

Abstract
One-hundred-fifty-three biliary cancers, including 70 intrahepatic cholangiocarcinomas (ICC), 57 extrahepatic cholangiocarcinomas (ECC) and 26 gallbladder carcinomas (GBC) were assessed for mutations in 56 genes using multigene next-generation sequencing. Expression of EGFR and mTOR pathway genes was investigated by immunohistochemistry. At least one mutated gene was observed in 118/153 (77%) cancers. The genes most frequently involved were KRAS (28%), TP53 (18%), ARID1A (12%), IDH1/2 (9%), PBRM1 (9%), BAP1 (7%), and PIK3CA (7%). IDH1/2 (p=0.0005) and BAP1 (p=0.0097) mutations were characteristic of ICC, while KRAS (p=0.0019) and TP53 (p=0.0019) were more frequent in ECC and GBC. Multivariate analysis identified tumour stage and TP53 mutations as independent predictors of survival. Alterations in chromatin remodeling genes (ARID1A, BAP1, PBRM1, SMARCB1) were seen in 31% of cases. Potentially actionable mutations were seen in 104/153 (68%) cancers: i) KRAS/NRAS/BRAF mutations were found in 34% of cancers; ii) mTOR pathway activation was documented by immunohistochemistry in 51% of cases and by mutations in mTOR pathway genes in 19% of cancers; iii) TGF-ß/Smad signaling was altered in 10.5% cancers; iv) mutations in tyrosine kinase receptors were found in 9% cases. Our study identified molecular subgroups of cholangiocarcinomas that can be explored for specific drug targeting in clinical trials.
Keywords: cholangiocarcinoma, next-generation sequencing, molecular subclassification, target therapy, multigene mutational panels
INTRODUCTION
Cholangiocarcinoma is a phenotypical and clinical heterogeneous collection of biliary tract malignancies, classified according to the World Health Organization (WHO) as intrahepatic (ICC) or extrahepatic cholangiocarcinomas (ECC) [1, 2]. The former arise in the substance of the liver, the latter in large extrahepatic ducts, i.e. hepatic ducts and common bile duct. Gallbladder carcinomas (GBC) also have biliary epithelial differentiation. Clinically, both cholangiocarcinomas and GBC have very poor prognosis. Surgical resection is the only potentially curative therapy, but most cases are inoperable [3-7]. In contrast to other solid tumours, no effective molecular targeted agent has been approved for biliary tract cancers, and patients have limited access to clinical trials [8-11].
Previous studies on molecular alterations in biliary tract cancers have focused on selected genes, including those altered in pancreatic adenocarcinoma (KRAS, TP53, CDKN2A and SMAD4) [12]. Mutations in PIK3CA, PTEN, AKT1, IDH1 and IDH2 have been reported in this class of tumours [13-20]. However, the prevalence of these alterations varies widely among studies. Two recent whole exome-sequencing studies of ICC revealed a key role for chromatin remodeling genes BAP1, ARID1A and PBRM1 in the development of these tumours [13, 21].
The validation of whole exome studies by sequencing analysis of hotspot mutations in larger and characterized series has been a fruitful approach in identifying potential targets for personalized therapy for several malignancies [22]. Next-generation sequencing (NGS) has been recently introduced and is the most sensitive approach to simultaneously characterize multiple genes starting from a limited amount of DNA, also DNA derived from formalin-fixed paraffin-embedded (FFPE) samples [13, 23-25].
In the present study, we assayed the mutational status of 56 cancer-related genes in 153 biliary tract cancers, using a targeted next-generation sequencing methodology, with the aim of identifying molecular subgroups driving the development of personalized therapy approaches for patients affected by these neoplasms.
RESULTS
Clinico-pathological characteristics of the series
Patients' demographic and clinico-pathological data are summarized in Table 1. Mean tumour size was 4.8±3.4 cm (median=6.5; range=0.5-20.0), and was significantly higher in ICC than ECC and GBC (p=9.77 E−12). Synchronous multinodular lesions were found in 31 cases (20.3%). Tumour grading was G1 in 20, G2 in 94, and G3 in 38 cases, while the remaining case was undifferentiated.
Table 1. Clinico-pathological features of 153 biliary carcinomas.
| Total (n=153) |
ICC (n=70) |
ECC (n=57) |
GBC (n=26) |
P-value* | ||
|---|---|---|---|---|---|---|
| Sex | 59F 94M | 28F 42M | 20F 37M | 11F 15M | 0.776 | |
| Age | 65.4±10.8 | 64.8±11.6 | 64.2±10.8 | 69.6±8.0 | 0.118 | |
| Dimension (cm) | 4.8±3.4 | 6.6±3.7 | 2.6±1.3 | 3.3±1.4 | 9.77 E-12 | |
| Multiple nodes | 31 (20.3%) |
28 (40.0%) |
3 (5.3%) |
- | 2.84 E-6 | |
| Grade | 1 | 20 (13.1%) |
7 (10.0%) |
8 (14.0%) |
5 (19.2%) |
0.630 |
| 2 | 94 (61.4%) |
45 (64.3%) |
36 (63.2%) |
13 (50.0%) |
||
| 3 | 38 (24.8%) |
18 (25.7%) |
12 (21.0%) |
8 (30.7%) |
||
| 4 | 1 (0.6%) |
0 (0.0%) |
1 (1.8%) |
0 (0.0%) |
||
| Presence of BiIIN | 49 (32.0%) |
11 (15.7%) |
24 (42.1%) |
14 (53.8%) |
0.0002 | |
| Vascular invasion | 107 (70.0%) |
51 (72.9%) |
38 (66.7%) |
18 (69.2%) |
0.773 | |
| Perineural invasion | 93 (60.8%) |
31 (44.3%) |
44 (77.2%) |
18 (69.2%) |
0.0005 | |
| Radicality of resection | R0 | 110 (71.9%) |
56 (80.0%) |
33 (57.9%) |
19 (73.1%) |
0.068 |
| R1 | 43 (28.1%) |
14 (20.0%) |
24 (42.1%) |
7 (26.9%) |
||
| HBV/HCV infection | 22 (14.4%) |
17 (24.3%) |
3 (5.3%) |
2 (7.7%) |
0.041 | |
| Cirrhosis | 13 (8.5%) |
10 (14.3%) |
3 (5.3%) |
0 (0.0%) |
0.005 | |
| Stage | I | 20 (13.1%) |
12 (17.1%) |
5 (8.8%) |
3 (11.5%) |
0.034 |
| II | 51 (33.3%) |
21 (30.0%) |
22 (38.6%) |
8 (30.7%) |
||
| III | 43 (28.1%) |
12 (17.1%) |
20 (35.1%) |
11 (42.3%) |
||
| IV | 39 (25.5%) |
25 (35.8%) |
10 (17.5%) |
4 (15.4%) |
Note: ICC, intrahepatic cholangiocarcinoma; ECC, extrahepatic cholangio-carcinoma; GBC, gallbladder carcinoma; BiIIN, biliary intraepithelial neoplasia
Fisher's exact test for categorical data, Kruskal-Wallis test for continuous variables.
