

ABSTRACT
Nuclear lamins form the major structural elements that comprise the nuclear lamina. Loss of nuclear structural integrity has been implicated as a key factor in the lamin A/C gene mutations that cause laminopathies, whereas the normal regulation of lamin A assembly and organization in interphase cells is still undefined. We assumed phosphorylation to be a major determinant, identifying 20 prime interphase phosphorylation sites, of which eight were high-turnover sites. We examined the roles of these latter sites by site-directed mutagenesis, followed by detailed microscopic analysis – including fluorescence recovery after photobleaching, fluorescence correlation spectroscopy and nuclear extraction techniques. The results reveal three phosphorylation regions, each with dominant sites, together controlling lamin A structure and dynamics. Interestingly, two of these interphase sites are hyper-phosphorylated in mitotic cells and one of these sites is within the sequence that is missing in progerin of the Hutchinson-Gilford progeria syndrome. We present a model where different phosphorylation combinations yield markedly different effects on the assembly, subunit turnover and the mobility of lamin A between, and within, the lamina, the nucleoplasm and the cytoplasm of interphase cells.
KEY WORDS: Intermediate filament, Lamin A, Phosphorylation, Sequestration, Signaling
INTRODUCTION
Nuclear lamins constitute an integral part of the nucleoskeleton. They belong to the large family of cytoskeletal intermediate filament (IF) proteins, which is characterized by a remarkable diversity with respect to their sequences, expression patterns and their abundance in various tissues. Mutations in the genes that encode IF proteins are now emerging as causative factors in a large number of human diseases (Eriksson et al., 2009; Omary, 2009). The most extensively mutated IF gene (LMNA) is that encoding lamin A/C. Hundreds of mutations in LMNA have been discovered to date, which cause a broad range of diseases, collectively known as the laminopathies. These diseases range from metabolic disturbances to myopathies and premature ageing disorders (Butin-Israeli et al., 2012; Capell and Collins, 2006).
From the evolutionary point of view, lamins are believed to be the progenitors of the entire IF protein family, because they share genomic and amino acid sequence homology with the cytoplasmic IFs that are found in invertebrates (for review, see Hutchison and Worman, 2004). There are A- and B-type lamins in mammals. The LMNA gene encodes the A-type lamins (lamin A, lamin C, lamin AΔ10 and lamin C2), whereas lamin B1 and lamin B2 are encoded by the LMNB1 and LMNB2, genes respectively (for review, see Dechat et al., 2008). Lamin expression is developmentally regulated – the B-type lamins are expressed in all vertebrate cells throughout development and differentiation, beginning in the unfertilized egg. It has recently been shown that the A-type lamins are also expressed, at very low levels compared with the B-type lamins, in early embryonic development and their expression increases as a function of cell differentiation (Burke and Stewart, 2002; Eckersley-Maslin et al., 2013). The major functions attributed to the nuclear lamins include the regulation of nuclear size, shape and mechanical properties (Dechat et al., 2008). Related to, or independently of, these structural roles, lamins are involved in DNA replication, DNA damage repair and transcriptional control (Dechat et al., 2008). All of these architectural and control functions could, potentially, be associated with the variable phenotypes of different laminopathies (Burke and Stewart, 2002; Eckersley-Maslin et al., 2013).
Little is known regarding the regulation of lamin assembly and structure within the nucleus. Although it is well established that phosphorylation is the most common and important mechanism that is responsible for the regulation of cytoskeletal IF assembly and disassembly (Eriksson et al., 2009; Pallari and Eriksson, 2006), the role of this post-translational modification in regulating the interphase lamin network is largely unexplored. Most studies of lamin phosphorylation have focused mainly on the roles of phosphorylation in cell division, following the discovery that the mitosis specific kinase Cdk1 targets both A- and B-type lamins during mitosis (Simon and Wilson, 2013). This phosphorylation drives their disassembly during nuclear envelope breakdown in the early stages of mitosis (Dessev et al., 1990; Heald and McKeon, 1990; Mall et al., 2012; Peter et al., 1990). In human lamin A/C, the Cdk1 sites are the N-terminal Ser22 and the C-terminal Ser392, often referred to as the ‘mitotic sites’. Early studies on lamin assembly in vitro have suggested that phosphorylation of Ser22 in human lamin A, or Ser16 in chicken lamin B2, is involved in the regulation of lamin dimer head-to-tail polymerization. The N-terminal site is more important, with respect to assembly, than the C-terminal Ser392 site (Goss et al., 1994; Peter et al., 1991). The phosphorylation of these two sites, however, might be insufficient to cause complete mitotic disassembly of the lamina in vivo, and it has been suggested that Cdk1 might be working synergistically with other mitotic kinase(s) to facilitate lamin depolymerization (Lüscher et al., 1991).
