiency had resulted in approximately 50% reduction of IFN-γ production in infected mice, which in turn was insufficient to combat the parasite [17]. Consequently and in agreement with our study, infected TLR-9-/- mice harbored more parasites as compared to WT animals. It is unclear, however, why the overwhelming (local) Th1 response in our report was still insufficient to control parasite replication. One reason could be that a down-regulated systemic expression of pro-inflammatory cytokines as shown by lower splenic IFN-γ and TNF-α and serum IL-6 might contribute to insufficient parasite control in T. gondii infected TLR-9-/- mice. Notably, expanding intestinal IL-10 producing FOXP3+ Treg populations exert important anti-inflammatory defense mechanisms against invading pathogens [23]. Given that WT, but not TLR-9-/- mice exhibited increased numbers of ileal FOXP3+ cells upon infection, reduced Treg numbers might be indicative for compromised ileal mucosal regulatory properties in the absence of TLR-9, which in turn contributes to a more devastating inflammatory scenario. Interestingly, a previous study revealed a TLR-9 dependent induction of intestinal α-defensins comprising a conserved heterogenous group of antimicrobial peptides upon T. gondii infection [24]. In infected TLR-9-/- mice, however, secretory granules of α-defensin producing Paneth cells failed to degranulate and, in turn, to elicit a potential local anti-parasitic effect [24]. Furthermore, we showed that in the absence of TLR-9 viable bacterial species originating from the commensal intestinal microbiota were more frequently capable of translocating through the inflamed epithelial barrier to extra-intestinal organs such as liver, spleen, kidney, and even cardiac blood. It is somewhat surprising, however, that even during more pronounced ileal disease TLR-9 deficient mice exhibited less extra-intestinal and systemic pro-inflammatory cytokine in liver, spleen, and serum as compared to WT mice at day 7 p.i.
Several factors might be responsible for the conflicting results reported by Minns et al. such as differences in T. gondii strain and numbers of applied cysts. Minns et al. perorally infected mice with 35 cysts of the 76 K strain, whereas in our study mice were challenged with three times more (i.e. 100) cysts of the ME49 strain. Hence, the induction of the Th1-type immunopathology was more pronounced in our present study due to the higher amount of applied T. gondii cysts. Furthermore, differences in the genetic background (complete versus incomplete backcrossings into the same genetic background), sex and age as well as housing conditions (e.g. diet, hygienic conditions in the animal facility) and the colonization status of the applied mice might also impact host resistance against parasitic infection [25-27]. A plethora of recent reports highlight the pivotal role of the intestinal microbiota in immunity and inflammation, and even subtle differences in the microbiota composition might have a major biological impact on the initiation and perpetuation of immunopathology [23,28-31]. This encouraged us to perform a comprehensive comparative survey of the intestinal microbiota composition in naïve and T. gondii infected TLR-9-/- as well as WT mice. Interestingly, naïve TLR-9-/- mice harbored approximately one order of magnitude lower enterobacteria (such as commensal E. coli) and Mouse Intestinal Bacteroides as compared to WT mice, whereas bifidobacteria were virtually absent in the former. One needs to take into consideration that bifidobacterial species are considered probiotic bacteria with anti-inflammatory capacities. In a previous in vitro study, for instance, CD4+ T cells co-cultured with bifidobacteria stimulated dendritic cells resulted in an increase of CD25+ FOXP3+ Tregs [32]. It is tempting to speculate that due to the absence of bifidobacteria in TLR-9-/- mice we did not observe increased numbers of ileal FOXP3+ Treg populations upon T. gondii infection, which additionally contributed to a compromized host resistance against the parasite. However, it is currently virtually impossible to decipher whether the observed immune responses are mainly due to signaling of DNA signals derived from the intestinal microbiota and/or the parasite.