# Chi-squared test with Monte Carlo simulation (2000 replicates).
Biliary intraepithelial neoplasia (BilIN) was present in 49/153 cases (32.0%), and its prevalence was significantly higher in ECC and GBC compared to ICC (ICC=15.7%; ECC=42.1%; GBC=53.8%; p=0.0002). Vascular and perineural invasion were present in 107 (69.9%) and 93 (60.8%) cases, respectively. Perineural invasion showed a significantly lower prevalence in ICC than ECC and GBC (ICC=44.3%; ECC=77.2%; GBC=69.2%; p=0.0005). Hepatitis virus infection (HBV and/or HCV) and cirrhosis prevalence were significantly higher in ICC patients (p=0.041 and p=0.005, respectively).
The pathologic stage of the 153 neoplasms was Stage I in 20, II in 51, III in 43, and IV in 39. ICC presented with more advanced stages at surgery compared to ECC and GBC (Table 1; p=0.034).
Next-generation sequencing of 56 genes dissects cholangiocarcinoma molecular heterogeneity
DNA from all samples was successfully amplified in multiplex PCR for the 56 genes and an adequate library for deep sequencing was obtained. The mean read length was 78 base pairs and a mean coverage of 1800x was achieved, with 87.1% target bases covered more than 100x. A minimum coverage of 20x was obtained in all cases.
At least one mutation was observed in 118/153 (77.1%) samples (Table 2, Figure 1); 60 cases (39.2%) showed concurrent mutations in different genes; 35 (22.9%) tumours showed no alterations in the 56 genes assayed. The most commonly mutated genes in the whole series were KRAS (28.1%), TP53 (18.3%), ARID1A (11.8%), IDH1/IDH2 (9.2%), PBRM1 (9.2%), BAP1 (7.2%), and PIK3CA (7.2%). Mutations in BRAF, KRAS, and TP53 were all confirmed at Sanger sequencing (Figure 2).
Table 2. Mutational status of 153 biliary tract carcinomas.
| Gene | Total | Type of mutation | ICC (n= 70) | ECC (n= 57) | GBC (n= 26) | P-value* | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| M | N | Fs | D | S | ||||||
| AKT1 | 2 | 2 | 2 | - | ||||||
| ALK | 1(0.7%) | 1 | 1(1.7%) | - | ||||||
| APC | 3(2.0%) | 2 | 1 | 1(1.4%) | 2(7.7%) | - | ||||
| ARID1A | 18(11.8%) | 10 | 5 | 1 | 1 | 1 | 8(11.4%) | 7(12.3%) | 3(11.5%) | 0.999 |
| BAP1 | 11(7.2%) | 7 | 1 | 3 | 10(14.3%) | 1(3.8%) | 0.0097 | |||
| BRAF | 3(1.9%) | 3 | 3(4.3%) | - | ||||||
| CDKN2A | 2(1.3%) | 1 | 1 | 1(1.4%) | 1(3.8%) | - | ||||
| CTNNB1 | 2(1.3%) | 1 | 1 | 2(3.5%) | - | |||||
| EGFR | 2(1.3%) | 2 | 1(1.7%) | 1(3.8%) | - | |||||
| ERBB2 | 1(0.7%) | 1 | 1(3.8%) | - | ||||||
| ERBB4 | 1(0.7%) | 1 | 1(1.4%) | - | ||||||
| FBXW7 | 3(2.0%) | 3 | 1(1.4%) | 2(3.5%) | - | |||||
| FGFR3 | 2(1.3%) | 1 | 1 | 2(2.8%) | - | |||||
| GNAS | 1(0.7%) | 1 | 1(1.7%) | - | ||||||
| IDH1 | 11(7.2%) | 11 | 11(15.7%) | 0.0021 | ||||||
| IDH2 | 3(2.0%) | 3 | 3(4.3%) | - | ||||||
| JAK3 | 1(0.7%) | 1 | 1(1.7%) | - | ||||||
| KDR | 5(3.3%) | 3 | 2 | 1(1.4%) | 2(3.5%) | 2(7.7%) | - | |||
| KIT | 2(1.3%) | 2 | 1(3.5%) | 1(3.8%) | - | |||||
| KRAS | 43(28.1%) | 43 | 11(15.7%) | 7(47.4%) | 5(19.2%) | 0.0019 | ||||
| MET | 1(0.7%) | 1 | 1(3.8%) | - | ||||||
| MLH1 | 1(0.7%) | 1 | 1(1.7%) | - | ||||||
| NRAS | 6(3.9%) | 6 | 5(9.3%) | 1(1.7%) | 0.421 | |||||
| PBRM1 | 14(9.2%) | 8 | 5 | 1 | 10(14.3%) | 2(3.5%) | 2(7.7%) | 0.182 | ||
| PIK3CA | 11(7.2%) | 11 | 4(5.7%) | 5(8.7%) | 2(7.7%) | 0.914 | ||||
| PIK3C2A | 9(5.9%) | 9 | 5(7.1%) | 4(7.0%) | 0.620 | |||||
| PIK3C2G | 8(5.2%) | 7 | 1 | 3(4.3%) | 5(8.7%) | 0.498 | ||||
| PTEN | 4(2.6%) | 4 | 1(1.4%) | 2(3.5%) | 1(3.8%) | - | ||||
| PTPN11 | 1(0.7%) | 1 | 1(1.7%) | 0(0.0%) | - | |||||
| RB1 | 1(0.7%) | 1 | 1(3.8%) | - | ||||||
| RET | 10.7%) | 1 | 1(1.7%) | - | ||||||
| SMAD4 | 9(5.9%) | 7 | 2 | 1(1.4%) | 6(10.5%) | 2(7.7%) | 0.179 | |||
| SMARCB1 | 2(1.3%) | 2 | 0(0.0%) | 2(7.7%) | - | |||||
| STK11 | 3(2.0%) | 2 | 1 | 1(1.4%) | 1(2.2%) | 1(3.8%) | - | |||
| TGFBR2 | 7(4.6%) | 6 | 1 | 3(4.3%) | 3(5.3%) | 1(3.8%) | 0.999 | |||
| TP53 | 28(18.3%) | 24 | 4 | 6(8.6%) | 10(17.5%) | 12(46.2%) | 0.0019 | |||
Note: ICC, intrahepatic cholangiocarcinoma; ECC, extrahepatic cholangiocarcinoma; GBC, gallbladder carcinoma; M, missense mutation; N, nonsense mutation; F, frameshift mutation; D, deletion; S, splice site alteration.
