

Abstract
Background
Shwachman–Diamond syndrome (SDS) is an autosomal recessive ribosomopathy caused mainly by compound heterozygous mutations in SBDS. Structural variation (SV) involving the SBDS locus has been rarely reported in association with the disease. We aimed to determine whether an SV contributed to the pathogenesis of a case lacking biallelic SBDS point mutations.
Case presentation
Whole exome sequencing was performed in a patient with SDS lacking biallelic SBDS point mutations. Array comparative genomic hybridization and Southern blotting were used to seek SVs across the SBDS locus. Locus-specific polymerase chain reaction (PCR) encompassing flanking intronic sequence was also performed to investigate mutation within the locus. RNA expression and Western blotting were performed to analyze allele and protein expression. We found the child harbored a single missense mutation in SBDS (c.98A > C; p.K33T), inherited from the mother, and an SV in the SBDS locus, inherited from the father. The missense allele and SV segregated in accordance with Mendelian expectations for autosomal recessive SDS. Complementary DNA and western blotting analysis and locus specific PCR support the contention that the SV perturbed SBDS protein expression in the father and child.
Conclusion
Our findings implicate genomic rearrangements in the pathogenesis of some cases of SDS and support patients lacking biallelic SBDS point mutations be tested for SV within the SBDS locus.
Keywords: Shwachman-Diamond syndrome, SBDS, Structural variation, Genomic rearrangement, Non-allelic homologous recombination, Low copy repeat, Whole exome sequencing, Copy number variation, Recessive disease
Background
Shwachman Diamond syndrome (SDS; OMIM 260400) is a rare autosomal recessive condition characterized by bone marrow dysfunction, typically neutropenia that can progress to aplastic anemia; and pancreatic exocrine insufficiency, which may improve beyond early childhood [1]. Skeletal abnormalities, such as metaphyseal chondrodysplasia and thoracic dystrophy, and short stature are also common. SBDS, located at 7q11.21, is the only gene associated with SDS to date [2]. Compound heterozygous mutations in SBDS can be identified in 75% to 98% of SDS patients [3,4]. The majority of mutated SBDS alleles, 74% in the pioneering study [2], are the result of recurrent gene conversion events between SBDS and the SBDSP1 pseudogene, which shares 97% nucleotide sequence identity with SBDS and resides in an inverted orientation at a locally duplicated genomic segment, which maps ~5.8 Mb in the telomeric direction. This inverted repeat genomic architecture is predicted to result in genomic instability by providing substrates for non-allelic homologous recombination (NAHR) and inversion structural variation (SV) [5-7]. The NAHR hypothesis is further supported by the high frequency that disease-associated alleles result from apparent gene conversion events, likely reflecting alternate resolution of a Holliday structure. Nevertheless, large structural variants have been rarely implicated in the pathogenesis of the disease; in fact, only a paternally inherited Alu-mediated deletion that removes exon 3 and part of flanking introns has been reported thus far [8].
SBDS is comprised of 5 exons, spanning 7.9 kb, has a 1.6 kb transcript and encodes a 250 amino acid protein, involved in ribosome biogenesis [2,9,10]. The two recurrent gene conversion mutations encode either a frameshift (p.84Cfs3) or nonsense (p.K62X) mutation [2]. Patients carrying compound heterozygous nonsense and/or frameshift mutations lack detectable SBDS protein by immunoblotting using SBDS antibody raised against the carboxy-terminal peptide [10]. Importantly, patients carrying compound heterozygous mutations, in which one leads to protein truncation and the other is a missense mutation, demonstrate markedly decreased SBDS protein expression in comparison to unaffected family members that carry only one heterozygous mutation. The SBDS protein expression in heterozygous carriers is often comparable to family members carrying non-altered SBDS alleles [11]. Thus, it has been suggested that the SDS disease phenotype is a consequence of expression of hypomorphic SBDS alleles [12].
We investigated a patient with SDS with severe disease manifestations who carried a single novel SBDS missense mutation. We uncovered an SV on the remaining allele, which resulted in altered SBDS expression and markedly decreased SBDS protein.
Case presentation
Methods
Research subjects
The proband with SDS was ascertained at the Texas Children’s Hospital Hematology Center. Informed consent for participation, sample collection and analysis was obtained using protocols H-17698 and H-23793, approved by the Institutional Review Board for Baylor College of Medicine and affiliated hospitals.
Lymphoblastoid cell lines
Epstein-Barr virus-transformed lymphoblastoid cell lines (LCLs) were generated from the proband (BAB3762), her parents (BAB3763 - mother, BAB3764 - father) and siblings (unaffected full sisters BAB5568, BAB5569, and BAB5570) by the Tissue Culture Core within the Intellectual and Developmental Disabilities Research Center at Baylor College of Medicine. Cells were grown in RPMI 1640 medium containing L-glutamine (Invitrogen) and 10% FBS (Invitrogen).
Array comparative genomic hybridization
Custom-designed array comparative genomic hybridization (aCGH) version 8.1, also containing probes for single nucleotide polymorphism (SNP) analysis, was performed by the Baylor College of Medicine Medical Genetics Laboratories (MGL, http://www.bcm.edu/geneticlabs/) (Agilent Technologies, Inc., Santa Clara, CA) [13-15]. In addition, a high-density custom tiling-path oligonucleotide microarray spanning the SBDS gene and flanking regions [(hg19): chr7:66,422,690-66,480,588] was designed using eArray (http://earray.chem.agilent.com/earray/). The average coverage was 1 probe per 391 bp. Probe labeling and hybridization were performed according to the manufacturer’s protocols with modifications as described [16].
