

Abstract
Objectives
Primary and acquired resistance to EGFR TKIs in EGFR mutant lung cancer occurs primarily through secondary mutations in EGFR or Met amplification. Drug resistance can also be mediated by expression of pluripotency transcription factors, such as OCT4, SOX2 and NANOG that decrease terminal differentiation. In this study, we investigated the expression and role of SOX2 in model systems of EGFR mutant tumors.
Materials and Methods
Immunoblotting or immunohistochemistry was used to assess expression of pluripotency transcription factors in lungs of transgenic mice or in human NSCLC cell lines. Expression of SOX2 was reduced by shRNA knockdown, and response to erlotinib and cellular proliferation were assessed.
Results and Conclusion
Induction of mutant EGFR in transgenic CCSP-rtTA/TetO-EGFRL858R/T790M mice correlated with increased OCT4 and SOX2 expression in lung tissue prior to tumor development. Established lung tumors retained SOX2 expression. To assess a role for SOX2 in tumorigenesis, a panel of NSCLC cell lines with activating EGFR mutations was assessed for SOX2 expression. Two of six cell lines with mutant EGFR showed detectable SOX2 levels, suggesting SOX2 expression did not correlate with EGFR mutation status. To assess the role of SOX2 in these cell lines, HCC827 and H1975 cells were infected with lentivirus containing SOX2 shRNA. Knockdown of SOX2 decreased proliferation in both cell lines and increased sensitivity to erlotinib in HCC827 cells. Because constitutive activation of the PI3K/Akt pathway is associated with EGFR TKI resistance, cells were treated with PI3K/AKT inhibitors and expression of SOX2 was examined. PI3K/Akt inhibitors decreased SOX2 expression in a time-dependent manner. These data suggest targeting SOX2 may provide therapeutic benefit in the subset of EGFR-mutant tumors with high constitutive levels of SOX2, and that until more direct means of inhibiting SOX2 are developed, PI3K/Akt inhibitors might be useful to inhibit SOX2 in EGFR TKI resistant tumors.
Keywords: SOX2, non-small cell lung cancer, EGFR, Akt
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer-related death worldwide and new therapeutic strategies are needed [1]. In lung cancer, drugs that target tumor-specific mutations in EGFR, MET or ALK are effective in a subset of patients [2, 3]; however, acquired resistance develops in nearly all patients. Thus the identification and development of strategies to prevent resistance are needed to achieve further improvements in survival. The EGFR TKI erlotinib improves progression-free survival in patients with activating mutations in EGFR over chemotherapy [4], and was recently approved by the FDA as a first line treatment in these patients. Resistance mechanisms to EGFR TKIs include mutations in EGFR that change affinity for EGFR TKI (T790M) that occur in 50% of patients, and MET amplification that occurs in 20% of patients [5-7]. Additional mechanisms of resistance include EMT, histologic transformation from adenocarcinoma to small cell lung cancer (SCLC) [8] and activation of IGFR signaling. Another mechanism of resistance believed to play a role in lung cancer is the acquisition of a dedifferentiated or ‘cancer stem cell’ (CSC) phenotype. The cancer stem cell hypothesis holds that only a rare subpopulation of tumor cells are capable of forming the full range of differentiated cells that make up a tumor. These cells can self renew, cycle slowly and exhibit drug resistance. In certain tumors the CSC population is believed fixed and a cellular hierarchy exists. In others, the phenotype appears to be plastic and may increase upon the acquisition of additional mutations or selective pressures in the tumor microenvironment.
Transcription factors such as SOX2, OCT4, NANOG, KLF4, LIN28 and c-MYC can reprogram somatic cells into pluripotent embryonic stem cells [9]. In lung cancer, overexpression of pluripotency genes contributes to drug resistance and poor prognosis [10-13].
The PI3K/Akt signaling pathway controls cell proliferation, survival, metabolism and drug resistance and is constitutively activated in a majority of human cancers, including NSCLC [14]. Activation of the PI3K/Akt pathway promotes resistance to EGFR TKI. Here, we investigate the role of pluripotency transcription factors and their relationship to Akt signaling and erlotinib sensitivity in mutant EGFR-driven lung cancer.