Fisher's exact test corrected for multiple comparisons was calculated If ≥ 6 mutated cases were observed.
Figure 1. Mutation and immunohistochemical landscape of 153 primary biliary carcinomas.

The series includes 70 intrahepatic cholangiocarcinomas (ICC), 57 extrahepatic cholangiocarcinomas (ECC), and 26 gallbladder carcinomas (GBC). Significantly mutated genes are listed vertically in decreasing order of prevalence of nonsilent mutation. Colored rectangles indicate mutation category observed in a given gene and tumour. Tumour classifications and molecular features are as indicated in the boxes on the right. Immunoistochemistry phenotypes and FISH analysis results are shown in the bottom tracks. White boxes indicate unknown status or missing data.
Figure 2. Representative examples of validation by Sanger sequencing of mutations identified using next generation sequencing.
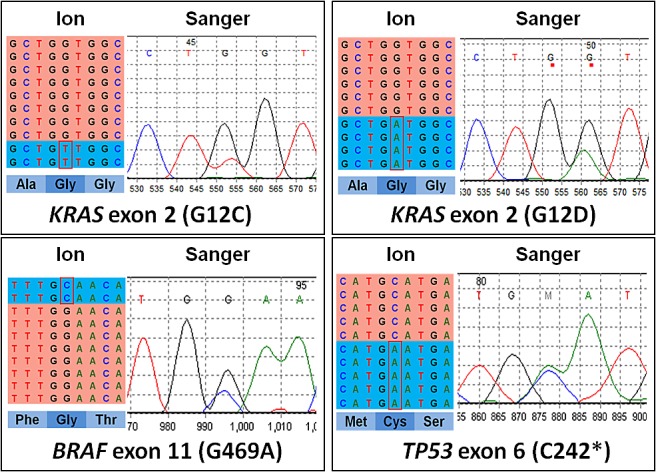
On the left of each sample is the representation of the results of next-generation sequencing where the reads are aligned to the reference genome as provided by the Integrative Genomics Viewer (IGV v.2.1, Broad Institute) software. On the right is the representation of the results of Sanger sequencing.
Mutations were differently distributed across the different tumour subtypes: IDH1/IDH2 (p=0.0005) were restricted to ICC and BAP1 mutations were all found in ICC (p=0.0097) with the exception of one GBC, while KRAS (p=0.0019) and TP53 (p=0.0019) were more represented in ECC and GBC, respectively (Table 2).
ICC were characterized by a high prevalence of IDH1/IDH2 mutations (20.0%) and the significant involvement of chromatin remodeling genes PBRM1 (14.3%), BAP1 (14.3%) and ARID1A (11.4%) (Figure 3), as described[32, 33]. BAP1 and IDH1 were mutually exclusive, whereas mutations in IDH2 were always associated to BAP1 mutations (3/3 cases). Eleven (15.7%) ICC had mutations in at least one of mTOR pathway genes: AKT1 (2.8%), PIK3CA (5.7%), PIK3C2A (7.1%), PIK3C2G (4.3%), and PTEN (1.4%). Mutations in tyrosine kinase receptors were uncommon, with the exception of TGFRB2 (4.3%). Of interest, most NRAS (5 of 6) and all BRAF (3 of 3) mutations clustered in ICC tumour subtype and were mutually exclusive with KRAS (15.7%). TP53 was mutated in 6 cases (8.6%). Low prevalence mutations were found in APC, CDKN2A, ERBB4, FBXW7, FGFR3, KDR/VEGFR2, SMAD4, and STK11.
Figure 3. Somatic mutations detected in chromatin remodeling genes ARID1A, BAP1, and PBRM1.

Schematic representation of ARID1A, BAP1, and PBRM1 genes with the indication of the site of the somatic mutations identified in our study. Genomic coordinates are shown at the bottom track for each gene. Gray arrow indicates gene transcriptional direction. In black are represented the exons for each gene. Vertically, in correspondence of genomic location, bar chart indicate the type and number of mutations. Bar chart color is specific for mutation type: red, non synonymous coding; green, deletion; blue, splice site; yellow, frameshift.
In ECC, KRAS was the most commonly mutated gene (47.4%), with codons 12, 13, 61 and 146 affected; one mutation was observed in NRAS, and none in BRAF. TP53 was the second most mutated gene (17.5%). Excluding ARID1A (12.3%), chromatin-remodeling genes were occasionally involved (PBRM1: 3.5%), whereas 24.6% of ECCs showed mTOR pathway gene mutations, including PIK3CA (8.7%), PIK3C2A (7.0%), PIK3C2G (8.7%), and PTEN (3.5%). SMAD4 mutations were observed in 6 cases (10.5%) and were mutually exclusive to TGFBR2 mutations that were found in 3 cases (5.3%). Low prevalence mutations were found in CTNNB1, FBXW7, and KDR/VEGFR2. The EGFR T790M mutation was observed in one case [34].
GBC showed a high prevalence of TP53 mutations (12/26, 46.2%), and in 6 cases TP53 mutation was the only alteration detected. KRAS was mutated in 19.2% of cases. Chromatin remodeling genes were mutated in 30.8% of cases: ARID1A, 11.5%; BAP1, 3.8%; PBRM1, 7.7%; SMARCB1, 7.7%. MTOR pathway genes were mutated in 11.5% of cases: PIK3CA (7.7%) and PTEN (3.8%).
mTOR pathway is dysregulated in all cholangiocarcinoma subtypes and Egfr is significantly overexpressed in intrahepatic cholangiocarcinomas
The results of immunohistochemistry are summarized in Table 3. We investigated mTOR pathway and Egfr expression in 113 neoplastic and 18 control cases. EGFR gene copy number was analyzed by FISH.