Illumina library construction and exome capture
Genomic DNA samples were constructed into Illumina paired-end libraries and processed through exome capture according to the manufacturer’s specifications with modifications as described in the BCM-HGSC Illumina Non-Barcoded Paired-End Capture Library Preparation protocol (https://www.hgsc.bcm.edu/sites/default/files/documents/BCM-HGSC_Illumina_Non-Barcoded_Paired-End_Capture_Library_Preparation.pdf).
Pre-capture libraries were prepared using Beckman robotic workstations (Biomek NXp and FXp models). Briefly, 5 μg of genomic DNA in 50 μl volume was sheared into fragments of approximately 400 bp in a Covaris plate with E210 system (Covaris, Inc. Woburn, MA) followed by end-repair, A-tailing and ligation of the Illumina non-barcoded PE adaptors. Pre-capture ligation mediated-PCR (LM-PCR) was performed for 7 cycles of amplification using the 2X SOLiD Library High Fidelity Amplification Mix (a custom product manufactured by Invitrogen). Purification was performed with Agencourt AMPure XP beads after enzymatic reactions. Following the final XP beads purification, quantification and size distribution of the pre-capture LM-PCR product was determined using the LabChip GX electrophoresis system (PerkinElmer).
For exome capture, pre-capture libraries (~1 μg) were individually hybridized in solution to the SeqCap EZ Exome 2.0 design (44 Mb, NimbleGen). Human COT1 DNA and 3’-ddC modified hybridization enhancing oligonucleotides were added into the hybridization to block repetitive genomic sequences and the adaptor sequences. Post-capture LM-PCR amplification was performed using the 2X SOLiD Library High Fidelity Amplification Mix with 14 cycles of amplification. After the final AMPure XP bead purification, quantity and size of the capture library was analyzed using the Agilent Bioanalyzer 2100 DNA Chip 7500. The efficiency of the capture was evaluated by performing a qPCR-based quality check on the four standard NimbleGen internal controls. Successful enrichment of the capture libraries was estimated to range from a 6 to 9 of ΔCt value over the non-enriched samples.
Illumina sequencing
Library templates were prepared for sequencing using Illumina’s cBot cluster generation system with TruSeq PE Cluster Generation Kits (Part No. PE-401-3001). Briefly, these libraries were denatured with sodium hydroxide and diluted to 3 pM in hybridization buffer in order to achieve a load density of ~800 K clusters/mm2. Each library was loaded in a single lane of a HiSeq flow cell, and each lane was spiked with 2% phiX control library for run quality control. The sample libraries then underwent bridge amplification to form clonal clusters, followed by hybridization with the sequencing primer. Sequencing runs were performed in paired-end mode using the Illumina HiSeq 2000 platform. Using the TruSeq SBS Kits (Part No. FC-401-3001), sequencing-by-synthesis reactions were extended for 101 cycles from each end. Sequencing runs generated approximately 200 million successful reads on each lane of a flow cell, yielding an average of 19 Gb per sample. With these sequencing yields, samples achieved an average of 91% of the targeted exome bases covered to a depth of 20X or greater.
Data analysis and variant calling
Illumina sequence analysis was performed using the HGSC Mercury analysis pipeline (https://www.hgsc.bcm.edu/software/mercury). First, the primary analysis software on the instrument produces .bcl files that are transferred off-instrument into the HGSC analysis infrastructure by the HiSeq Real-time Analysis module. Once the run is complete and all .bcl files are transferred, Mercury runs the vendor’s primary analysis software (CASAVA), which generates sequence reads and base-call confidence values (qualities). The next step is the mapping of reads to the NCBI36 Human reference genome (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/) using the Burrows-Wheeler aligner (BWA [17], http://bio-bwa.sourceforge.net/) and producing a BAM [18] (binary alignment/map) file. The third step utilizes GATK [19] (http://www.broadinstitute.org/gatk/) for quality recalibration and also includes BAM sorting, duplicate read marking, and realignment to improve in/del discovery. Next, the Atlas2 [20] suite (Atlas-SNP and Atlas-indel) is used to call variants and produce a variant call file (VCF [21]). Finally, annotation data is added to the VCF using a suite of annotation tools “Cassandra” [22] that brings together frequency, function, and other relevant information using AnnoVar with UCSC and RefSeq gene models, as well as a host of other internal and external data resources.
Probe design and Southern blot hybridization
A DNA probe was amplified by PCR from BAC clone RP11-347P19 (product size: 322 bp) using primers SBDS_KPN_F: 5’CCCATGCCAATCATTTCTCT3’ and SBDS_KPN_R: 5’GAGACGCACCGAGCTACC 3’, which target the 5’ flanking region of the gene SBDS. Genomic DNA was digested with KpnI or XbaI (New England BioLabs, Ipswich, MA) for 1 day at 37°C, followed by separation on a 0.7% agarose gel in 0.5X Tris–Borate–EDTA buffer. Hybridization was performed as described [23].
RNA extraction and cDNA preparation
Total RNA was extracted from LCLs using TRIzol reagent (Invitrogen Corp., Carlsbad, CA); this was DNaseI treated and purified using the RNeasy mini kit according to the manufacturer’s protocol (Qiagen, Valencia, CA). cDNA was synthesized from 1 μg of RNA using qScript cDNA Super Mix (Quanta Biosciences, Gaithersburg, MD).