Experimental chronic encephalitis develops within several weeks following intraperitoneal low dose (i.e. 1 to 10 cysts) T. gondii infection of mice [2,33,34]. To our surprise, in the acute ileitis model presented here overt inflammatory changes were not restricted to the small intestinal tract, but could also be observed within the central nervous system affecting both, the brain cortex and meninges as early as one week following peroral high dose T. gondii infection in WT as well as TLR-9-/- mice. Remarkably, brains of T. gondii infected TLR-9-/- mice were even more distinctly affected by the induced pro-inflammatory immune responses as compared to WT control animals given that higher numbers of inflammatory foci and higher abundance of F4/80+ cells comprising recruited macrophages, inflammatory monocytes, and residential microglia were detectable. Interestingly, intracerebral parasitic loads and mRNA expression levels of pro-inflammatory cytokines such as IFN-γ, TNF-α, and IL-6 were similar in mice of either genotype at day 7 p.i. Even though we did not address the underlying mechanisms of this collateral damage within the hyper-acute intestinal inflammatory disease, several factors might explain the concomitant intracerebral inflammatory response. It is well described that pro-inflammatory cytokines and active molecules such as IFN-γ, TNF-α, IL-6, and reactive oxygen species are up-regulated during acute T. gondii infection [1,2,8,35]. TNF-α is mainly produced by Ly6C + inflammatory monocytes that regulate parasite control [35]. Importantly, systemic TNF-α has been shown to activate cerebral microglia in infection-induced encephalopathy [36] and murine neurocysticercosis [37], and subsequently up-regulates other inflammatory mediators and neurotransmitters. Furthermore, matrix-degrading mediators such as matrix metalloproteinases (MMP), e.g. gelatinases, are up-regulated in the small intestinal tract during acute T. gondii induced ileitis [22] and ischemic injury [38]. As a consequence, immune cells become activated [39,40] and further biologically active TNF-α and IL-6 are released from the surfaces of macrophages [25,41,42] which might lead to blood brain barrier breach and consequent influx of immune cells from the blood to the CNS [38]. Enhanced intracerebral F4/80+ monocyte recruitment might exacerbate oxidative stress to the brain parenchyma to perpetuate the inflammatory response [36].
Conclusion
TLR-9 impacts the intestinal microbiota composition and mediates immunopathological responses during acute ileitis following peroral high dose T. gondii infection. Furthermore, acute T. gondii induced ileitis is accompanied by distinct TLR-9 dependent inflammatory sequelae in extra-intestinal compartments including the brain as early as 7 days p.i. In the absence of TLR-9 local (i.e. small intestinal) as well as intracerebral inflammatory changes are more pronounced, whereas systemic pro-inflammatory immune responses are down-regulated upon T. gondii infection.
Methods
Ethics statement
All animal experiments were conducted according to the European Guidelines for animal welfare (2010/63/EU) with approval of the commission for animal experiments headed by the “Landesamt für Gesundheit und Soziales” (LaGeSo, Berlin; registration number G0146/10). Animal welfare was monitored twice daily by assessment of clinical conditions and weight loss of mice.
Mice and induction of acute ileitis
TLR-9-/- mice (in C57BL/6 background; described elsewhere [43]) and wildtype controls were bred and housed under specific pathogen-free (SPF) conditions in the Forschungseinrichtung für Experimentelle Medizin (FEM, Charité – University Medicine Berlin, Germany). For induction of acute ileitis, age matched 3 months old female mice were infected perorally by gavage with 100 T. gondii cysts (ME49 strain) from homogenized brains of intraperitoneally infected NMRI mice in a volume of 0.3 mL phosphate-buffered saline (PBS), as described previously [9,10,44].
Sampling procedures and histopathology
Mice were sacrificed by isofluran treatment (Abbott, Germany). Cardiac blood and tissue samples from brain, spleen, liver, kidneys, mesenteric lymph nodes, and ileum were removed under sterile conditions. Small intestinal samples from each mouse were collected in parallel for histopathological, immunohistochemical, microbiological, and immunological analyses. Immunohistopathological changes were determined in samples derived from ileum and brain that were immediately fixed in 5% formalin and embedded in paraffin. Sections (5 μm) were stained with hematoxylin and eosin (H&E), examined by light microscopy (magnification 100 × and 400 ×) and histopathological changes quantitatively assessed applying respective histopathological scoring systems.