Table 3. EGFR immunohistochemical and gene copy number status, and mTOR pathway immunohistochemical profiling.
| Gene | Total | ICC | ECC | GBC | P-value* | ||
|---|---|---|---|---|---|---|---|
| Immunohistochemistry | EGFR | 0 | 37 (32.7%) |
17 (29.8%) |
17 (42.5%) |
3 (18.8%) |
0.025 |
| 1 | 36 (31.9%) |
12 (21.0%) |
16 (40.0%) |
8 (50.0%) |
|||
| 2 | 25 (22.1%) |
15 (26.4%) |
6 (15.0%) |
4 (25.0%) |
|||
| 3 | 15 (13.3%) |
13 (22.8%) |
1 (2.5%) |
1 (6.2%) |
|||
| PTEN | 0 | 88 (77.9%) |
39 (68.4%) |
34 (85.0%) |
15 (93.8%) |
0.085 | |
| 1 | 25 (22.1%) |
18 (31.6%) |
6 (15.0%) |
1 (6.2%) |
|||
| ph-mTOR | 0 | 55 (48.7%) |
26 (45.6%) |
21 (52.5%) |
8 (50.0%) |
0.785 | |
| 1 | 58 (51.3%) |
31 (54.4%) |
19 (47.5%) |
8 (50.0%) |
|||
| ph-p70S6 | 0 | 45 (39.8%) |
29 (50.8%) |
15 (37.5%) |
1 (6.3%) |
0.015 | |
| 1 | 68 (60.2%) |
28 (49.2%) |
25 (62.5%) |
15 (93.7%) |
|||
| ph-4EBP1 | 0 | 67 (59.3%) |
34 (59.6%) |
25 (62.5%) |
8 (50.0%) |
0.785 | |
| 1 | 46 (40.7%) |
23 (40.4%) |
15 (37.5%) |
8 (50.0%) |
|||
| FISH | EGFR amplification | 6 (5.3%) |
4 (7.0%) |
0 (0.0%) |
2 (12.5%) |
0.900 | |
Note: ICC, intrahepatic cholangiocarcinoma; ECC, extrahepatic cholangiocarcinoma; GBC, gallbladder carcinoma.
Fisher's exact test corrected for multiple comparisons.
A significant over-expression of the activated forms of mTOR and its effectors p70S6K and 4EBP1 was seen in most cancers with no significant differences among subtypes, but for p70S6K (Table 3). Of interest, the expression of phosphorylated ph-mTOR was significantly associated to the expression of the activated downstream effectors ph-4EBP1 and ph-p70S6K (p=0.05 and p=0.00012, respectively). Pten was significantly down-regulated in the whole series, particularly in ECC of common bile duct and in GBC: a weak cytoplasmic/nuclear immunolabeling was observed in most cases.
Egfr expression was significantly altered in biliary tract tumours, with different subtype specific profiles. Forty cancers (35.4%) labeled for Egfr with sharp membranous pattern and signals ranging from very strong to moderate (Table 3, Figure 1 and Figure 4). Strong (3+) overexpression was observed only in the ICC subtype. At FISH analysis, 3/113 (2.7%) tumours showed EGFR amplification (Figure 1).
Figure 4. Immunohistochemical profiles of Egfr and mTOR pathway in cholangiocarcinomas.

Representative examples of immunohistochemical staining in cholangiocarcinoma samples. The prevalence of positive cases within the different tumour types is shown. Original magnfications 20x.
There was no significant association between both EGFR and mTOR pathway immunophenotype and mutational status.
TP53 mutation is an independent prognostic factor in cholangiocarcinoma
Survival data were available in 125 cases (ICC=51; ECC=50; GBC=24). Median survival was 31 months and 79 (63.2%) subjects were followed to their deaths from disease.
At univariate analysis, the most significant predictors of cancer outcome were tumour stage (p=0.0001), TP53 (p=0.0043) and KRAS (p=0.0162) mutations (Figure 5). Considering together KRAS/BRAF alterations, tumors characterized by mutations in KRAS/BRAF genes were associated to a worse patients' prognosis (p=0.0054). ICC showed a better outcome than ECC (p=0.018). No correlation emerged for any of the other clinicopathologic variables considered: sex, age, grade, vascular/perineural invasion.
Figure 5. Overall survival according to pathological and mutational features.

Overall survival of 125 cholangiocarcinomas is significantly affected by tumour stage (p=0.0001) (A), tumour location (p=0.0176) (B), TP53 (p=0.0043) (C) and KRAS (p=0.0162) (D) mutational status. Vertical axis indicates percent survival; horizontal axis shows time expressed in months. Kaplan–Meier and log-rank statistics were used to determine levels of significance.
Cox multivariate analysis including tumour subtype, stage, grade, vascular and perineural invasion, IDH1/2 mutations, KRAS and TP53 mutations, identified only Stage III (p=0.005; OR 4.27; 95%C.I. 1.54-11.8), Stage IV (p=0.003; OR 4.85; 95%C.I. 1.68-14.0), and TP53 mutations (p=0.002; OR 2.26; 95%C.I. 1.35-3.78) as being significantly associated with cancer-related death (Table 4).
Table 4. Multivariate survival analysis of 125 cholangiocarcinomas; median survival was 31 months and 79 subjects died of disease.
| Variable | Odds-ratio | 95% C.I. | P-value |
|---|---|---|---|
| Stage = I | 1 | - | - |
| Stage = II | 1.57 | 0.58-4.25 | 0.371 |
| Stage = III | 4.27 | 1.54-11.8 | 0.005 |
| Stage = IV | 4.85 | 1.68-14.0 | 0.003 |
| Vascular invasion = yes | 1.64 | 0.93-2.88 | 0.087 |
| Perineural invasion = yes | 0.63 | 0.36-1.08 | 0.093 |
| TP53 = mutated | 2.26 | 1.35-3.78 | 0.002 |
| KRAS = mutated | 1.51 | 0.91-2.51 | 0.110 |
| Excluded variables | |||
| Grade = 1 | 1 | - | - |
| Grade = 2 | 1.38 | 0.57-3.37 | 0.474 |
| Grade = 3 | 1.49 | 0.58-3.82 | 0.404 |
| Class = ICC | 1 | - | - |
| Class = ECC | 1.31 | 0.70-2.41 | 0.390 |
| Class = GBC | 0.67 | 0.30-1.49 | 0.323 |
| IDH1/2 = mutated | 1.26 | 0.50-3.17 | 0.631 |
Note: ICC, intrahepatic cholangiocarcinoma; ECC, extrahepatic cholangiocarcinoma; GBC, gallbladder carcinoma.
DISCUSSION
The results of our next-generation mutational survey of 56 cancer genes in 153 biliary tree carcinomas can be summarized as follows: i) the vast majority (77.1%) of cancers harbours a driver-gene mutation; ii) the diverse sites of origin in the biliary tree have significantly different molecular profiles, and each site of origin also shows molecular heterogeneity; iii) targetable pathway alterations are present in 68% (104/153) of cancers, defining molecular cancer subclasses; iv) specific alterations are eligible for the investigational development of prognostic and non-invasive follow-up markers.
The vast majority of cancers (118/153, 77.1%) harboured at least one driver-gene mutation, and 39.2% (60/153 cases) showed concurrent mutations in two or more genes. KRAS was the most frequently mutated gene (28.1%), followed by TP53 (18.3%), as reported in prior studies [9, 14, 35-38]. The recently described frequent involvement of the chromatin remodeling genes ARID1A, PBRM1 and BAP1 [9, 13] was also confirmed in our series, being found in 11.8%, 9.2% and 7.2% of cases, respectively. Univariate survival analysis showed that KRAS mutations were associated to a worse patients' prognosis confirming the univariate analysis previously described by Andersen [42]. Multivariate survival analysis identified tumour stage and TP53 gene mutations as independent predictors of poor survival.