Short- and long-range PCR amplification, PCR digestion and Sanger sequencing
Each of the five SBDS exons were amplified using primers described in Woloszynek, et al. [11], except for exon 3 which was amplified using primers exon3f1 5’ GATTGTAGTGAGCCGAGATCATACT 3’ and exon3r1 5’CTCCATCCAGTTACTCATTTTTTATG 3’. The amplicons were Sanger sequenced in both forward and reverse directions.
To assay for the presence of short insertions or deletions in the SBDS gene or flanking regions, we used primer Del_Fb 5’GTGTCAATTTTCCCCATGCT3’ in combination with primer SBDS_KPN_R to produce a 13.1 kb PCR product, which spans the entire SBDS gene plus flanking regions [(hg19): chr7:66,448,791-66,461,897]. SBDSP1 is not amplified using those primers. The long-range PCR was performed using TaKaRa LA Taq (Clontech, Mountain View, CA). Long-range PCR products were digested using restriction enzymes KpnI and SacI (New England BioLabs, Ipswich, MA) for 2 hours at 37°C, followed by separation on a 1% agarose gel in 0.5X Tris–Borate–EDTA buffer. Amplification of the SBDS transcript was performed using cDNA obtained as described above and primers SBDS_3utr_F1 5’GCAGCATGTTCAATGAAAGGTAA3’ and SBDS_5utr_R1 5’ CCTGCCAGACACACTGTGA3’ to generate a 1.4 kb PCR product, which was sequenced by bidirectional Sanger sequencing.
Extraction of protein and western blot analysis
Lymphoblastoid cells were harvested at a density of approximately 5×105 cells/mL, re-suspended in 200 μL of RIPA buffer + 1% Set III protease inhibitors (VWR) and 1% PMSF (Sigma) and incubated for 30 minutes on ice. The lysate was collected by centrifugation at 21,000 g for 30 minutes at 4°C. Fifty μg of lysate was fractionated by 10% SDS-PAGE. Following transfer to nitrocellulose membrane, the SBDS protein was detected using rabbit α-SBDS antibody (Abcam catalog# ab128946) at 1:5,000 dilution and α-rabbit IgG-IR800 secondary antibody (Li-Cor catalog# 926–32211) at 1:5,000 dilution. The membrane was also probed for β-actin using α-β-actin antibody (Sigma cat# A5441) at 1:5000 dilution and α-mouse IgG-IR800 secondary (Li-Cor Cat# 926–32210) at 1:5,000 dilution.
Results
Clinical report
The patient was a white/Hispanic female who was previously evaluated at age 4 years by our immunology and genetics services for recurrent infections, short stature and skeletal abnormalities. She was noted to have the unusual finding of normal serum immunoglobulins with a virtual absence of circulating B cells. Additional clinical features were suggestive of Shwachman Diamond syndrome, although it was noted she carried only a single missense change, which was not previously reported and possibly deleterious, in the SBDS gene. Her case was described in a brief report [24]. Here, we elaborate on her clinical features and clinical course and further explore an underlying molecular etiology.
In addition to her evaluations by the immunology and genetics services, she was referred to our hematology center at age 4 years for progressive pancytopenia, with macrocytic anemia. Her absolute neutrophil count fluctuated between <100 to >1,500 cells/μl over a period of 2 ½ years prior to initiating granulocyte-colony stimulating factor (G-CSF). Her bone marrow showed maturing trilineage hematopoiesis with moderate to, eventually, marked hypocellularity (10% cellularity). Additional evaluations demonstrated an elevated hemoglobin F (4.9%), chromosome breakage studies with mitomycin C and diepoxybutane (DEB) within the normal range, and lymphocyte and granulocyte telomere lengths at ~ 40th and <1st percentiles for age, respectively. She became both red blood cell and platelet transfusion-dependent and remained neutropenic despite administration of G-CSF at a dose of 10 μg/kg/day, eventually developing a pseudomonal soft tissue abscess while on G-CSF. She underwent bone marrow transplantation (BMT) for her bone marrow failure with an histocompatibility locus antigen (HLA)-identical sibling donor using a reducing intensity conditioning regimen and remained engrafted without significant transplant-related complications 2 ½ years after BMT, the time of this manuscript preparation.
The patient had no history of diarrhea or steatorrhea; however, her serum pancreatic isoamylase (ascertained at age 6 years at The Hospital for Sick Children, Toronto) was below the age-adjusted normal range (7 U/L, lower limits of normal 13 U/L), consistent with an SDS pancreatic exocrine deficiency state. Her trypsinogen (93.2 ng/ml) was above normal limits for age (46.5 ng/ml). Despite these findings, her pancreas appeared normal by ultrasound, computed tomography (CT) and magnetic resonance imaging (MRI) (Figure 1a–c), when last obtained at age approximately 6 years. This is an unusual observation, since pancreatic lipomatosis is typically demonstrated by imaging in patients with SDS [25]. In addition, she manifested hyperlipasemia without symptoms of acute or chronic pancreatitis, possibly secondary to macrolipasemia.
Figure 1.