Ileal histopathology was determined as described previously [9].
Brain histopathology (max. 10 points; according to [33-35] with minor modifications): The numbers of inflammatory foci per HPF (100 × magnification) were assessed in cortex and meninges (separately) and the sum of the resulting scores indicated (maximum score of 10). 0, healthy brain structure with no inflammatory foci; 1, single inflammatory foci [1-3]; 2, inflammatory foci [3-6]; 3, inflammatory foci [7-10]; 4, inflammatory foci [11-15]; 5, inflammatory foci (>15).
Immunohistochemistry
In situ immunohistochemical analyses of 5 μm thin ileal paraffine sections were performed as described previously [43,45-47]. Primary antibodies against cleaved caspase-3 (Asp175, Cell Signaling, USA, 1:200), CD3 (M20, Santa Cruz, 1:1000), myeloperoxidase-7 (MPO-7, # A0398, Dako, 1:10000), F4/80 (# 14–4801, clone BM8, eBioscience, 1:50), and FOXP3 (FJK-16 s, eBioscience, 1:100) were used. For each animal, the average number of positively stained cells within at least six high power fields (HPF, 0.287 mm2; 400 × magnification) was determined microscopically by two independent double-blinded investigators.
Ileal T. gondii DNA detection and cytokine measurement
Ileal ex vivo biopsies were cut longitudinally and washed in PBS. In approximately 1 cm2 of homogenized ileal tissue T. gondii DNA was measured as described previously [22]. Spleen, liver, mesenteric lymph nodes or strips of approximately 1 cm2 ileal tissue were placed in 24-flat-bottom well culture plates (Nunc, Wiesbaden, Germany) containing 500 μL serum-free RPMI 1640 medium supplemented with penicillin (100 U/mL) and streptomycin (100 μg/mL; PAA Laboratories). After 18 h incubation at 37°C, culture supernatants or serum samples were tested for IFN-γ, TNF-α, IL-6, and IL-10 by the Mouse Inflammation Cytometric Bead Assay (CBA; BD Biosciences) on a BD FACSCanto II flow cytometer (BD Biosciences). Nitric oxide (NO) was determined by Griess reaction as described earlier [9].
Lymphocyte isolation, sorting and flow cytometry
Mesenteric lymph nodes were removed and subsequently minced through a 70 μm filter. For intracellular staining, cells were stimulated for 4 h with 50 ng/mL 12-O-tetradecanoylphorbol-13 acetate (Sigma, Missouri, USA), 750 ng/mL ionomycin (Sigma) and Golgi Stop (BD Biosciences, San Diego, USA) at 37°C. Stainings and cell sorting with anti-CD3 (clone 145-2C11, isotype hamster IgG1; BD Pharmingen), anti-CD4 (clone GK1.5 isotype rat IgG2b; BD Pharmingen), anti-CD69 (clone H1.2 F3, isotype Hamster IgG1*, Lamda 3; BD Pharmingen), and anti-IFN-γ (clone 145-2C11, isotype hamster IgG1; BD Pharmingen) were performed. Cells were analyzed with a FACSCalibur or LSR II Flow Cytometer (BD Biosciences).
Molecular analysis of the intestinal microbiota
DNA from fecal samples was extracted as described previously [9]. Briefly, DNA extracts and plasmids were quantified using Quant-iT PicoGreen reagent (Invitrogen, UK) and adjusted to 1 ng per μL. Then, main bacterial groups abundant in the murine conventional intestinal microbiota were assessed by quantitative RT-PCR with group-specific 16S rRNA gene primers (Tib MolBiol, Germany) as described previously [43,45,48]. The number of 16S rRNA gene copies per ng DNA of each sample was determined and frequencies of respective bacterial groups calculated proportionally to the eubacterial (V3) amplicon.