The different sites of origin showed significantly diverse molecular characteristics. IDH1/IDH2 mutations were restricted to ICC (p=0.0005) and, with the exception of one GBC, BAP1 mutations were all found in ICC (p=0.0097). ECC and GBC were characterized by a high prevalence of KRAS (p=0.0019) and TP53 (p=0.0019) mutations, respectively.
The standard of care of biliary tract cancer is based on the combination of cisplatin and gemcitabine [39]. To date, clinical trials with targeted therapies for advanced biliary tract cancers have failed to produce significant benefits [11], and ongoing studies are exploring the combination of chemotherapy with novel MAPK/ERK Kinase (MEK) and mTOR inhibitors [40]. However, neither previous nor ongoing studies have considered evaluating tumour response against genetic alterations. Our identification of molecular subclasses with specific drug actionable pathway alterations in 104/153 (68.0%) tumours may tailor the design of trials based on the molecular selection of patients, irrespective of the site of origin, where actionable signaling pathways include tyrosine-kinase receptors (TKR), RAS/RAF/MAPK/ERK, mTOR, and TGF-ß.
Mutations in tyrosine kinase receptors (ALK, EGFR, ERBB2, ERBB4, FGFR3, MET, KIT, KDR/VEGFR2) potentially amenable to target therapies were found in 9.2% of cases, with a higher prevalence in GBC (6/26 cases, 23.1%) than in ICC (4/70 cases, 5.7%) and ECC (4/57, 7.0%). In spite of a relatively high prevalence of Egfr overexpression detected by immunohistochemistry (35.4% of cases with 2+ or 3+), only two cases had EGFR mutations and three had gene amplification, confirming the low prevalence reported in the literature in cholangiocarcinomas unassociated with chronic liver disease [14, 38, 41-44].
Mutations in components of the RAS pathway (KRAS, NRAS, BRAF) were observed in 34% of the whole series. In particular KRAS was the most frequently mutated gene in the 153 tumours (28.1%). KRAS mutations have been described in most of prior studies [9, 14, 35-38]. KRAS/NRAS/BRAF mutations were mutually exclusive, and the highest mutation prevalence in RAS pathway was observed in ECC (49.1%) vs. ICC (27.1%) and GBC (19.2%). Of note, RAS mutations sensitize tumours to MEK inhibitors, highlighting the importance of these mutations in the use of targeted therapies [11, 40].
MTOR pathway relevance in biliary tract cancers is suggested by our immunohistochemical detection of activated forms of mTor and its downstream effectors in 51.3% of the cancers. The molecular basis of this activation in a proportion of cases is the mutation in one of the genes involved in this pathway (i.e., AKT, FBXW7, PIK3CA, PIK3C2A, PIK3C2G, PTEN). This suggests that mTOR inhibitors might play a role in this molecular subgroup of patients.
TGF-ß/Smad signaling was altered in 16/153 cases (10.5%), 9 of which were ECC (9/57, 15.8%). Our finding supports previous studies demonstrating the involvement of this pathway in cholangiocarcinomas[45, 46]. Our study may have underestimated the involvement of this pathway, as only the mutational status of TGFBR2 and SMAD4 genes was investigated, and the latter is frequently inactivated by mechanisms different from intragenic mutations, such as homozygous deletions, which would not be detected by the techniques used in this study.
Alterations in chromatin remodeling genes (ARID1A, BAP1, IDH1, IDH2, PBRM1, SMARCB1) were found in 30.7% (47/153) of cancers in our series, confirming recent reports on the significant involvement of these genes in cholangiocarcinoma [9, 13]. Mutations in these genes appear to be either specific to ICC, as is the case of IDH1 and IDH2 (p=0.0005), or cluster within this cancer type as is the case for ARID1A, BAP1, and PBRM1 that were found in 34.3% (24/70) of ICC. The open challenge is now to translate knowledge of the targeting of these genes to improved patient care through either the development of new disease specific markers or of therapy targets. Of interest, IDH1/2 mutated cancers accumulate 2-hydroxyglutarate in tumour tissue and release the molecule in blood, and the measurement of 2-hydroxyglutarate might be used as both a surrogate biomarker for IDH1/2 mutational status and a non-invasive test for the assessment of tumour burden in ICC [47].
Our study is limited by the number of genes analyzed; 35 cancers (22.9%) showed no alterations in the 56 genes assayed, including 50 genes from a commercial panel and a custom panel exploring 6 recently discovered cholangiocarcinoma genes [9, 13]. Mechanisms different from intragenic mutations, such as amplifications, deletions, translocations, and epigenetic anomalies should also be addressed.
In conclusion, we demonstrate that specific molecular alterations are associated to different cholangiocarcinomas categories and that potentially drug actionable pathways are evident in 68% of cases. These data further support the pathological and molecular heterogeneity characterizing biliary tree carcinomas. In currently designed clinical trials, cholangiocarcinomas are grouped together irrespective of their intrahepatic or extrahepatic site of origin. Our study shows that ICC and ECC should be considered separately, since they have different molecular characteristics. However, in the advent of molecular designed clinical trials, it would be appropriate to consider them together but only for the proportion of ICC and ECC sharing common molecular alterations.
We also show that a high-throughput next-generation sequencing analysis can be successfully applied using low amounts of DNA from routinely processed paraffin tissues. Such a time- and cost-effective analysis is the basis to significantly improve the development of personalized treatments for cholangiocarcinoma patients, and their early access to innovative drugs.
MATERIALS AND METHODS
Cases
A retrospective series (1990-2011) of 153 surgically-resected primary biliary cancers were retrieved from the FFPE archives of the ARC-Net Biobank at Verona University Hospital under the local ethics committee approval (n. prog. 1959). All cases were re-classified according to WHO 2010 [1], and included 70 ICC, 57 ECC and 26 GBC. Staging was according to AJCC/UICC 7th edition[26]. Matched normal liver was used to determine the somatic/germline nature of mutations.
In 113 cases (57 ICC, 40 ECC, 16 GBC), sufficient material for the construction of 1-mm cores tissue microarrays (TMAs) was available. Eighteen non-neoplastic controls (8 normal biliary duct and 10 chronic cholecystitis) were included in the TMAs. Three tissue cores per case were analyzed.
DNA extraction and qualification
DNA was prepared after enrichment for neoplastic cellularity to at least 70% using manual microdissection of 10 consecutive 4-μm FFPE sections, purified using the QIAamp DNA FFPE Tissue Kit (Qiagen), and qualified as reported elsewhere [24, 27].