In addition to the disassembly of lamins in mitosis, there are some studies that delineate phosphorylation-dependent lamin functions in interphase cells. For example, site specific phosphorylation of B-type lamin polymers either alters their assembly state (Goss et al., 1994; Hocevar et al., 1993) or inhibits their nuclear import (Hennekes et al., 1993). There is also evidence that Ser403 and Ser404, adjacent to the nuclear localization signal sequence in lamin A, are phosphorylated by protein kinase C (PKC) and that this regulates lamin nuclear targeting. It has also been proposed that phosphorylation of Ser525 regulates either lamina assembly or its interaction with chromatin (Haas and Jost, 1993; Leukel and Jost, 1995). Tyrosine phosphorylation of lamins has also been described (Otto et al., 2001), and numerous phosphorylation sites have been identified in global, high-throughput or large-scale proteomic studies (Beausoleil et al., 2004; Kim et al., 2005; Olsen et al., 2006). However, the existence, and the functional significance, of the majority of lamin phosphorylation sites remain uncharacterized.
There is scarce information on the specific roles of lamin phosphorylation during interphase, the discovery of a large number of laminopathies with phenotypes that indicate a disturbance in transcription and/or signaling, suggests that the lamins, especially the A-type lamins, interact with numerous signaling pathways. To determine the sites and roles of these interactions, we aimed to identify the most important bona fide in vivo lamin A/C phosphorylation sites, by using mass spectrometry techniques, and to characterize their roles in regulating lamin A organization and assembly states within the nucleus. In this way, we identify 20 sites that are phosphorylated during interphase, all restricted to three specific regions. Eight of these sites are characterized as high-turnover sites for phosphorylation (for detailed information on the classification of these sites, see supplementary material Figs S1 and S2). To determine the functions of these latter sites, we generated and expressed lamin A containing either phosphomimetic or phosphorylation-deficient amino acid substitutions and expressed them in HeLa cells. Our findings demonstrate that site-specific phosphorylation of lamin A is a major determinant of the assembly state, nuclear localization, mobility and cytoplasmic transport of nuclear lamins during interphase.
RESULTS
Identification of in vivo phosphorylation sites on lamin A/C
Numerous phosphorylation sites have been described for lamins; however, to establish the bona fide in vivo phosphorylation sites of lamin A/C, and to determine how abundantly the sites are phosphorylated, we used a combination of advanced mass spectrometry and 32P-labeling. One approach was label-free quantitative phosphopeptide analysis of lamin A/C by TiO2 affinity chromatography and liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Table 1). In addition, we employed two-dimensional phosphopeptide mapping (2D-PPM) in combination with MALDI-TOF MS (Kochin et al., 2006) of lamin A and lamin C prepared from lysates of in vivo 32P-labeled asynchronous HeLa cells. This approach yielded an overview of both the number of individual identified phosphorylation sites and their relative importance, as indicated by labeling intensity (supplementary material Fig. S1). To more sensitively identify sites with low levels of phosphorylation and high-turnover sites, cells were incubated in the presence of the type-1 and type-2A phosphatase inhibitor calyculin A. To distinguish the differences between the interphase and mitotic phosphorylation sites, these analyses were carried out on lamin A/C derived from asynchronous interphase cells as well as on cell cultures from which mitotic cells were mechanically removed in order to maximize the interphase cell population.
Table 1. Lamin A/C phosphorylation sites identified by LC-MS/MS from interphase Hela cells.