Materials and Methods
Cell culture
Human NSCLC cell lines with activating EGFR mutations, PC9 (exon 19 deletion), HCC827 and HCC827 GR5 (E746_A750 deletion), H820 (L858R) and H1975 (L858R) were used. H1975 and H820 cells also contain T790M mutation at exon 20 of the EGFR gene. HCC827 GR5 cells are EGFR TKI resistant and have MET amplification [7]. All cells were maintained in RPMI 1640 containing 5% or 10% fetal bovine serum (FBS), 100 units/mL of penicillin, and 100 mg/mL of streptomycin at 37°C in a 5% CO2 atmosphere.
SOX2 shRNA knockdown
pLKO.1 lentivirus containing constitutive or doxycycline-inducible shRNA against Sox2 and scrambled control construct (Sigma) were packaged in 293FT cells using PsPAX2 and pMDG.2 packaging plasmids from Addgene. Cells were grown to 70–80% confluence in a T25 flask and transfected with plasmids using Lipofectamine 2000 reagent (Invitrogen, Grand Island, NY). Media was changed following overnight infection, and viral supernatants were collected after 24h and stored at -80°C for future infections.
Immunoblotting
Cells were plated at a density of 5 × 105 per well in six-well plates. The following day, cells were treated with drug or equal volume of DMSO for the indicated times. Cell extracts were prepared by adding 2× Laemmli sample buffer supplemented with a phosphatase and protease inhibitor cocktail [15]. Lysates were sonicated and the protein concentration was quantified using the bicinchoninic acid protein assay. Equivalent protein was loaded and proteins were separated by SDS-PAGE then transferred to 0.45-μm nitrocellulose membranes. Equivalent loading was confirmed by staining membranes with fast green as previously described [15]. Membranes were blocked for 1 h in blocking buffer (5% milk, 1× TBS, 0.1% Tween 20) and placed in primary antibody (5% bovine serum albumin, 1× TBS, 0.1% Tween 20) overnight at 4°C. The following day, membranes were washed thrice in wash buffer (1× TBS, 0.1% Tween 20). Primary antibodies against P-Akt, Oct4 (#2788), P-EGFR (Y1068) (#3777), total EGFR (#2232), EGFR L858R (#3197), P-stat3 Y705 (#9145), SOX2 (#3728) from Cell Signaling (Danvers, MA) and an alpha-tubulin antibody #T5168 from Sigma (St. Louis, MO) were detected using horseradish peroxidase–linked secondary antibodies and visualized with the enhanced chemiluminescent detection system (GE Healthcare Biosciences, Pittsburgh, PA). Immunoblot experiments were performed at least thrice.
Immunohistochemistry
Immunohistochemistry was performed on lungs harvested from Dox- treated mice. Lung tissues were fixed in formalin, embedded in paraffin blocks and sectioned to slides. Briefly, slides were heated at 65°C and then placed through xylene and ethanol washes. Antigen retrieval was performed using target retrieval solution (Dako, Carpinteria, CA) and a decloaking chamber. Sections were incubated with (1:50) SOX2 antibody (Cell Signaling #3728) overnight at 4°C. Detection was done using a VECTASTAIN Elite ABC kit (Vector Laboratories).
Proliferation assay
The effect of erlotinib on proliferation was determined using the sulforhodamine B (SRB) assay. Cells were plated at a density of 5,000 per well in 96-well plates in RPMI 1640 + 5% FBS and allowed to grow for 24 h in a humidified 5% CO2 incubator at 37°C. Cells were incubated with drugs for 48h. Following drug treatments cells were fixed with 10% trichloroacetic acid for 1h at 4°C. Cells then were washed 5 times with distilled and de-ionized water. After air drying, cells were stained with a 0.4% (w/v) solution of SRB in 1% acetic acid. Cells were then washed with 1% acetic acid 5 times to remove the unbound dye. To redissolve the dye, 10 mM Tris solution was added and the absorbance was read at 515 nm. Cell survival was calculated as a percentage of the value of DMSO-treated controls minus the value of untreated cells on day 0. Experiments were done in triplicate and each drug concentration was evaluated in sextuplet wells for any given experiment.