Next-Generation Sequencing of Multiplex PCR Amplicons
Two multigene panels were used: the 50-gene Ion AmpliSeq Cancer Hotspot panel v2 (Life Technologies) and an AmpliSeq custom panel targeting 6 genes not included in the commercial panel. The first explores selected regions of 50 cancer- genes: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR/VEGFR2, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, VHL. Details on target regions of the commercial panel are at http://www.lifetechnologies.com. The custom panel targets 6 genes selected upon the results of published ICC exome sequencing: ARID1A, BAP1, PBRM1, PIK3C2A, PIK3C2G, TGFBR2[13]. Details of the custom panel are in Supplementary Table 1.
Twenty nanograms of DNA were used for each multiplex PCR amplification. Emulsion PCR was performed with the OneTouch2 system (Life Technologies). The quality of the obtained libraries was evaluated by the Agilent 2100 Bioanalyzer on-chip electrophoresis (Agilent Technologies). Sequencing was run on the Ion Torrent Personal Genome Machine (PGM, Life Technologies) loaded with 316 (50-gene panel) or 318 chips (6-gene panel). Data analysis, including alignment to the hg19 human reference genome and variant calling, was done using the Torrent Suite Software v.3.6 (Life Technologies). Filtered variants were annotated using the SnpEff software v.3.1. Alignments were visually verified with the Integrative Genomics Viewer; IGV v.2.2, Broad Institute.
DNA Sanger Sequencing
To validate the mutations detected by deep sequencing, BRAF (exon 11), KRAS (exon 2), and TP53 (exons 2, 5, 6, 7, 8) were analyzed by Sanger sequencing [28].
Immunohistochemistry
The immunohistochemical expression of Egfr (Dako), Pten (Abnova Corporation), and of the phosphorylated forms of mTOR (Ser2448, clone 49F9; Cell Signaling) and its downstream effectors 4EBP1 (Thr37/46, clone 236B4; Cell Signaling) and p70S6K (Thr389, clone 1A5; Cell Signaling) was examined on consecutive 4-μm FFPE TMA sections. Appropriate positive and negative controls were run concurrently.
Egfr expression was scored according to the EGFR pharmDx protocol (Dako): 0, no staining or membrane staining in ≤10% cancer cells; 1+, faint and partial membrane staining in >10% cancer cells; 2+, moderate and complete membrane staining in >10% cancer cells; 3+, strong and complete membrane staining in >10% cancer cells. Cases were then classified in two groups Egfr-positive (2+ and 3+) or Egfr-negative (0 and 1+).
Pten expression was considered positive if more than 50% of neoplastic cells showed a moderate/strong nuclear and cytoplasmic immunoreaction[29].
Staining for phosphorylated markers (ph-mTOR, ph-4EBP1, and ph-p70S6K) was considered positive when tumour cells showed cytoplasmic and/or nuclear staining with equal to stronger intensity compared with that of endothelial cells [30].
Fluorescent in situ hybridization (FISH)
The EGFR gene copy number status was assessed applying the Vysis EGFR/CEP7 Probe Kit (Vysis/Abbott Molecular). At least 50 representative nuclei per specimen were scored and EGFR-amplification was defined as described [31].
Statistical analysis
Kruskal-Wallis test, Chi-squared test with Monte Carlo simulation, and Fisher's exact test corrected for multiple comparisons were used as appropriate. For comparison of Kaplan-Meier survival curves, Mantel-Cox log-rank test was used; for multivariate survival analysis, stepwise Cox proportional hazards regression was used; selection of the best model was performed using the “backward elimination” algorithm. For all the analyses a p-value below 0.05 was considered significant. Graphs and univariate analyses were performed using GraphPad Prism® version 5.00 for Mac (GraphPad Software, San Diego California USA), multivariate Cox regression was done with R v. 3.0.2, using survival library v.2.37-4.
SUPPLEMENTARY TABLE
Acknowledgments
The study has been supported by: an unrestricted grant of GlaxoSmithKline (WEUSKOP5847), AIRC grants n. 12182, 11930 and 6421, National Cancer Institutes (USA) grant CA62924, FP7 EU project CAM-PaC no. 602783, the Italian Cancer Genome Project grant from the Italian Ministry of Research (FIRB - RBAP10AHJB) and FIMP-Ministry of Health (CUP_J33G13000210001).
Abbreviations
- ECC
extrahepatic cholangiocarcinomas
- FFPE
formalin-fixed paraffin-embedded
- GBC
gallbladder carcinomas
- ICC
intrahepatic cholangiocarcinomas
- NGS
next-generation sequencing
Footnotes
MS, MF, AR, VC, AM, LDW, RHH, GT, FdB, AS: ideation of the study, planning, primary writers. MF, AT, PC, CV, MC: histopathological and immunohistochemical study and evaluation. MF: sample microdissection. MS, NS, DA, GM: preparation of DNA, quality control and sequencing including data analysis and interpretation. MS, AM: bioinformatic analysis of next-gen sequencing. AR, NS: sample choice and preparation, collection and assembly of clinical-pathological data. CI, RTL, DM, AG, CB, GT, FdB: clinical-pathological data analysis and interpretation of results related to the peculiar tumour type and clinical implications. All authors participated in writing and approved the final, submitted manuscript.