Bacterial translocation
For qualitative detection of bacterial translocation, entire MLNs, liver, spleen, kidneys, and cardiac blood were transferred into a thioglycolate broth each and incubated for maximum seven days at 37°C [49]. Bacterial growth was monitored daily by turbidity assessment. Aliquots of turbid broths were cultivated on respective solid media under aerobic, microaerobic, and obligate anaerobic conditions. Bacterial species identification was performed as described earlier [9].
Cerebral cytokine and parasitic DNA detection
Brain tissue preparation and measurement of pro-inflammatory cytokine mRNA expression by quantitative real time-PCR were performed as described previously [34]. Perfused brain tissue samples were snap-frozen and kept at -80°C. 30 mg of brain tissue were used for nucleic acid purification using the spin column based AllPrep DNA/RNA/Protein Mini Kit (QIAgen, Hilden, Germany) and following the manufacturer’s instructions. On-membrane DNase I digestion (peqGOLD, Erlangen, Germany) was performed during RNA purification. RNA and DNA purity and concentration were determined by absorbance at 230, 260 and 280 nm in a NanoDrop spectrophotometer (Fisher Scientific, Germany).
Semi-quantitative real time-PCR analyses were performed to determine parasite loads in brains. FastStart Essential DNA Green Master (Roche, Grenzach-Wyhlen, Germany) was used with 90 ng genomic DNA in a reaction volume of 20 μL. Triplicate reactions were developed in a LightCycler® 480 Instrument II (Roche, Grenzach-Wyhlen, Germany). After an initial activation step (95°C for 10 min), 45 amplification cycles were run, comprising of denaturation at 95°C for 15 sec, annealing at 60°C for 15 sec and elongation at 72°C for 15 sec. The following primers were manufactured by Tib MolBiol (Berlin, Germany) and used at a final concentration of 0.3 μM: Toxoplasma gondii B1: (Forward) 5’- TCCCCTCTgCTggCgAAAAgT-3’ and (Reverse) 5’-AgCgTTCgTggTCAACTATCgATTg-3’ [50]. Mus musculus argininosuccinate lyase (ASL) gene: (Forward) 5’-TCTTCgTTAgCTggCAACTCACCT-3’ and (Reverse) 5’-ATgACCCAgCAgCTAAgCAgATCA-3’ [51]. Parasite loads (target: Toxoplasma gondii, B1 gene) were measured relative to mouse cell number (reference: Mus musculus, argininosuccinate lyase (ASL) gene), that is the target/reference ratio calculated with the LightCycler® 480 Software release 1.5.0 (Roche, Grenzach-Wyhlen, Germany).
To determine relative gene expression, SuperScript® III Platinum® One-Step Quantitative RT-PCR System (life technologies, Darmstadt, Germany) was used with 300 ng total RNA in a reaction volume of 10 μL. Triplicate reactions were developed in a LightCycler® 480 Instrument II (Roche, Grenzach-Wyhlen, Germany). Reverse transcription was performed for 15 min at 50°C followed by 2 min at 95°C. Subsequently, 45 amplification cycles were run, comprising of denaturation at 95°C for 15 sec and annealing/elongation at 60°C for 30 sec. TaqMan® Gene Expression Assays (life technologies, Darmstadt, Germany) were used for amplification of the house-keeping gene HPRT, IFN-γ TNF-α, and IL-6. HPRT expression was chosen as reference for normalization and target/reference ratios were calculated with the LightCycler® 480 Software release 1.5.0 (Roche, Grenzach-Wyhlen, Germany). Resulting data were further normalized on values of control groups.
Statistical analysis
Medians, means and levels of significance were determined using Mann–Whitney U-Test. Two-sided probability (P) values ≤0.05 were considered significant. All experiments were performed three times.
Competing interests
The authors have declared that no competing interests exist.
Authors’ contributions
Conceived and designed the experiments: IRD, MMH. Performed the experiments: AAK, MA, AF, LM, DS, IRD, MMH. Analyzed the data: AAK, AF, LM, DS, OL, IRD, MMH. Contributed reagents/materials/analysis tolls: SB, AAK, OL, UBG. Wrote the paper: SB, AAK, AF, IRD, MMH. All authors read and approved the final manuscript.