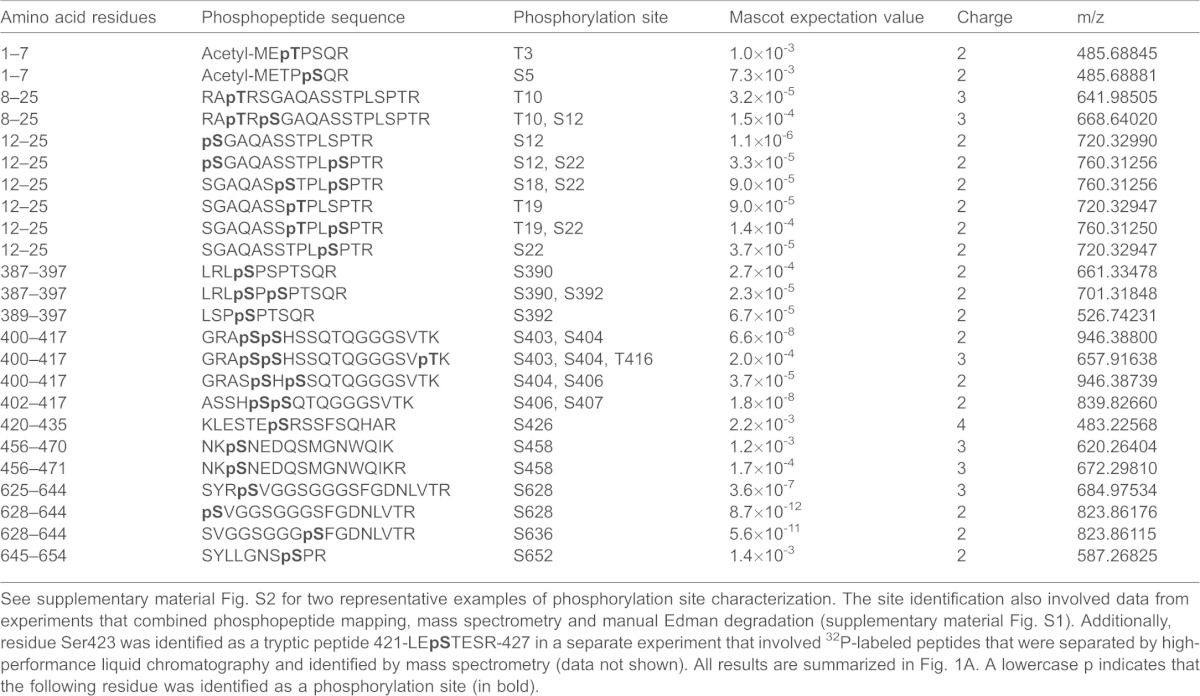
See supplementary material Fig. S2 for two representative examples of phosphorylation site characterization. The site identification also involved data from experiments that combined phosphopeptide mapping, mass spectrometry and manual Edman degradation (supplementary material Fig. S1). Additionally, residue Ser423 was identified as a tryptic peptide 421-LEpSTESR-427 in a separate experiment that involved 32P-labeled peptides that were separated by high-performance liquid chromatography and identified by mass spectrometry (data not shown). All results are summarized in Fig. 1A. A lowercase p indicates that the following residue was identified as a phosphorylation site (in bold).
Table 1 shows 24 phosphopeptides that were identified by LC-MS/MS analysis. Supplementary material Fig. S2A,B shows representative mass spectra, illustrating the identification of two important interphase sites that have previously been identified in mitotic cells (see below). The number of phosphopeptides that were identified by LC-MS/MS corresponded well to the number of individual phosphopeptides observed in the 2D-PPM (supplementary material Fig. S1). The phosphopeptide mapping revealed that only a fraction of these peptides contained high-turnover sites (supplementary material Fig. S1), a result supported by the mass-spectrometry-based estimation of the occupancy rate of individual phosphorylation sites (supplementary material Fig. S2C). Both data sets, obtained by 2D-PPM and LC-MS/MS, corresponded well with each other, in terms of identifying the predominant phosphorylation sites. Both data sets were employed to assign the principal phosphorylation sites, which are illustrated in Fig. 1A. Because the data from interphase cells identified previously established mitotic sites, we wanted to ensure that there was no contamination by mitotic cells in the interphase extracts. Therefore, we checked whether the mitotic markers phosphorylated Cdc27 (detected as a bandshift) and phosphorylated Ser10 on histone 3 (detected with a phospho-specific antibody) were present in extracts from interphase HeLa cells, compared with that of mitotic cells that had been separated from the interphase cells by using the mitotic shake-off technique. Western blotting using antibodies against these mitotic markers yielded positive signals in mitotic lysates only (supplementary material Fig. S2D), confirming the successful separation of interphase cells from mitotic cells and minimizing the likelihood that mitotic extracts contaminated those from interphase cells.