REFERENCES
- 1.Bosman FT. WHO classification of tumours of the digestive system. Lyon: International Agency for Research on Cancer; 2010. World Health Organization. and International Agency for Research on Cancer. [Google Scholar]
- 2.Patel T. Cholangiocarcinoma--controversies and challenges. Nat Rev Gastroenterol Hepatol. 2011;8:189–200. doi: 10.1038/nrgastro.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guglielmi A, Ruzzenente A, Campagnaro T, Pachera S, Valdegamberi A, Nicoli P, Cappellani A, Malfermoni G, Iacono C. Intrahepatic cholangiocarcinoma: prognostic factors after surgical resection. World J Surg. 2009;33:1247–1254. doi: 10.1007/s00268-009-9970-0. [DOI] [PubMed] [Google Scholar]
- 4.Ribero D, Pinna AD, Guglielmi A, Ponti A, Nuzzo G, Giulini SM, Aldrighetti L, Calise F, Gerunda GE, Tomatis M, Amisano M, Berloco P, Torzilli G, Capussotti L. Surgical Approach for Long-term Survival of Patients With Intrahepatic Cholangiocarcinoma: A Multi-institutional Analysis of 434 Patients. Arch Surg. 2012;147:1107–1113. doi: 10.1001/archsurg.2012.1962. [DOI] [PubMed] [Google Scholar]
- 5.Ribero D, Nuzzo G, Amisano M, Tomatis M, Guglielmi A, Giulini SM, Aldrighetti L, Calise F, Gerunda GE, Pinna AD, Capussotti L. Comparison of the prognostic accuracy of the sixth and seventh editions of the TNM classification for intrahepatic cholangiocarcinoma. HPB (Oxford) 2011;13:198–205. doi: 10.1111/j.1477-2574.2010.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guglielmi A, Ruzzenente A, Campagnaro T, Pachera S, Valdegamberi A, Capelli P, Pedica F, Nicoli P, Conci S, Iacono C. Does intrahepatic cholangiocarcinoma have better prognosis compared to perihilar cholangiocarcinoma? J Surg Oncol. 2010;101:111–115. doi: 10.1002/jso.21452. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Li J, Xia Y, Gong R, Wang K, Yan Z, Wan X, Liu G, Wu D, Shi L, Lau W, Wu M, Shen F. Prognostic nomogram for intrahepatic cholangiocarcinoma after partial hepatectomy. J Clin Oncol. 2013;31:1188–1195. doi: 10.1200/JCO.2012.41.5984. [DOI] [PubMed] [Google Scholar]
- 8.Hezel AF, Deshpande V, Zhu AX. Genetics of biliary tract cancers and emerging targeted therapies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:3531–3540. doi: 10.1200/JCO.2009.27.4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, McPherson JR, Allen GE, Ng CC, Wong BH, Myint SS, Rajasegaran V, Heng HL, Gan A, Zang ZJ, Wu Y, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet. 2012;44:690–693. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- 10.Marsh Rde W, Alonzo M, Bajaj S, Baker M, Elton E, Farrell TA, Gore RM, Hall C, Nowak J, Roy H, Shaikh A, Talamonti MS. Comprehensive review of the diagnosis and treatment of biliary tract cancer 2012. Part I: diagnosis-clinical staging and pathology. J Surg Oncol. 2012;106:332–338. doi: 10.1002/jso.23028. [DOI] [PubMed] [Google Scholar]
- 11.Sia D, Tovar V, Moeini A, Llovet JM. Intrahepatic cholangiocarcinoma: pathogenesis and rationale for molecular therapies. Oncogene. 2013;32:4861–4870. doi: 10.1038/onc.2012.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fassan M, Baffa R, Kiss A. Advanced precancerous lesions within the GI tract: the molecular background. Best Pract Res Clin Gastroenterol. 2013;27:159–169. doi: 10.1016/j.bpg.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, Offerhaus GJ, Roa JC, Roberts LR, Gores GJ, Popescu I, Alexandrescu ST, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet. 2013;45:1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voss JS, Holtegaard LM, Kerr SE, Fritcher EG, Roberts LR, Gores GJ, Zhang J, Highsmith WE, Halling KC, Kipp BR. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Human pathology. 2013;44:1216–1222. doi: 10.1016/j.humpath.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, Kwak EL, Clark JW, Ryan DP, Deshpande V, Dias-Santagata D, Ellisen LW, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu RF, Sun JP, Zhang SR, Zhu GS, Li LB, Liao YL, Xie JM, Liao WJ. KRAS and PIK3CA but not BRAF genes are frequently mutated in Chinese cholangiocarcinoma patients. Biomed Pharmacother. 2011;65:22–26. doi: 10.1016/j.biopha.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Chuang SC, Lee KT, Tsai KB, Sheen PC, Nagai E, Mizumoto K, Tanaka M. Immunohistochemical study of DPC4 and p53 proteins in gallbladder and bile duct cancers. World J Surg. 2004;28:995–1000. doi: 10.1007/s00268-004-7447-8. [DOI] [PubMed] [Google Scholar]
- 18.Tannapfel A, Sommerer F, Benicke M, Weinans L, Katalinic A, Geissler F, Uhlmann D, Hauss J, Wittekind C. Genetic and epigenetic alterations of the INK4a-ARF pathway in cholangiocarcinoma. J Pathol. 2002;197:624–631. doi: 10.1002/path.1139. [DOI] [PubMed] [Google Scholar]
- 19.Parwani AV, Geradts J, Caspers E, Offerhaus GJ, Yeo CJ, Cameron JL, Klimstra DS, Maitra A, Hruban RH, Argani P. Immunohistochemical and genetic analysis of non-small cell and small cell gallbladder carcinoma and their precursor lesions. Mod Pathol. 2003;16:299–308. doi: 10.1097/01.MP.0000062656.60581.AA. [DOI] [PubMed] [Google Scholar]
- 20.Yanagisawa N, Mikami T, Saegusa M, Okayasu I. More frequent beta-catenin exon 3 mutations in gallbladder adenomas than in carcinomas indicate different lineages. Cancer Res. 2001;61:19–22. [PubMed] [Google Scholar]
- 21.Chan-On W, Nairismagi ML, Ong CK, Lim WK, Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL, Ooi L, Chung A, Chow P, Cheow PC, Lee SY, Choo SP, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013;45:1474–1478. doi: 10.1038/ng.2806. [DOI] [PubMed] [Google Scholar]
- 22.Hadd AG, Houghton J, Choudhary A, Sah S, Chen L, Marko AC, Sanford T, Buddavarapu K, Krosting J, Garmire L, Wylie D, Shinde R, Beaudenon S, Alexander EK, Mambo E, Adai AT, et al. Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J Mol Diagn. 2013;15:234–247. doi: 10.1016/j.jmoldx.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Luchini C, Capelli P, Fassan M, Simbolo M, Mafficini A, Pedica F, Ruzzenente A, Guglielmi A, Corbo V, Scarpa A. Next-Generation Histopathologic Diagnosis: A Lesson From a Hepatic Carcinosarcoma. J Clin Oncol. 2014 doi: 10.1200/JCO.2012.47.5855. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24.Simbolo M, Gottardi M, Corbo V, Fassan M, Mafficini A, Malpeli G, Lawlor RT, Scarpa A. DNA qualification workflow for next generation sequencing of histopathological samples. PLoS One. 2013;8:e62692. doi: 10.1371/journal.pone.0062692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fassan M, Simbolo M, Bria E, Mafficini A, Pilotto S, Capelli P, Bencivenga M, Pecori S, Luchini C, Neves D, Turri G, Vicentini C, Montagna L, Tomezzoli A, Tortora G, Chilosi M, et al. High-throughput mutation profiling identifies novel molecular dysregulation in high-grade intraepithelial neoplasia and early gastric cancers. Gastric Cancer. 2013 doi: 10.1007/s10120-013-0315-1. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 26.Farges O, Fuks D, Le Treut YP, Azoulay D, Laurent A, Bachellier P, Nuzzo G, Belghiti J, Pruvot FR, Regimbeau JM. AJCC 7th edition of TNM staging accurately discriminates outcomes of patients with resectable intrahepatic cholangiocarcinoma: By the AFC-IHCC-2009 study group. Cancer. 2011;117:2170–2177. doi: 10.1002/cncr.25712. [DOI] [PubMed] [Google Scholar]