Animal study
Animal studies were conducted under a protocol approved by the Animal Care and Use Committee of the National Cancer Institute. The generation of bi-transgenic mice harboring the CCSP-rtTA and TetO-regulated EGFRL858R+T790M transgenes has been previously described [16]. To induce expression of mutant EGFR, mice were fed doxycycline-impregnated food pellets (NIH-31 with 650 ppm doxycycline, Harlan-Teklad, Madison, WI). Beginning at 6 weeks of age, groups of 3 mice received doxycycline (DOX) food or regular cereal diet. Mice were sacrified after 3 or 9 weeks. At sacrifice, lungs were removed and inflated with 10% neutral-buffered formalin. Lungs were embedded in paraffin and sectioned for immunohistochemistry.
Results
To determine whether pluripotency genes play a role in mutant EGFR-driven lung tumorigenesis, we examined the expression of OCT4 and SOX2 in lungs of CCSP-rtTA/TetO-EGFRL858R+T790M mice by immunoblotting analysis. Mice fed doxycycline (dox) in their diet displayed increased expression of phospho- and mutant-EGFR in whole lung lysates compared to mice that received a normal diet (Figure 1A). Doxycycline also induced lung tumors after 3 weeks of treatment, whereas mice on a regular diet had normal lung phenotypes (data not shown). The levels of phosphorylated AKT and FOXO were evaluated as readouts of EGFR signaling and were increased in the groups that received doxycycline. Furthermore, SOX2 and OCT4 expression increased with dox administration compared to the control diet. To investigate if EGFR mutant lung tumors expressed SOX2, lungs of mice that received dox were stained for SOX2 by immunohistochemistry (IHC). IHC of mouse lungs showed that tumors displayed both nuclear and cytoplasmic SOX2 (Figure 1B). In addition, cells lining bronchioles stained positive for SOX2, whereas alveolar cells were negative. This finding is consistent with SOX2 expression in clara, goblet and ciliated cells, as previously reported [17]. To determine if SOX2 and OCT4 expression were maintained in tumors when EGFR signaling was decreased, mice were given doxycycline to induce tumors, then switched to a normal diet for 2 and 5 days. EGFR expression and activation were rapidly reduced when dox was withdrawn, which correlated with decreased SOX2 and OCT4 expression (data not shown). These data show that EGFR signaling correlates with SOX2 expression in lung tumors.
Figure 1.
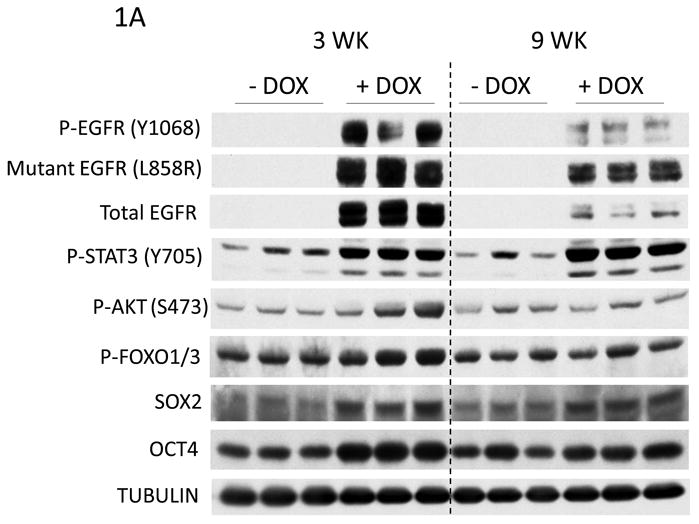
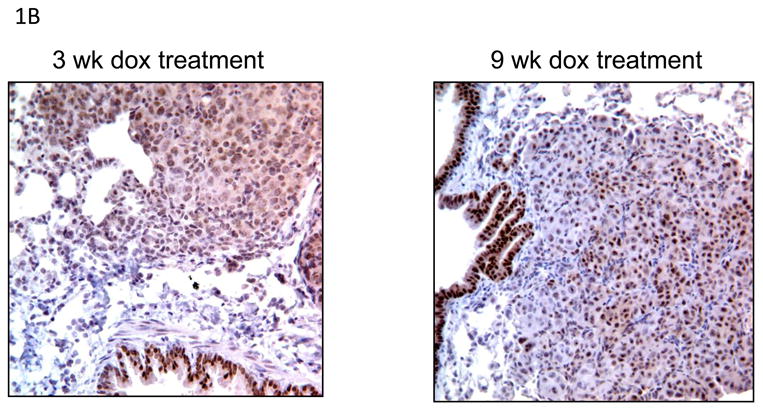
Elevated EGFR signaling correlates with SOX2 expression in lung tissues in vivo. A) Immunoblots of whole lung lysates of CCSP-rtTA/TetO- EGFRL858R+T790M mutant mice treated with DOX for 3 and 9 weeks. B) Immunohistochemical staining of CCSP-rtTA/TetO-egfrL858R+T790m mouse lungs show SOX2 expression in tumors after 3 and 9 weeks of DOX administration.