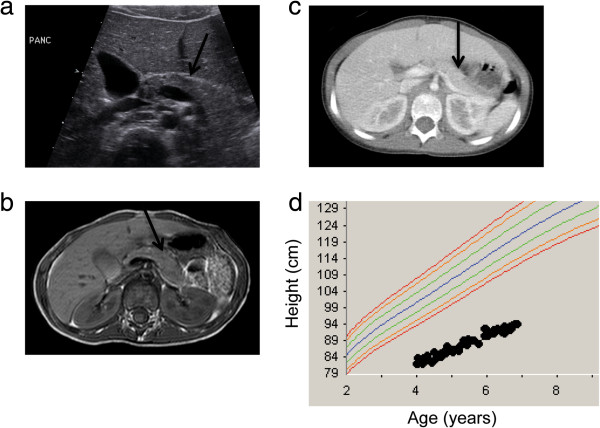
Clinical features of SDS proband. a. Transverse abdominal ultrasound image obtained at 5 years 11 months of age revealing normal size, contour, and echogenicity of the pancreas, indicated by arrow. b. Axial conventional spin echo T1-weighted MR image obtained at 5 years 11 months of age showing normal signal intensity of the pancreatic parenchyma with no evidence of atrophy or fatty infiltration of the pancreas, indicated by arrow. c. Axial contrast-enhanced CT image obtained at 6 years 5 months of age demonstrating normal attenuation of the pancreatic parenchyma with no evidence of atrophy or fatty infiltration of the pancreas, indicated by arrow. d. The proband’s growth curve showing markedly short stature. The bottom-most curve normal curve represents the 5th percentile for girls.
Her medical history was also notable for a full term delivery with intrauterine growth retardation (birth weight 2.24 kg), a prolonged postnatal admission for poor feeding, global developmental delay, extreme short stature (<1st percentile) (Figure 1d), marked pectus carinatum [24], metaphyseal chondrodysplasia, tracheomalacia with recurrent lower respiratory tract infections and acute otitis media, severe myopia and chronic hypomagnesemia. Her parents each reported being healthy. She had three healthy full siblings and a sibling stillborn at 5 months gestation.
Molecular genetics
A diagnosis of SDS was rendered based on established clinical criteria of (1) a hematological cytopenia of any given lineage on at least 2 occasions over at least 3 months and (2) exocrine pancreas dysfunction [26]. Bone abnormalities supported the diagnosis. Her SBDS sequence analysis revealed a novel, heterozygous missense variant in SBDS exon 1, c.98A > C (p.K33T), mapping to the N-terminal domain (domain I); a region in which most of the SBDS disease-causing mutations map [12]. Although this particular variant was novel, a mutation altering the same codon, c.97A > G (p.K33E), was previously reported in a patient with SDS [12]. The mapping of the p.K33E mutation onto the Archaeoglobus fulgidus SBDS protein orthologue crystal structure led to the prediction that it altered surface epitopes [12]. Furthermore, cells from a SDS patient carrying the p.K33E mutant protein had altered ribosomal profiles and impaired association of the 40S and 60S subunits; causality was further demonstrated by in vitro studies of ribosomal subunit joining using cells expressing the p.K33E variant [9]. Thus, the c.98A > C (p.K33T) variant detected in our patient was deemed likely deleterious. Notably, however, c.98A > C (p.K33T) was the only SBDS variant detected.
The inability to identify biallelic mutations at the SBDS locus prompted us to explore potentially causative genetic and genomic variation for her disease. Array comparative genomic hybridization with SNP analysis was performed at Baylor College of Medicine Medical Genetics Laboratories as a clinical diagnostic evaluation using a custom-designed array and revealed no copy number variation (CNV) or regions of copy neutral absence of heterozygosity (AOH). Whole exome sequencing of peripheral blood DNA from the proband/parental trio confirmed the presence of the heterozygous missense mutation in SBDS exon 1, c.98A > C (p.K33T) and established that it was inherited from the mother. Inspection for deleterious novel or rare single nucleotide variants that were de novo or inherited from the father failed to yield any potential variants in candidates that might potentially impact ribosome biology and, therefore, might result in a condition due to digenic or oligogenic inheritance.
Sanger sequencing of the three unaffected siblings indicated that they, too, inherited the missense mutant allele from the mother and analysis of SBDS cDNA derived from EBV-transformed LCLs demonstrated that the missense mutant allele was expressed in all but one of the carrier family members (Figure 2a). Remarkably, one of the carrier sister’s (BAB5569) LCLs expressed exclusively the paternally inherited allele, whereas the proband’s (BAB3762) LCLs expressed exclusively the maternally inherited allele, in this assay on clonally derived cells (Figure 2a). These results are consistent with prior reports indicating that the SBDS locus might be subjected to differential allele expression in LCLs [27].To determine if the differential allele expression would lead to altered levels of the SBDS protein, we analyzed SBDS levels in the family’s LCLs by western blotting. Notably, both the unaffected father (BAB3764) and the proband’s cells’ SBDS levels were decreased relative to the other family members, including sibling BAB5569, all of which demonstrated levels of SBDS protein similar to each other and an unrelated normal wild type control (Figure 2b).
Figure 2.
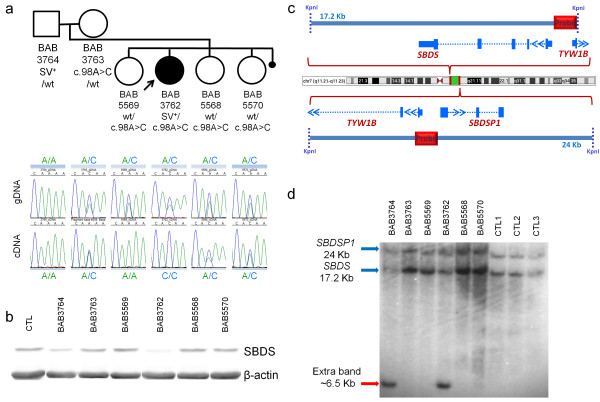
Segregation of private mutations spanning SBDS in family HOU1479. Missense mutation c.98A > C was maternally inherited by all four children whereas the SV is present only in the father and the patient with SDS. a. Top: Pedigree of family HOU1479 with information about carrier status of both mutations in each family member. Bottom: SBDS allele status with corresponding chromatograms of exon 1 Sanger sequenced using genomic DNA (gDNA) or cDNA prepared from RNA extracted from LCLs. b. Western blot analysis of SBDS in LCL whole cell extracts prepared from an unrelated, unaffected control (CTL), the proband and her family members as indicated. Beta-actin blotting was performed for a protein loading control. c. Genomic organization of SBDS and SBDSP1 with identification of the region of the probe used for Southern blot in d. d. Southern blotting of SBDS and SBDSP1 in samples from family HOU1479 and three unrelated, unaffected controls (CTL1-3) using KpnI restriction enzyme.