Authors’ information
Ildikò R Dunay and Markus M Heimesaat shared last authorship.
Contributor Information
Stefan Bereswill, Email: stefan.bereswill@charite.de.
Anja A Kühl, Email: anja.kuehl@charite.de.
Marie Alutis, Email: marie.alutis@charite.de.
André Fischer, Email: andre.fischer@charite.de.
Luisa Möhle, Email: luisa.moehle@med.ovgu.de.
Daniela Struck, Email: daniela.struck@charite.de.
Oliver Liesenfeld, Email: oliver.liesenfeld@charite.de.
Ulf B Göbel, Email: ulf.goebel@charite.de.
Ildikò R Dunay, Email: ildikodunay@gmail.com.
Markus M Heimesaat, Email: markus.heimesaat@charite.de.
Acknowledgments
This work was supported by grants from the German Research Foundation (DFG) to UBG, SB and AF (SFB633, TP A7), MMH (SFB633, TP B6), AAK (SPF633, TP Z1), IRD (DFG DU 1112/3-1, SFB854) and from the German Federal Ministery of Education and Research (BMBF) to SB (“Lab in a hanky” projects TP 1.1 and TP 8.2). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank Michaela Wattrodt, Ursula Rüschendorf, Ines Puschendorf, Alexandra Bittroff-Leben, Silvia Schulze, Gernot Reifenberger, Uwe Lohmann, and the staff of the animal research facility for excellent technical assistance, animal breeding and genotyping of mice. We are grateful to Simone Spieckermann for immunohistochemistry staining of tissue sections.
References
- Liesenfeld O, Kosek J, Remington JS, Suzuki Y. Association of CD4+ T cell-dependent, interferon-gamma-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J Exp Med. 1996;184:597–607. doi: 10.1084/jem.184.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz M, Liesenfeld O, Heimesaat MM. Immunology of Toxoplasma gondii. Immunol Rev. 2011;240:269–285. doi: 10.1111/j.1600-065X.2010.00992.x. [DOI] [PubMed] [Google Scholar]
- Khan IA, Schwartzman JD, Matsuura T, Kasper LH. A dichotomous role for nitric oxide during acute Toxoplasma gondii infection in mice. Proc Natl Acad Sci U S A. 1996;94:13955–13960. doi: 10.1073/pnas.94.25.13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesenfeld O, Kang H, Park D, Nguyen TA, Parkhe CV, Watanabe H, Abo T, Sher A, Remington JS, Suzuki Y. TNF-alpha, nitric oxide and IFN-gamma are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite Immunol. 1999;21:365–376. doi: 10.1046/j.1365-3024.1999.00237.x. [DOI] [PubMed] [Google Scholar]
- Mennechet FJL, Kasper H, Rachinel N, Li W, Vandewalle A, Buzoni-Gatel D. Lamina propria CD4+ T lymphocytes synergize with murine intestinal epithelial cells to enhance proinflammatory response against an intracellular pathogen. J Immunol. 2002;168:2988–2996. doi: 10.4049/jimmunol.168.6.2988. [DOI] [PubMed] [Google Scholar]
- Vossenkamper A, Struck D, Alvarado-Esquivel C, Went T, Takeda K, Akira S, Pfeffer K, Alber G, Lochner M, Forster I, Liesenfeld O. Both IL-12 and IL-18 contribute to small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii, but IL-12 is dominant over IL-18 in parasite control. Eur J Immunol. 2004;34:3197–3207. doi: 10.1002/eji.200424993. [DOI] [PubMed] [Google Scholar]
- Buzoni-Gatel D, Schulthess J, Menard LC, Kasper LH. Mucosal defences against orally acquired protozoan parasites, emphasis on Toxoplasma gondii infections. Cell Microbiol. 2006;8:535–544. doi: 10.1111/j.1462-5822.2006.00692.x. [DOI] [PubMed] [Google Scholar]
- Liesenfeld O. Oral infection of C57BL/6 mice with Toxoplasma gondii: a new model of inflammatory bowel disease? J Infect Dis. 2002;185(Suppl 1):S96–S101. doi: 10.1086/338006. [DOI] [PubMed] [Google Scholar]