Fig. 1.

Phosphoproteomics of lamin A/C identifies three regions for phosphate exchange with highly conserved phosphorylation motifs. (A) The regions that are phosphorylated in lamin A/C are confined to three regions (top panel, green circles) – 1, the N-terminal head preceding the α-helical rod domain; 2, the proximal C-terminal region preceding the Ig-fold motif; 3, the far C-terminal region. Region 3 is only present in lamin A. The amino acid sequence of each region is also shown. The underlined residues in region 3 are lacking in progerin. Phosphorylated residues are highlighted in red; high-turnover sites are indicated by green bars. The numbers indicate the amino acid residue number. NLS, nuclear localization signal; L1 is a linker domain between coil 1A and 1B domains, L12 is a linker domain between coil 1B and 2A domains, L2 is a linker domain between coil 2A and 2B domains. (B) ClustalW2 multiple sequence alignment of lamin A/C from nine species shows that the phosphorylation motifs are highly conserved (highlighted in green).
The data revealed three main regions on lamin A that were phosphorylated in interphase cells (Fig. 1A). The first region spanned the entire N-terminus, with seven identified phosphorylation sites, two of which (Ser12 and Ser22) were predominant high-turnover sites. Ser22 has been previously identified as one of the major Cdk1 targets that is phosphorylated during mitosis (Dessev et al., 1991; Enoch et al., 1991; Heald and McKeon, 1990; Ward and Kirschner, 1990). The second region lies between the end of the α-helical central rod domain and the Ig-fold, and contained ten sites, including five high-turnover sites (Ser390, Ser392, Ser404, Ser407, Ser423). One of these sites, Ser392, is also a known mitotic site (Goss et al., 1994; Heald and McKeon, 1990; Peter et al., 1991). Our results indicated this to also be an important interphase site as well, because it displayed the highest occupancy rate in the mass spectrometry analysis (supplementary material Fig. S2C). Two sites upstream of the nuclear localization signal (Ser403 and Ser404) have been shown previously to be involved in nuclear transport (Haas and Jost, 1993). Ser22 and Ser392 were observed as being high-turnover sites, regardless of whether the cells were isolated from asynchronous populations or from populations in which the mitotic cells had been mechanically removed (as mentioned above), the fractionation was confirmed by western blotting for mitotic markers (supplementary material Fig. S2D). The third region contained three sites, with one high-turnover site for phosphorylation at Ser628, located at the far C-terminus of lamin A. This region also contained another important site for post-translational modification – the CAAX motif, which determines the farnesylation of prelamin A. It should be noted that all four sites in the third region, including the high-turnover site Ser628, are within the 50-amino-acid stretch that is lacking in progerin – i.e. the mutant lamin A that is expressed in the most commonly encountered form of Hutchinson-Gilford progeria syndrome (HGPS) (Eriksson et al., 2003). As perhaps expected, the 2D-PPM pattern, and the phosphorylation sites that we identified on lamin A and lamin C, were strikingly similar (supplementary material Fig. S1B,C), with the exception of the third region, which is not present in lamin C owing to alternative splicing.
All of the most prominent high-turnover phosphorylation sites are highly conserved among A-type lamins, including evolutionarily distant species (Fig. 1B). In fact, most of the phosphorylation motifs showed a high degree of conservation, supporting the assumption that they play important roles in regulating lamin functions.