The differences in the SBDS protein levels between the mother and siblings, who are heterozygous for the p.K33T mutation, and the father and the proband led us to suspect that the proband and father shared an additional SBDS molecular alteration not yet revealed by the aforementioned methods. To explore this possibility further, we carried out a variety of molecular approaches: i) additional aCGH, employing a customized high-resolution array spanning the SBDS gene and flanking regions, to detect large deletions or duplications; ii) Southern blotting to interrogate the SBDS locus; iii) long-range PCR specific for the SBDS gene and flanking regions (PCR product of 13.1 kb) followed by digestion using KpnI and SacI in order to detect abnormal size bands relative to samples without the rearrangement; and iv) short-range PCR followed by Sanger sequencing of the SBDS exons to detect small deletions and duplications or insertions; all approaches which might uncover genomic rearrangements or SVs.
High-resolution aCGH of the SBDS gene showed no evidence of CNV. Remarkably, however, Southern blot analysis using KpnI and a probe specific for the 5’ intergenic region of SBDS (Figure 2c) revealed an additional band, approximately 6.5 kb in length, shared by the father and proband and not present in any of the other family members or 11 additional controls (Figure 2d and data not shown). In addition, the father and proband’s samples demonstrated a relative reduction in the SBDS 17.2 kb signal (Figure 2d). The presence of a genomic rearrangement, likely an insertion of unknown genomic sequence rather than a point mutation, was supported by additional Southern blotting using a different restriction enzyme (XbaI) which produced an extra band of ~ 7.0 kb in both the father and proband (Figure 3).
Figure 3.
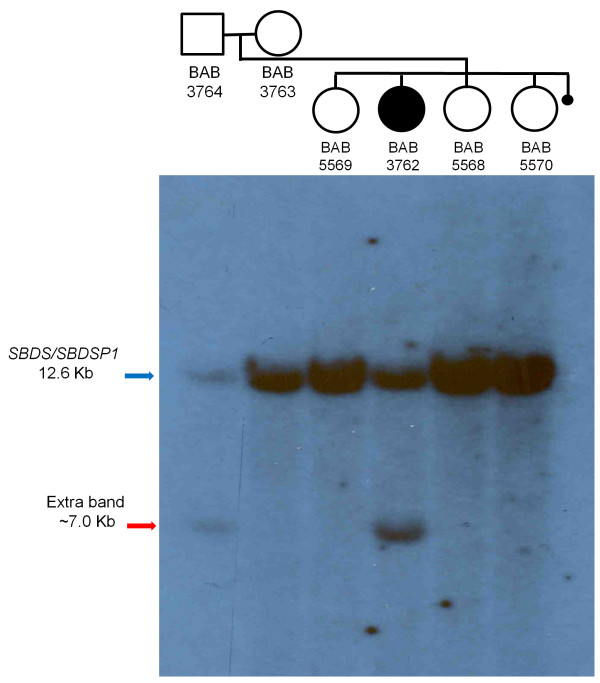
Southern blotting of SBDS and SBDSP1 in samples from family HOU1479 using Xba I restriction enzyme.
Short- and long-range PCR helped to delineate the nature of such an insertion further. Both approaches failed to reveal band sizes different in the patient and paternal samples from those expected (Figure 4a and data not shown). In addition, digestion of the long-range PCR product of 13.1 kb that includes SBDS with KpnI and SacI showed a similar pattern in all samples (Figure 4a). However, in contrast to the detection of both maternal and paternal polymorphic SNPs in the 5’ untranslated region and introns 1 and 3, short range PCR and Sanger sequencing failed to demonstrate the paternal genotype in the patient (BAB3762) for two polymorphic SNPs in intron 2 (Figure 4b). Taken together, these results are consistent with a lack of PCR amplification of the paternal allele that had been revealed by Southern blotting (Figure 4b) due to an insertion resulting in an amplification product larger than what could be afforded by the DNA polymerases used here (data not shown).
Figure 4.