To examine the relationship of SOX2 to EGFR mutation status, SOX2 levels were analyzed in a panel of human lung cancer cells with activating EGFR mutations that are either EGFR TKI sensitive (PC9, HCC827) or resistant (H1975, H820, HCC827 GR5, H1650) (Figure 2). (A549 and H522 are Kras mutant NSCLC cell lines that were used as positive controls.) Of the six EGFR mutant cell lines, only H1975 and HCC827 cells had detectable levels of SOX2 by immunoblotting analysis. Therefore, SOX2 expression did not appear to correlate with EGFR mutation status or TKI sensitivity in these cell lines.
Figure 2.
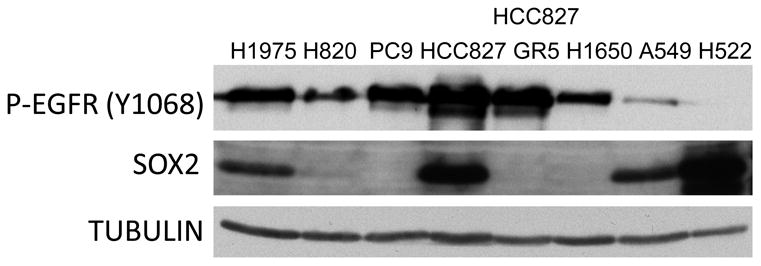
Basal levels of SOX2 vary in human NSCLC cell lines. Immunoblot analysis of human NSCLC cell lines with activating mutations in EGFR (H1975, H820, PC9, HCC827, HCC827 GR5, H1650) or Kras (A549, H522).
To examine the effect of EGFR signaling on SOX2 in human cell lines, EGFR mutant cell lines were treated with erlotinib (Figure 3A). At 3 days, erlotinib decreased p-EGFR and p-Akt levels in HCC827 cells, and this correlated with a decrease in SOX2, OCT4, and NANOG expression. In contrast, erlotinib had a minimal or no impact on p-EGFR and p-Akt in H1975 cells, likely due to the presence of a T790M mutation. In H1975 cells, expression of the three pluripotentcy markers increased with 5 μM erlotinib. At 25 μM, expression of p-EGFR and SOX2 were decreased. Phospho-Akt was not decreased by erlotinib in H1975 cells. These experiments show that erlotinib can decrease p-EGFR and SOX2 in EGFR TKI resistant and sensitive cells in a dose dependent manner.
Figure 3.