Nuclear organization and dynamics of lamin A with phosphomimetic or phosphorylation-deficient amino acid substitutions
Due to the large number of lamin A/C phosphorylation sites, we chose to further investigate the eight high phosphate-turnover sites to determine whether or not they play a role in lamin A assembly or organization. To this end, we expressed either phosphomimetic (Ser to Asp) or phosphorylation-deficient (Ser to Ala) single mutations at residues Ser12, Ser22, Ser390, Ser392, Ser423 and Ser628. Ser404 and Ser407 were modified either as single mutants or as a double mutant. In these experiments, GFP-tagged wild-type lamin A and GFP-tagged wild-type lamin A with amino acid substitutions were expressed in HeLa cells and subsequently fixed and examined by using confocal microscopy to compare fluorescence intensity levels, both in the nucleoplasm and in the lamina (see Materials and Methods). It should be noted that all of the lamin constructs coding for the wild-type and mutant proteins were expressed from a cDNA that encoded prelamin A (which then undergoes post-translational modification in the cell to mature lamin A, as described in Materials and Methods).
As predicted, GFP–lamin A localized predominantly to the lamina with a small fraction residing in the nucleoplasm (Fig. 2). By contrast, several of the phosphomimetic substitutions of specific serine residues caused significantly different distributions of GFP–lamin A, as reflected by the intensity ratio of nucleoplasmic to lamina fluorescence (N∶L) in fixed cells (Fig. 2B). The most prominent effect was obtained with the expression of the phosphomimetic substitution of Ser22 in lamin A (lamin A S22D), which resulted in the largest increase in the N∶L due to its increase in the nucleoplasm (Fig. 2). Lamin A S392D also displayed a similar statistically significant increase in the N∶L. There was no change in the distribution of lamin A S22A compared with wild-type GFP–lamin A, but lamin A S390A and lamin A S392A showed a significant decrease in the N∶L, caused by either an increase in the lamina and/or a decrease in the nucleoplasm. The remaining substitutions showed no significant changes in their N∶L. Single substitutions at Ser404 and Ser407 did not yield any differences in their N∶L (data not shown); however, in light of the evidence that both of these sites regulate the nuclear localization signal (NLS) and the transport of lamins into the nucleus (Leukel and Jost, 1995), the effects of double substitutions were assayed. Only the lamin A S404D S407D double mutation showed a significant increase in the N∶L (Fig. 2).
Fig. 2.
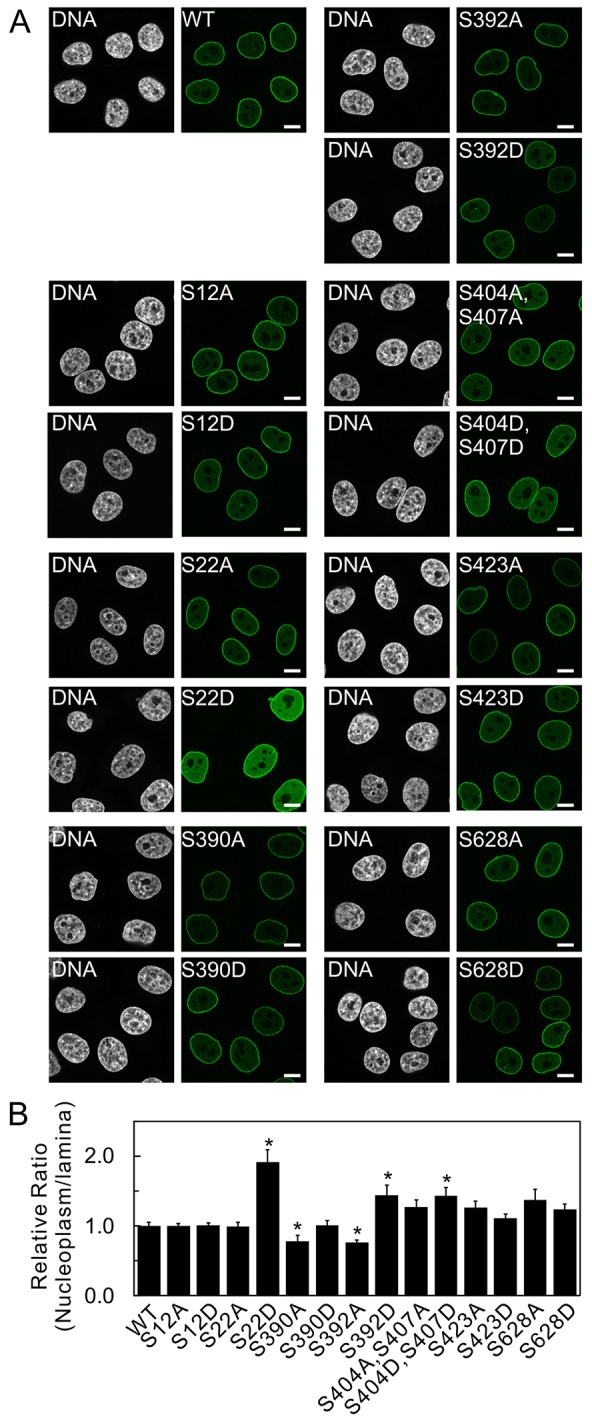
Site-directed mutagenesis introducing phosphorylation-deficient and phosphomimetic substitutions identifies the phosphorylation sites with the most profound effects on the subcellular localization of lamin A. (A) Confocal microscopy was employed to determine the subcellular localization of wild-type (WT) GFP–lamin A, compared with that of GFP–lamin A comprising phosphomimetic Asp or phosphorylation-deficient Ala replacements. The following mutants were analyzed: GFP–lamin A S12A, S12D, S22A, S22D, S390A, S390D, S392A, S392D, S423A, S423D, S628A, S628D, and the double mutants S404A