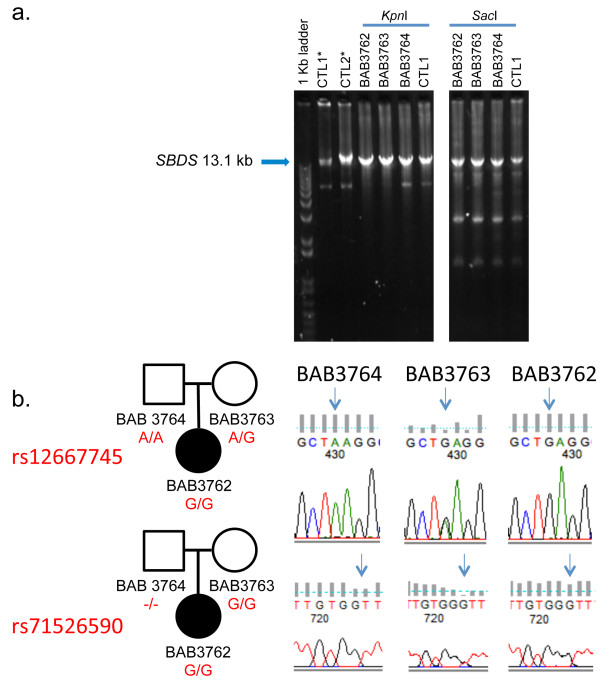
Additional studies of the genomic segment that includes SBDS supports the presence of an insertion of unknown origin within the paternal allele. a. Agarose gel analysis of a 13.1 kb long-range PCR product spanning SBDS and flanking region, the region targeted by the Southern blotting studies (Figures 2 and 3). DNA from patient (BAB3762), parents (BAB3763- mother, BAB3764-father) and a normal controls (CTL1 and CTL2) were amplified using primers DelFb + KpnR, and then digested with either KpnI or SacI. KpnI did not digest the 13.1 kb PCR product, consistent with lack of amplification of the paternal allele. Consistently, SacI digestion of the 13.1 kb PCR product showed an identical pattern in samples and controls. b. Sanger sequencing of intron 2 amplified along with exon 2 using short range PCR revealed inconsistent segregation of a paternal genotype for two polymorphic SNPs in the patient (BAB3762). *Non-digested PCR product.
Discussion
Our findings implicate a genomic rearrangement at the SBDS locus in the pathogenesis of the SDS phenotype in our patient and extend the underlying molecular genetic causes of this disease. The molecular nature of the putative genomic alteration was not determined by our study, but we speculate that it is likely a DNA insertion of at least 2.8 kb that introduced new KpnI and XbaI sites distal to the probe annealing site (Figure 5a). Insertion of genomic segments can account for at least 42% of SVs, as observed in eight individual genomes cloned in fosmid libraries and subjected to the paired end sequence approach [28]; 20% of those selected for complete sequencing result from retrotranspositional events [28]. Retrotransposition along with insertional translocation events are especially challenging to uncover in CNV studies due to the current lack of appropriate technical tools for high-throughput screening. Additional difficulty is provided by insertion of sequences within low-copy repeat (LCR) or segmental duplications regions, which can be associated with as many as 71% of the CNVs present in an individual genome [29]. Here, we speculate that an unknown segment is inserted in the genomic interval demarcated by the 5’ flanking region to intron 1 or 2 of the SBDS locus (Figure 5a), which results in a null allele or leads to an altered regulation of SBDS. Both of the latter alternatives are consistent with the decrease in protein expression experimentally observed in the LCLs from the carriers reported herein. Whereas an SV present within the pseudogene SBDSP1 locus can not be ruled out at this point, it is less likely because there is no common probe target region with the band size obtained upon genomic digestion with KpnI (Figure 5b).
Figure 5.
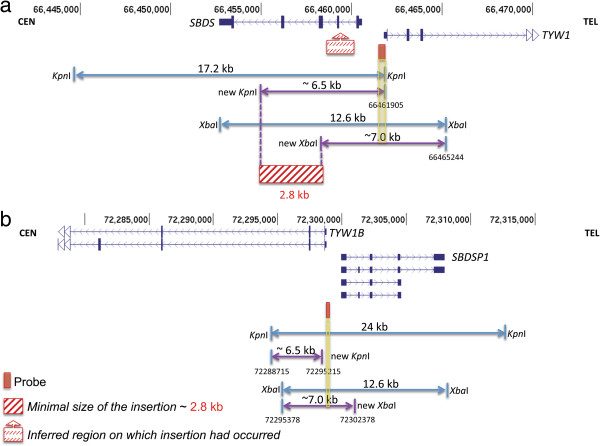
Approximate location of the inherited SV at the SBDS locus based on Southern blotting assays using KpnI and XbaI restriction enzymes. a. Size and location of the expected segments obtained using KpnI and XbaI restriction enzymes based on the reference genome (blue arrows) and extra segments obtained exclusively in SV carriers (BAB3762 and BAB3764 - purple arrows). The nature of the SV is currently unknown although the Southern blotting results are consistent with a genomic insertion of at least 2.8 kb (represented by a crosshatch red box) somewhere between SBDS 5’flanking region and intron 3 (represented by a red box with an arrow on top). Red box: Southern blotting probe. Yellow rectangle highlights a common probe target region between the reference genome and the genome of the SV carriers. b. An alternative hypothesis of SV present within the pseudogene. Genomic coordinates as of GRCh37/ hg19 to SBDS and SBDSP1 loci are represented on top of each region. CEN: centromere; TEL: telomere.
Conclusions
Our findings lead us to hypothesize that genomic rearrangements contribute to the underlying molecular cause of some cases of SDS without biallelic SBDS mutations. In addition to insertion of genomic segments, other structural alterations such as large deletions, duplications or inversions are anticipated due to the peculiar genomic architecture of the SBDS/SBDSP1 loci. Each of the loci resides embedded in a pair of inverted-oriented LCRs consisting of ~301 kb and ~271 kb, respectively, that map ~5.8 Mb apart; the presence of such large LCRs may render the 7q11.21 region prone to instability [5,30]. For instance, intrachromosomal inverted-oriented LCRs may be substrates for NAHR, which can result in segmental inversion of the genomic region in between the LCRs, as exemplified by the H1 and H2 inverted haplotypes at the disease-associated 17q21.31 region [31-33] and recombinant/hybrid LCRs. We predicted previously that formation of hybrid LCRs by NAHR using gene and pseudogenes as substrates could lead to disruption of important disease-associated genes including SBDS[30]. Indeed, NAHR between inverted LCRs present within the IDS gene and its telomeric pseudogene IDSP1 is responsible for approximately 13% of mucopolysaccharidosis type II (Hunter syndrome; MIM# 309900) cases [34] and inversion between inverted-oriented LCRs disrupting the factor VIII gene (F8) accounts for >45% of severe hemophilia A (MIM# 306700) cases. Similar events (inversions, large deletions and duplications) have not been reported in SDS patients thus far; we speculate this is likely due to the challenge of studying genes embedded within LCRs. Currently, it is estimated that up to 25% of patients with SDS do not carry SBDS biallelic mutations [3,4], therefore, a search for genomic rearrangements within that locus may help unveil an important but underappreciated contributor to this disease.