- Heimesaat MM, Bereswill S, Fischer A, Fuchs D, Struck D, Niebergall J, Jahn HK, Dunay IR, Moter A, Gescher DM, Schumann RR, Göbel UB, Liesenfeld O. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- Heimesaat MM, Fischer A, Jahn HK, Niebergall J, Freudenberg JM, Blaut M, Liesenfeld O, Schumann RR, Göbel UB, Bereswill S. Exacerbation of murine ileitis by Toll-like receptor 4 mediated sensing of lipopolysaccharide from commensal Escherichia coli. Gut. 2007;56:941–948. doi: 10.1136/gut.2006.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C, Duncan SH, Bereswill S, Heimesaat MM. The induction of colitis and ileitis in mice is associated with marked increases in intestinal concentrations of stimulants of TLRs 2, 4, and 5. PLoS One. 2011;5:e9125. doi: 10.1371/journal.pone.0009125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Rutz M, Metzger J, Gellert T, Luppa P, Lipford GB, Wagner H, Bauer S. Toll-like receptor 9 binds single-stranded CpG-DNA in a sequence- and pH-dependent manner. Eur J Immunol. 2004;34:2541–2550. doi: 10.1002/eji.200425218. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Kim NI, Oh YK, Kim YJ, Youn J, Ahn MJ. CpG oligodeoxynucleotides induce IL-8 expression in CD34+ cells via mitogen-activated protein kinase-dependent and NF-kappaB-independent pathways. Int Immunol. 2005;17:1525–1531. doi: 10.1093/intimm/dxh345. [DOI] [PubMed] [Google Scholar]
- Minns LA, Menard LC, Foureau DM, Darche S, Ronet C, Mielcarz DW, Buzoni-Gatel D, Kasper LH. TLR9 is required for the gut-associated lymphoid tissue response following oral infection of Toxoplasma gondii. J Immunol. 2006;176:7589–7597. doi: 10.4049/jimmunol.176.12.7589. [DOI] [PubMed] [Google Scholar]
- Yarovinsky F. Innate immunity to Toxoplasma gondii infection. Nat Rev Immunol. 2014;14:109–121. doi: 10.1038/nri3598. [DOI] [PubMed] [Google Scholar]
- Andrade WA, Souza Mdo C, Ramos-Martinez E, Nagpal K, Dutra MS, Melo MB, Bartholomeu DC, Ghosh S, Golenbock DT, Gazzinelli RT. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe. 2013;13:42–53. doi: 10.1016/j.chom.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson A, Pifer R, Behrendt CL, Hooper LV, Yarovinsky F. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe. 2009;6:187–196. doi: 10.1016/j.chom.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Sher A, Yap G, Park D, Neyer LE, Liesenfeld O, Fort M, Kang H, Gufwoli E. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J Immunol. 2000;164:5375–5382. doi: 10.4049/jimmunol.164.10.5375. [DOI] [PubMed] [Google Scholar]
- Munoz M, Heimesaat MM, Danker K, Struck D, Lohmann U, Plickert R, Bereswill S, Fischer A, Dunay IR, Wolk K, Loddenkemper C, Krell HW, Libert C, Lund LR, Frey O, Holscher C, Iwakura Y, Ghilardi N, Ouyang W, Kamradt T, Sabat R, Liesenfeld O. Interleukin (IL)-23 mediates Toxoplasma gondii-induced immunopathology in the gut via matrixmetalloproteinase-2 and IL-22 but independent of IL-17. J Exp Med. 2009;206:3047–3059. doi: 10.1084/jem.20090900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foureau DM, Mielcarz DW, Menard LC, Schulthess J, Werts C, Vasseur V, Ryffel B, Kasper LH, Buzoni-Gatel D. TLR9-dependent induction of intestinal alpha-defensins by Toxoplasma gondii. J Immunol. 2010;184:7022–7029. doi: 10.4049/jimmunol.0901642. [DOI] [PubMed] [Google Scholar]