- 27.Zamo A, Bertolaso A, van Raaij AW, Mancini F, Scardoni M, Montresor M, Menestrina F, van Krieken JH, Chilosi M, Groenen PJ, Scarpa A. Application of microfluidic technology to the BIOMED-2 protocol for detection of B-cell clonality. J Mol Diagn. 2012;14:30–37. doi: 10.1016/j.jmoldx.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Scarpa A, Sikora K, Fassan M, Rachiglio AM, Cappellesso R, Antonello D, Amato E, Mafficini A, Lambiase M, Esposito C, Bria E, Simonato F, Scardoni M, Turri G, Chilosi M, Tortora G, et al. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PLoS One. 2013;8:e80478. doi: 10.1371/journal.pone.0080478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M, Piemonti L, Capurso G, Di Florio A, delle Fave G, Pederzoli P, Croce CM, Scarpa A. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28:245–255. doi: 10.1200/JCO.2008.21.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Endo M, Yamamoto H, Setsu N, Kohashi K, Takahashi Y, Ishii T, Iida K, Matsumoto Y, Hakozaki M, Aoki M, Iwasaki H, Dobashi Y, Nishiyama K, Iwamoto Y, Oda Y. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res. 2013;19:450–461. doi: 10.1158/1078-0432.CCR-12-1067. [DOI] [PubMed] [Google Scholar]
- 31.Corbo V, Beghelli S, Bersani S, Antonello D, Talamini G, Brunelli M, Capelli P, Falconi M, Scarpa A. Pancreatic endocrine tumours: mutational and immunohistochemical survey of protein kinases reveals alterations in targetable kinases in cancer cell lines and rare primaries. Ann Oncol. 2012;23:127–134. doi: 10.1093/annonc/mdr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:512–522. doi: 10.1038/nrgastro.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertuccio P, Bosetti C, Levi F, Decarli A, Negri E, La Vecchia C. A comparison of trends in mortality from primary liver cancer and intrahepatic cholangiocarcinoma in Europe. Ann Oncol. 2013;24:1667–1674. doi: 10.1093/annonc/mds652. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 35.Kipp BR, Fritcher EG, Clayton AC, Gores GJ, Roberts LR, Zhang J, Levy MJ, Halling KC. Comparison of KRAS mutation analysis and FISH for detecting pancreatobiliary tract cancer in cytology specimens collected during endoscopic retrograde cholangiopancreatography. J Mol Diagn. 2010;12:780–786. doi: 10.2353/jmoldx.2010.100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Dell MR, Huang JL, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, Rossi RM, Zhu AX, Land H, Bardeesy N, Hezel AF. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72:1557–1567. doi: 10.1158/0008-5472.CAN-11-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deshpande V, Nduaguba A, Zimmerman SM, Kehoe SM, Macconaill LE, Lauwers GY, Ferrone C, Bardeesy N, Zhu AX, Hezel AF. Mutational profiling reveals PIK3CA mutations in gallbladder carcinoma. BMC Cancer. 2011;11:60. doi: 10.1186/1471-2407-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang S, Chun SM, Hong SM, Sung CO, Park H, Kang HJ, Kim KP, Lee YJ, Yu E. High throughput molecular profiling reveals differential mutation patterns in intrahepatic cholangiocarcinomas arising in chronic advanced liver diseases. Mod Pathol. 2013 doi: 10.1038/modpathol.2013.194. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 39.Valle JW, Furuse J, Jitlal M, Beare S, Mizuno N, Wasan H, Bridgewater J, Okusaka T. Cisplatin and gemcitabine for advanced biliary tract cancer: a meta-analysis of two randomised trials. Ann Oncol. 2014;25:391–398. doi: 10.1093/annonc/mdt540. [DOI] [PubMed] [Google Scholar]
- 40.Faris JE, Zhu AX. Targeted therapy for biliary tract cancers. J Hepatobiliary Pancreat Sci. 2012;19:326–336. doi: 10.1007/s00534-011-0496-0. [DOI] [PubMed] [Google Scholar]
- 41.Gwak GY, Yoon JH, Shin CM, Ahn YJ, Chung JK, Kim YA, Kim TY, Lee HS. Detection of response-predicting mutations in the kinase domain of the epidermal growth factor receptor gene in cholangiocarcinomas. J Cancer Res Clin Oncol. 2005;131:649–652. doi: 10.1007/s00432-005-0016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andersen JB, Spee B, Blechacz BR, Avital I, Komuta M, Barbour A, Conner EA, Gillen MC, Roskams T, Roberts LR, Factor VM, Thorgeirsson SS. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology. 2012;142:1021–1031 e1015. doi: 10.1053/j.gastro.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leone F, Cavalloni G, Pignochino Y, Sarotto I, Ferraris R, Piacibello W, Venesio T, Capussotti L, Risio M, Aglietta M. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin Cancer Res. 2006;12:1680–1685. doi: 10.1158/1078-0432.CCR-05-1692. [DOI] [PubMed] [Google Scholar]
- 44.Reinersman JM, Johnson ML, Riely GJ, Chitale DA, Nicastri AD, Soff GA, Schwartz AG, Sima CS, Ayalew G, Lau C, Zakowski MF, Rusch VW, Ladanyi M, Kris MG. Frequency of EGFR and KRAS mutations in lung adenocarcinomas in African Americans. J Thorac Oncol. 2011;6:28–31. doi: 10.1097/JTO.0b013e3181fb4fe2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coulouarn C, Cavard C, Rubbia-Brandt L, Audebourg A, Dumont F, Jacques S, Just PA, Clement B, Gilgenkrantz H, Perret C, Terris B. Combined hepatocellular-cholangiocarcinomas exhibit progenitor features and activation of Wnt and TGFbeta signaling pathways. Carcinogenesis. 2012;33:1791–1796. doi: 10.1093/carcin/bgs208. [DOI] [PubMed] [Google Scholar]
- 46.Araki K, Shimura T, Suzuki H, Tsutsumi S, Wada W, Yajima T, Kobayahi T, Kubo N, Kuwano H. E/N-cadherin switch mediates cancer progression via TGF-beta-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br J Cancer. 2011;105:1885–1893. doi: 10.1038/bjc.2011.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borger DR, Goyal L, Yau TC, Poon RT, Ancukiewicz M, Deshpande V, Christiani DC, Liebman HM, Yang H, Kim H, Ellwood-Yen K, Faris JE, Iafrate AJ, Kwak EL, Clark JW, Allen JN, et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-13-2649. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.