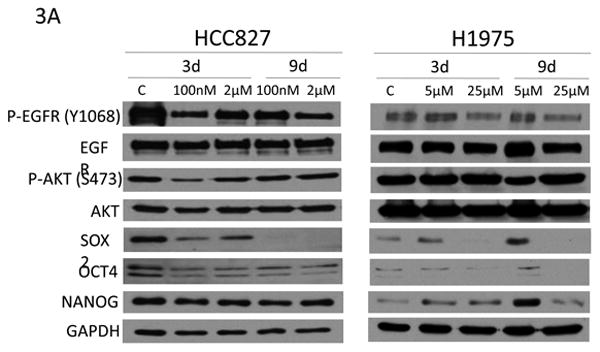
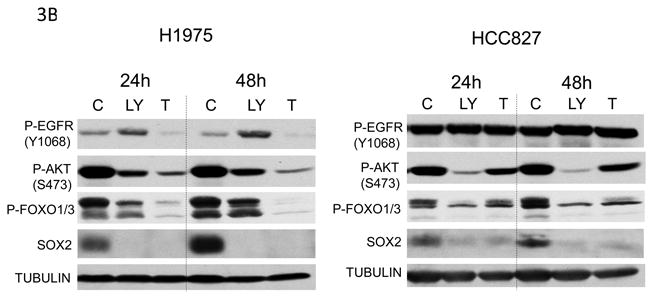
A) Erlotinib alters the expression of SOX2 and OCT4 in EGFR mutant NSCLC cell lines. Immunoblot analysis of H1975 and HCC827 cells treated with varying doses of erlotinib for 3 and 9 days. C=vehicle control. B) PI3K/AKT pathway inhibitors decrease SOX2 expression in NSCLC cell lines. Immunoblot analysis of H1975 (left) and HCC827 (right) cells treated with DMSO (C), 10μM LY294002 (LY) or 10μM tricibine (T) for the indicated times.
The PI3K/AKT pathway increases cell survival and therapeutic resistance in cancer [18], and Akt has been shown to phosphorylate and stabilize SOX2 [19]. To determine if SOX2 is under the control of the PI3K/AKT pathway in these cells, H1975 and HCC827 cells were treated with a PI3K inhibitor (LY294002) or an AKT inhibitor (triciribine) (Figure 3B). Phosphorylation of AKT and FOXO decreased with inhibitor treatment, which correlated with decreased SOX2 expression. This suggests that SOX2 is under the control of PI3K/AKT pathway in these cells.
To more directly evaluate the role of SOX2 expression in EGFR mutant lung cancer, cells that had high SOX2 levels were infected with lentivirus containing doxycycline-inducible shRNA to SOX2 or dox-inducible scrambled shRNA. Following antibiotic selection, cell proliferation was evaluated by counting cells grown in the presence or absence of dox over time (Figure 4A). In the presence of dox, the shSOX2 RNA infected cells grew more slowly, but cells infected with the scrambled shRNA grew at the same rate, indicating SOX2 plays a role in proliferation. Next, we tested the erlotinib sensitivity of NSCLC cells infected with lentivirus to scrambled (scr) or SOX2 shRNA. Stable SOX2 knockdown increased erlotinib sensitivity in HCC827, but had little impact on growth inhibition by erlotinib in H1975 cells (Figure 4B). This shows that the ability of SOX2 to control sensitivity to erlotinib is context dependent. This may be relevant to H1975 cells since they also have Met amplification as well as a T790M mutation in EGFR. Collectively, our data suggest that SOX2 is under the control of the PI3K/Akt pathway in EGFR mutant lung cancer cells and contributes to proliferation in EGFR mutant lung tumors with high SOX2 levels.
Figure 4.