- Heimesaat MM, Dunay IR, Fuchs D, Trautmann D, Fischer A, Kühl AA, Loddenkemper C, Siegmund B, Batra A, Bereswill S, Liesenfeld O. The distinct roles of MMP-2 and MMP-9 in acute DSS colitis. Eur J Microbiol Immunol (Bp) 2011;1:302–310. doi: 10.1556/EuJMI.1.2011.4.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deloris Alexander A, Orcutt RP, Henry JC, Baker JJR, Bissahoyo AC, Threadgill DW. Quantitative PCR assays for mouse enteric flora reveal strain-dependent differences in composition that are influenced by the microenvironment. Mamm Genome. 2006;17:1093–1104. doi: 10.1007/s00335-006-0063-1. [DOI] [PubMed] [Google Scholar]
- Ge Z, Feng Y, Taylor NS, Ohtani M, Polz MF, Schauer DB, Fox JG. Colonization dynamics of altered Schaedler flora is influenced by gender, aging, and Helicobacter hepaticus infection in the intestines of Swiss Webster mice. Appl Environ Microbiol. 2006;72:5100–5103. doi: 10.1128/AEM.01934-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydora BC, Macfarlane SM, Walker JW, Dmytrash AL, Churchill TA, Doyle J, Fedorak RN. Epithelial barrier disruption allows nondisease-causing bacteria to initiate and sustain IBD in the IL-10 gene-deficient mouse. Inflamm Bowel Dis. 2007;13:947–954. doi: 10.1002/ibd.20155. [DOI] [PubMed] [Google Scholar]
- Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL, Dunne WMJR, Allen PM, Stappenbeck TS. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe. 2011;9:390–403. doi: 10.1016/j.chom.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- Sartor RB. Genetics and environmental interactions shape the intestinal microbiome to promote inflammatory bowel disease versus mucosal homeostasis. Gastroenterology. 2010;139:1816–1819. doi: 10.1053/j.gastro.2010.10.036. [DOI] [PubMed] [Google Scholar]
- O’Mahony D, Murphy S, Boileau T, Park J, O’Brien F, Groeger D, Konieczna P, Ziegler M, Scully, Shanahan PF, Kiely B, O’Mahony L. Bifidobacterium animalis AHC7 protects against pathogen-induced NF-kappaB activation in vivo. BMC Immunol. 2010;11:63. doi: 10.1186/1471-2172-11-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunay IR, Heimesaat MM, Bushrab FN, Muller RH, Stocker H, Arasteh K, Kurowski M, Fitzner R, Borner K, Liesenfeld O. Atovaquone maintenance therapy prevents reactivation of toxoplasmic encephalitis in a murine model of reactivated toxoplasmosis. Antimicrob Agents Chemother. 2004;48:4848–4854. doi: 10.1128/AAC.48.12.4848-4854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohle L, Parlog A, Pahnke J, Dunay IR. Spinal cord pathology in chronic experimental Toxoplasma gondii infection. Eur J Microbiol Immunol (Bp) 2014;4:65–75. doi: 10.1556/EuJMI.4.2014.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunay IR, Damatta A, Fux B, Presti R, Greco S, Colonna M, Sibley LD. Gr1 (+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GB. Encephalopathy of infection and systemic inflammation. J Clin Neurophysiol. 2013;30:454–461. doi: 10.1097/WNP.0b013e3182a73d83. [DOI] [PubMed] [Google Scholar]
- Mishra BB, Mishra PK, Teale JM. Expression and distribution of Toll-like receptors in the brain during murine neurocysticercosis. J Neuroimmunol. 2006;181:46–56. doi: 10.1016/j.jneuroim.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty M, Bowie AG. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol. 2011;81:825–837. doi: 10.1016/j.bcp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Basset C, Holton J. Inflammatory bowel disease: is the intestine a Trojan horse? Sci Prog. 2002;85:33–56. doi: 10.3184/003685002783238861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinesi M, Treves C, Bonanomi AG, Milla M, Bagnoli S, Zuegel, Steinmeyer UA, Stio M. Down-regulation of adhesion molecules and matrix metalloproteinases by ZK 156979 in inflammatory bowel diseases. Clin Immunol. 2010;136:51–60. doi: 10.1016/j.clim.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Naito Y, Takagi T, Kuroda M, Katada K, Ichikawa H, Kokura S, Yoshida N, Okanoue NT, Yoshikawa T. An orally active matrix metalloproteinase inhibitor, ONO-4817, reduces dextran sulfate sodium-induced colitis in mice. Inflamm Res. 2004;53:462–468. doi: 10.1007/s00011-004-1281-1. [DOI] [PubMed] [Google Scholar]