Competing interests
JRL holds stock ownership in 23andMe, Inc. and Ion Torrent Systems, Inc., and is a co-inventor on multiple United States and European patents related to molecular diagnostics. MB is founder and chief executive officer of Codified Genomics. All other authors have no conflicts of interest. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered in the Medical Genetics Laboratories (http://www.bcm.edu/geneticlabs/).
Authors’ contributions
CMBC conducted high-density CGH arrays, long-range PCR, RT-PCR, experimental design and data analysis. LWZ assisted with long-range PCR, RT-PCR, data analysis and conducted Southern blotting assays. NJN assisted with high-density CGH arrays, long-range PCR, RT-PCR. SJ, DMM and RAG conducted WES experiments, DM and MB performed genomic sequence data analysis. CLW conducted western and Southern blotting assays, assisted with sample collection and data analysis. WI performed the pancreatic isoamylase and trypsinogen assays. RPG interpreted the diagnostic imaging. JRL was involved in research design and data analyses. AAB coordinated clinical studies and subject recruitment, research design and data analyses. CMBC, JRL and AAB prepared the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Claudia M B Carvalho, Email: cfonseca@bcm.edu.
Luciana W Zuccherato, Email: lucianawerneck@yahoo.com.br.
Christopher L Williams, Email: clwillia@txch.org.
Nicholas J Neill, Email: Nicholas.Neill@bcm.edu.
David R Murdock, Email: dmurdock@gmail.com.
Matthew Bainbridge, Email: bainbrid@bcm.edu.
Shalini N Jhangiani, Email: jhangian@bcm.edu.
Donna M Muzny, Email: donnam@bcm.edu.
Richard A Gibbs, Email: agibbs@bcm.edu.
Wan Ip, Email: wan.ip@sickkids.ca.
Robert Paul Guillerman, Email: rpguille@texaschildrens.org.
James R Lupski, Email: jlupski@bcm.edu.
Alison A Bertuch, Email: abertuch@bcm.edu.
Acknowledgments
We thank the families for their participation in the study. The pancreatic isoamylase and trypsinogen assays were performed in the laboratory of Peter Durie, MD, at The Hospital for Sick Children, Toronto, Canada. This work was supported in part by US National Institute of Neurological Disorders and Stroke (NINDS) grant R01 NS058529 and US National Human Genome Research Institute (NHGRI) grant U54HG006542 to JRL. The Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), a Young Investigator fellowship from the Science without Borders Program grant 402520/2012-2 to CMBC The content is solely the responsibility of the authors and does not necessarily represent the official views of the NINDS, NHGRI, NIH or CNPq.
References
- Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: an update. Hematol Oncol Clin North Am. 2013;15(1):117–128. doi: 10.1016/j.hoc.2012.10.003. ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;15(1):97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- Donadieu J, Fenneteau O, Beaupain B, Beaufils S, Bellanger F, Mahlaoui N, Lambilliotte A, Aladjidi N, Bertrand Y, Mialou V, Perot C, Michel G, Fouyssac F, Paillard C, Gandemer V, Boutard P, Schmitz J, Morali A, Leblanc T, Bellanne-Chantelot C. Associated investigators of the French Severe Chronic Neutropenia Registry. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. 2012;15(9):1312–1319. doi: 10.3324/haematol.2011.057489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers TW, Alders M, Tool AT, Mellink C, Roos D, Hennekam RC. Hematologic abnormalities in Shwachman Diamond syndrome: lack of genotype-phenotype relationship. Blood. 2005;15(1):356–361. doi: 10.1182/blood-2004-11-4371. [DOI] [PubMed] [Google Scholar]
- Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;15(3):211–220. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;15(10):417–422. doi: 10.1016/S0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;15(2):74–82. doi: 10.1016/S0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- Costa E, Duque F, Oliveira J, Garcia P, Goncalves I, Diogo L, Santos R. Identification of a novel AluSx-mediated deletion of exon 3 in the SBDS gene in a patient with Shwachman-Diamond syndrome. Blood Cells Mol Dis. 2007;15(1):96–101. doi: 10.1016/j.bcmd.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Burwick N, Coats SA, Nakamura T, Shimamura A. Impaired ribosomal subunit association in Shwachman-Diamond syndrome. Blood. 2012;15(26):5143–5152. doi: 10.1182/blood-2012-04-420166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menne TF, Goyenechea B, Sanchez-Puig N, Wong CC, Tonkin LM, Ancliff PJ, Brost RL, Costanzo M, Boone C, Warren AJ. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;15(4):486–495. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- Woloszynek JR, Rothbaum RJ, Rawls AS, Minx PJ, Wilson RK, Mason PJ, Bessler M, Link DC. Mutations of the SBDS gene are present in most patients with Shwachman-Diamond syndrome. Blood. 2004;15(12):3588–3590. doi: 10.1182/blood-2004-04-1516. [DOI] [PubMed] [Google Scholar]
- Shammas C, Menne TF, Hilcenko C, Michell SR, Goyenechea B, Boocock GR, Durie PR, Rommens JM, Warren AJ. Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond Syndrome. J Biol Chem. 2005;15(19):19221–19229. doi: 10.1074/jbc.M414656200. [DOI] [PubMed] [Google Scholar]
- Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang SH, Yang Y, Wiszniewska J, Nowakowska BA, del Gaudio D, Xia Z, Simpson-Patel G, Immken LL, Gibson JB, Tsai AC, Bowers JA, Reimschisel TE, Schaaf CP, Potocki L, Scaglia F, Gambin T, Sykulski M, Bartnik M, Derwinska K, Wisniowiecka-Kowalnik B, Lalani SR, Probst FJ, Bi W, Beaudet AL. et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;15(12):1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, Lupski JR, Chinault AC, Beaudet AL. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;15(6):422–432. doi: 10.1097/01.GIM.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- Wiszniewska J, Bi W, Shaw C, Stankiewicz P, Kang SH, Pursley AN, Lalani S, Hixson P, Gambin T, Tsai CH, Bock HG, Descartes M, Probst FJ, Scaglia F, Beaudet AL, Lupski JR, Eng C, Wai Cheung S, Bacino C, Patel A. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur J Hum Genet. 2014;15(1):79–87. doi: 10.1038/ejhg.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, Clark G, Lee J, Proud M, Stocco A, Rodriguez DL, Kozel BA, Sparagana S, Roeder ER, McGrew SG, Kurczynski TW, Allison LJ, Amato S, Savage S, Patel A, Stankiewicz P, Beaudet AL, Cheung SW, Lupski JR. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;15(5):332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;15(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome Project Data Processing S: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;15(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;15(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis D, Yu J, Evani US, Jackson AR, Paithankar S, Coarfa C, Milosavljevic A, Gibbs RA, Yu F. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinforma. 2012;15:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R. 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics. 2011;15(15):2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, Reid JG, Fink JK, Morgan MB, Gingras MC, Muzny DM, Hoang LD, Yousaf S, Lupski JR, Gibbs RA. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;15(87):87re83. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Inoue K, Cheung SW, Shaw CA, Stankiewicz P, Lupski JR. Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum Mol Genet. 2006;15(14):2250–2265. doi: 10.1093/hmg/ddl150. [DOI] [PubMed] [Google Scholar]
- Shah SS, Bacino CA, Sheehan AM, Shearer WT. Diagnosis of primary immunodeficiency: let your eyes do the talking. J Allergy Clin Immunol. 2009;15(6):1363––1364 e1361. doi: 10.1016/j.jaci.2009.10.049. [DOI] [PubMed] [Google Scholar]
- Berrocal T, Simon MJ, Al-Assir I, Prieto C, Pastor I, de Pablo L, Lama R. Shwachman-Diamond syndrome: clinical, radiological and sonographic findings. Pediatr Radiol. 1995;15(5):356–359. doi: 10.1007/BF02021702. [DOI] [PubMed] [Google Scholar]
- Dror Y, Donadieu J, Koglmeier J, Dodge J, Toiviainen-Salo S, Makitie O, Kerr E, Zeidler C, Shimamura A, Shah N, Cipolli M, Kuijpers T, Durie P, Rommens J, Siderius L, Liu JM. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci. 2011;15:40–55. doi: 10.1111/j.1749-6632.2011.06349.x. [DOI] [PubMed] [Google Scholar]
- Lo HS, Wang Z, Hu Y, Yang HH, Gere S, Buetow KH, Lee MP. Allelic variation in gene expression is common in the human genome. Genome Res. 2003;15(8):1855–1862. doi: 10.1101/gr.1006603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, Haugen E, Zerr T, Yamada NA, Tsang P, Newman TL, Tuzun E, Cheng Z, Ebling HM, Tusneem N, David R, Gillett W, Phelps KA, Weaver M, Saranga D, Brand A, Tao W, Gustafson E, McKernan K, Chen L, Malig M. et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;15(7191):56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Kidd JM, Eichler EE. Human copy number polymorphic genes. Cytogenet Genome Re. 2008;15(1–4):234–243. doi: 10.1159/000184713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittwald P, Gambin T, Gonzaga-Jauregui C, Carvalho CM, Lupski JR, Stankiewicz P, Gambin A. Inverted low-copy repeats and genome instability–a genome-wide analysis. Hum Mutat. 2013;15(1):210–220. doi: 10.1002/humu.22217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, Schinzel A, Baumer A, Anderlid BM, Schoumans J, Knoers NV, van Kessel AG, Sistermans EA, Veltman JA, Brunner HG, de Vries BB. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;15(9):999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genome structural variation and sporadic disease traits. Nat Genet. 2006;15(9):974–976. doi: 10.1038/ng0906-974. [DOI] [PubMed] [Google Scholar]
- Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, Curley R, Cumming S, Dunn C, Kalaitzopoulos D, Porter K, Prigmore E, Krepischi-Santos AC, Varela MC, Koiffmann CP, Lees AJ, Rosenberg C, Firth HV, de Silva R, Carter NP. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet. 2006;15(9):1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- Bondeson ML, Dahl N, Malmgren H, Kleijer WJ, Tonnesen T, Carlberg BM, Pettersson U. Inversion of the IDS gene resulting from recombination with IDS-related sequences is a common cause of the Hunter syndrome. Hum Mol Genet. 1995;15(4):615–621. doi: 10.1093/hmg/4.4.615. [DOI] [PubMed] [Google Scholar]