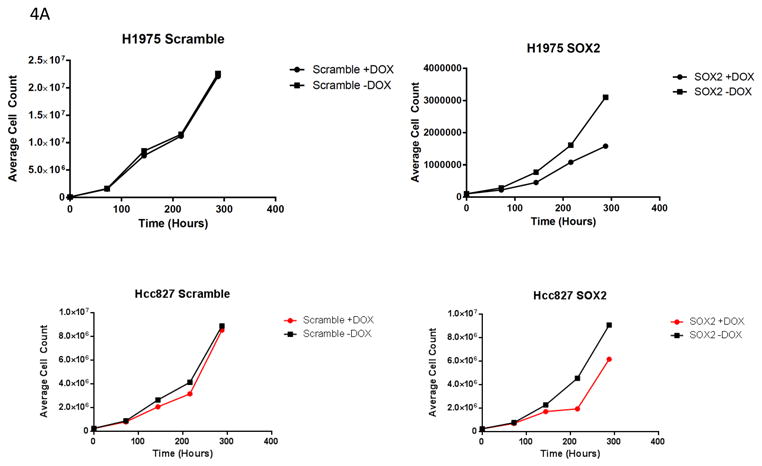
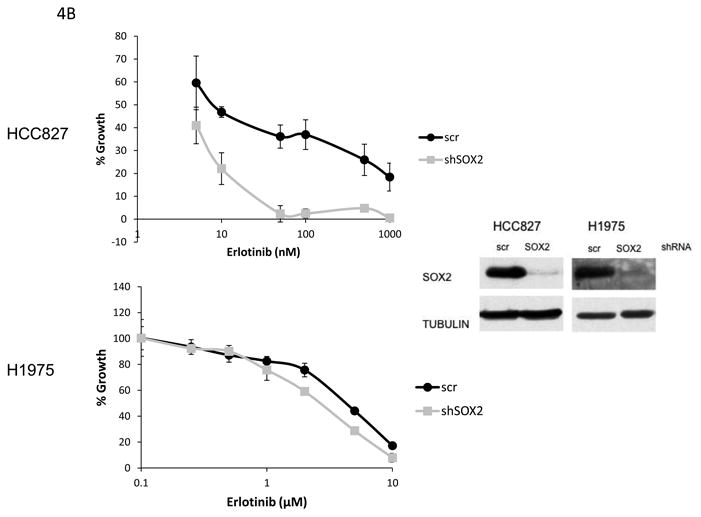
Effect of SOX2 knockdown in EGFR mutant lung cancer cell lines. A) Sox2 knockdown decreases proliferation in HCC827 and H1975 cells. B) Sox2 knockdown increases sensitivity to erlotinib in HCC827 cells.
Discussion
Tumor genotype is an important determinant of lung cancer initiating cells [20]. Although the cell of origin for Kras mutant lung tumors has been proposed [21], the cell or origin for EGFR mutant lung cancer remains poorly defined [20]. Here we investigated the expression of pluripotency transcription factors in a mouse model of mutant EGFR-driven lung cancer. We show that SOX2 and OCT4 expression correlate with EGFR signaling in lung tissue of mutant CCSP-rtTA/TetO-EGFRL858R/T790M mice, and that mutant EGFR-driven lung tumors in this model express SOX2. In human NSCLC cell lines, EGFR mutation status or TKI sensitivity did not correlate with SOX2 expression. For example, whereas HCC827 cells had high SOX2 expression, MET-amplified HCC827 GR5 cells that were made resistant to EGFR TKIs had low levels of SOX2. Interestingly, MET expression has been inversely correlated with SOX2 expression in human EGFR mutant lung cancers [22]. Therefore, acquisition of MET amplification might confer SOX2 independence and might be a negative predictor of SOX2 expression in EGFR mutant NSCLC.
In this study, knockdown of SOX2 slowed the proliferation of HCC827 and H1975 cells suggesting that SOX2 plays a role in proliferation. This finding is similar to the role of SOX2 in other histologic types of lung cancer. For example, SOX2 is amplified and overexpressed in 23% of squamous cell carcinomas and 27% of small cell lung cancer [23-26]; in both subtypes SOX2 promotes proliferation, whereas in squamous cell carcinoma SOX2 overexpression also promotes migration and anchorage-independence [23]. Because SOX2 expression has been linked to drug resistance [27-29], we examined the effect of SOX2 expression on erlotinib sensitivity in these cells. SOX2 knockdown increased the sensitivity to erlotinib in EGFR TKI sensitive HCC827 cells, but had a much smaller effect on erlotinib sensitivity in EGFR TKI resistant H1975 cells.
What mechanisms could explain the context dependency and complexity of regulation of SOX2 by EGFR? In cancer SOX2 is regulated by androgen, TGF beta, chemotherapy or hypoxia [30-33], and there is evidence that EGFR and SOX2 can regulate each other. In neural progenitor cells, EGFR signaling causes SOX2 to bind to the EGFR promoter that drives its expression [34] and EGFR-Hedgehog/Gli signaling induces SOX2 expression in pancreatic cancer initiating cells [35]. In NSCLC, Singh et al showed that EGFR is upstream of SOX2 in the side population [36], and several studies have suggested SOX2 expression plays a role in lung tumor-initiating cells [37] [12] [38, 39]. However, it is unclear if EGFR controls SOX2 in differentiated tumor cells, since in our study only 2 cell lines with constitutively active EGFR had high SOX2 expression. Because PC9, H820, H1650 all had low SOX2 expression, it is possible that mutations in other pathways related to pluripotency may drive the increased SOX2 expression in HCC827 and H1975 cells. According to the Cancer Cell line encyclopedia [40], these cell lines harbor mutations in members of the notch, sonic hedgehog and nuclear receptor pathways; (HCC827-mutations in NCOA3, MAML3, PTCH1 and H1975-mutations in NCOA3 [41], GLI1, Notch4 and MAML2+3).