- Wang M, Qin X, Mudgett JS, Ferguson TA, Senior RM, Welgus HG. Matrix metalloproteinase deficiencies affect contact hypersensitivity: stromelysin-1 deficiency prevents the response and gelatinase B deficiency prolongs the response. Proc Natl Acad Sci U S A. 1999;96:6885–6889. doi: 10.1073/pnas.96.12.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimesaat MM, Nogai A, Bereswill S, Plickert R, Fischer A, Loddenkemper C, Steinhoff U, Tchaptchet S, Thiel E, Freudenberg MA, Göbel UB, Uharek L. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut. 2010;59:1079–1087. doi: 10.1136/gut.2009.197434. [DOI] [PubMed] [Google Scholar]
- Heimesaat MM, Plickert R, Fischer A, Göbel UB, Bereswill S. Can microbiota transplantation abrogate murine colonization resistance against Campylobacter jejuni? Eur J Microbiol Immunol (Bp) 2013;3:36–43. doi: 10.1556/EuJMI.3.2013.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereswill S, Fischer A, Plickert R, Haag LM, Otto B, Kühl AA, Dashti JL, Zautner AE, Munoz M, Loddenkemper C, Gross U, Göbel UB, Heimesaat MM. Novel murine infection models provide deep insights into the “Menage a Trois” of Campylobacter jejuni, microbiota and host innate immunity. PLoS One. 2011;6:e20953. doi: 10.1371/journal.pone.0020953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag LM, Fischer A, Otto B, Plickert R, Kühl AA, Göbel UB, Bereswill S, Heimesaat MM. Campylobacter jejuni induces acute enterocolitis in gnotobiotic IL-10-/- mice via Toll-like-receptor-2 and -4 signaling. PLoS One. 2012;7:e40761. doi: 10.1371/journal.pone.0040761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimesaat MM, Haag LM, Fischer A, Otto B, Kühl AA, Göbel UB, Bereswill S. Survey of extra-intestinal immune responses in asymptomatic long-term Campylobacter jejuni-infected mice. Eur J Microbiol Immunol (Bp) 2013;3:174–182. doi: 10.1556/EuJMI.3.2013.3.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch S, Held J, Fischer A, Heimesaat MM, Kühl AA, Bereswill S, Hartmann S. Small intestinal nematode infection of mice is associated with increased enterobacterial loads alongside the intestinal tract. PLoS One. 2013;8:e74026. doi: 10.1371/journal.pone.0074026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimesaat MM, Boelke S, Fischer A, Haag LM, Loddenkemper C, Kühl AA, Göbel UB, Bereswill S. Comprehensive postmortem analyses of intestinal microbiota changes and bacterial translocation in human flora associated mice. PLoS One. 2012;7:e40758. doi: 10.1371/journal.pone.0040758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EH, Wille-Reece U, Dzierszinski F, Hunter CA. A critical role for IL-10 in limiting inflammation during toxoplasmic encephalitis. J Neuroimmunol. 2005;165:63–74. doi: 10.1016/j.jneuroim.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Butcher BA, Fox BA, Rommereim LM, Kim SG, Maure KJ, Yarovinsky F, Herbert DR, Bzik DJ, Denker EY. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 2011;7:e1002236. doi: 10.1371/journal.ppat.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]