Although our experiments were performed using pre-clinical models that may not be representative of human disease, our results suggest SOX2 may be a therapeutic target in EGFR mutant lung cancers that overexpress SOX2. Drugs designed to target transcription factors are rare, and blocking upstream signaling pathways that regulate them might be a practical approach to reduce their activity. In this regard, our results demonstrate that the PI3K/AKT pathway regulates SOX2 and that inhibitors of this pathway may be useful to decrease SOX2 expression in EGFR TKI sensitive or resistant NSCLC.
Acknowledgments
Funding: This work was supported by the Intramural Program of the National Cancer Institute [ZIA SC010292].
Footnotes
Conflict of interest statement.- SOX2 is expressed and promotes proliferation in a subset of EGFR mutant lung adenocarcinomas.
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal ABF, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;2:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 5.Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. Plos Medicine. 2005;2:225–235. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 8.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aoi T, Yae K, Nakagawa M, et al. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321:699–702. doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- 10.Rousseaux S, Debernardi A, Jacquiau B, et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013;5:186ra166. doi: 10.1126/scitranslmed.3005723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakatsugawa M, Takahashi A, Hirohashi Y, et al. SOX2 is overexpressed in stem-like cells of human lung adenocarcinoma and augments the tumorigenicity. Laboratory Investigation. 2011;91:1796–1804. doi: 10.1038/labinvest.2011.140. [DOI] [PubMed] [Google Scholar]
- 13.Chiou SH, Wang ML, Chou YT, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70:10433–10444. doi: 10.1158/0008-5472.CAN-10-2638. [DOI] [PubMed] [Google Scholar]
- 14.Bartholomeusz C, Gonzalez-Angulo AM. Targeting the PI3K signaling pathway in cancer therapy. Expert Opinion on Therapeutic Targets. 2012;16:121–130. doi: 10.1517/14728222.2011.644788. [DOI] [PubMed] [Google Scholar]
- 15.Canman CE, Wolff AC, Chen CY, et al. The p53-dependent G1 cell cycle checkpoint pathway and ataxia-telangiectasia. Cancer Res. 1994;54:5054–5058. [PubMed] [Google Scholar]
- 16.Regales L, Balak MN, Gong Y, et al. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS One. 2007;2:e810. doi: 10.1371/journal.pone.0000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tompkins DH, Besnard V, Lange AW, et al. Sox2 is required for maintenance and differentiation of bronchiolar Clara, ciliated, and goblet cells. PLoS One. 2009;4:e8248. doi: 10.1371/journal.pone.0008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resistance Updates. 2002;5:234–248. doi: 10.1016/s1368-7646(02)00120-6. [DOI] [PubMed] [Google Scholar]
- 19.Jeong CH, Cho YY, Kim MO, et al. Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells. 2010;28:2141–2150. doi: 10.1002/stem.540. [DOI] [PubMed] [Google Scholar]
- 20.Curtis SJ, Sinkevicius KW, Li D, et al. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell. 2010;7:127–133. doi: 10.1016/j.stem.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 22.Cai YR, Zhang HQ, Qu Y, et al. Expression of MET and SOX2 genes in non-small cell lung carcinoma with EGFR mutation. Oncol Rep. 2011;26:877–885. doi: 10.3892/or.2011.1349. [DOI] [PubMed] [Google Scholar]
- 23.Bass AJ, Watanabe H, Mermel CH, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238–1242. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hussenet T, Dali S, Exinger J, et al. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS One. 2010;5:e8960. doi: 10.1371/journal.pone.0008960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan P, Kadara H, Behrens C, et al. Sex Determining Region Y-Box 2 (SOX2) Is a Potential Cell-Lineage Gene Highly Expressed in the Pathogenesis of Squamous Cell Carcinomas of the Lung. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudin CM, Durinck S, Stawiski EW, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian T, Zhang Y, Wang S, et al. Sox2 enhances the tumorigenicity and chemoresistance of cancer stem-like cells derived from gastric cancer. J Biomed Res. 2012;26:336–345. doi: 10.7555/JBR.26.20120045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carina V, Zito G, Pizzolanti G, et al. Multiple Pluripotent Stem Cell Markers in Human Anaplastic Thyroid Cancer: The Putative Upstream Role of SOX2. Thyroid. 2013;23:829–837. doi: 10.1089/thy.2012.0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagerstrand D, He XB, Lindh MB, et al. Identification of a SOX2-dependent subset of tumor- and sphere-forming glioblastoma cells with a distinct tyrosine kinase inhibitor sensitivity profile. Neuro-Oncology. 2011;13:1178–1191. doi: 10.1093/neuonc/nor113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kregel S, Kiriluk KJ, Rosen AM, et al. Sox2 is an androgen receptor-repressed gene that promotes castration-resistant prostate cancer. PLoS One. 2013;8:e53701. doi: 10.1371/journal.pone.0053701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pirozzi G, Tirino V, Camerlingo R, et al. Epithelial to mesenchymal transition by TGFbeta-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS One. 2011;6:e21548. doi: 10.1371/journal.pone.0021548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X, Ghisolfi L, Keates AC, et al. Induction of cancer cell stemness by chemotherapy. Cell Cycle. 2012;11:2691–2698. doi: 10.4161/cc.21021. [DOI] [PubMed] [Google Scholar]
- 33.Iida H, Suzuki M, Goitsuka R, Ueno H. Hypoxia induces CD133 expression in human lung cancer cells by up-regulation of OCT3/4 and SOX2. Int J Oncol. 2012;40:71–79. doi: 10.3892/ijo.2011.1207. [DOI] [PubMed] [Google Scholar]
- 34.Hu QK, Zhang LR, Wen JH, et al. The Egf Receptor-Sox2-Egf Receptor Feedback Loop Positively Regulates the Self-Renewal of Neural Precursor Cells. Stem Cells. 2010;28:279–286. doi: 10.1002/stem.246. [DOI] [PubMed] [Google Scholar]
- 35.Eberl M, Klingler S, Mangelberger D, et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol Med. 2012;4:218–233. doi: 10.1002/emmm.201100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh S, Trevino J, Bora-Singhal N, et al. EGFR/Src/Akt signaling modulates Sox2 expression and self-renewal of stem-like side-population cells in non-small cell lung cancer. Mol Cancer. 2012;11:73. doi: 10.1186/1476-4598-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S, Xu Y, Chen Y, et al. SOX2 gene regulates the transcriptional network of oncogenes and affects tumorigenesis of human lung cancer cells. PLoS One. 2012;7:e36326. doi: 10.1371/journal.pone.0036326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiang R, Liao D, Cheng T, et al. Downregulation of transcription factor SOX2 in cancer stem cells suppresses growth and metastasis of lung cancer (vol 104, pg 1410, 2011) British Journal of Cancer. 2011;104:1931–1931. doi: 10.1038/bjc.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu C, Xie D, Yu SC, et al. beta-Catenin/POU5F1/SOX2 Transcription Factor Complex Mediates IGF-I Receptor Signaling and Predicts Poor Prognosis in Lung Adenocarcinoma. Cancer Res. 2013;73:3181–3189. doi: 10.1158/0008-5472.CAN-12-4403. [DOI] [PubMed] [Google Scholar]
- 40.Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Percharde M, Lavial F, Ng JH, et al. Ncoa3 functions as an essential Esrrb coactivator to sustain embryonic stem cell self-renewal and reprogramming. Genes Dev. 2012;26:2286–2298. doi: 10.1101/gad.195545.112. [DOI] [PMC free article] [PubMed] [Google Scholar]