S407A and S404D S407D. These GFP constructs were transiently expressed in HeLa cells for 48 h, the cells were then fixed. Pairs of images, of the same field, are shown – GFP–lamin A is shown in green and DNA, stained with Hoechst 33258, is shown in white. GFP–lamin A-S22D accumulated in the nucleoplasm, whereas WT GFP–lamin A and S22A were, predominantly, localized to the lamina. Scale bars: 10 µm. (B) Quantification of lamin A/C distribution in the nucleoplasm compared with that in the lamina was determined by measuring the fluorescence intensities of the lamina and the nucleoplasm by using Zeiss LSM software. The relative ratios of the signals in the nucleoplasm to the signals in the lamina were calculated as a measure of nuclear distribution (see Materials and Methods). The ratio for WT was set to a value of 1, and the nucleoplasm-to-lamina ratios of the substituted lamin A proteins were normalized to this value. Error bars indicate the s.e.m. *P<0.005 compared with that of WT GFP–lamin A.
The analyses above demonstrate that both Ser22 and Ser392 are high-turnover sites for phosphorylation during interphase. To further confirm the finding that Ser22 is phosphorylated in interphase nuclei, HeLa cells were labeled for phosphorylated lamin A/C at residue Ser22. The signal was not stimulated in any way, and the cells were not incubated in the presence of phosphatase inhibitor calyculin A, in order to prevent an increase in the phosphorylation signal. The results showed that interphase nuclei exhibited bright punctate fluorescence within the nucleoplasm, with little, if any, staining in the lamina (Fig. 3). Mitotic cells displayed, as expected, a stronger overall signal, with a completely different labeling pattern, indicative of depolymerized lamins that had been released from the nuclear lamina. These results were confirmed by western blotting of interphase and mitotic cells. The interphase fraction showed an obvious phosphorylated Ser22 lamin A/C signal (supplementary material Fig. S2D). The mitotic fraction, again as expected, also showed an apparent phosphorylated Ser22 lamin A/C signal, which was stronger than the interphase signal (supplementary material Fig. S2D). As stated above, the successful fractionation of interphase cells from mitotic cells was validated by using the specific mitotic markers of phosphorylated Cdc27 and phosphorylated Ser10 histone 3 (supplementary material Fig. S2D). Because it is well established that the phosphorylation by Cdk1 of both Ser22 and Ser392 drives the disassembly of the lamina before mitotic spindle assembly (Dessev et al., 1990; Heald and McKeon, 1990; Mall et al., 2012; Peter et al., 1990), we also performed experiments with mutations at both of these sites (Fig. 4). Interestingly, the N∶L in the nuclei that expressed lamin A S22D S392D appeared to reflect the same significant increase in nucleoplasmic fluorescence as lamin A S22D alone, the combination of S22D S392A showed a lesser increase in nucleoplasmic fluorescence (Fig. 4B). These results suggest that the nucleoplasmic location of lamin A is predominantly attributable to the phosphorylation of residue Ser22, but that its nucleoplasmic localization is further facilitated when Ser392 is phosphorylated.
Fig. 3.
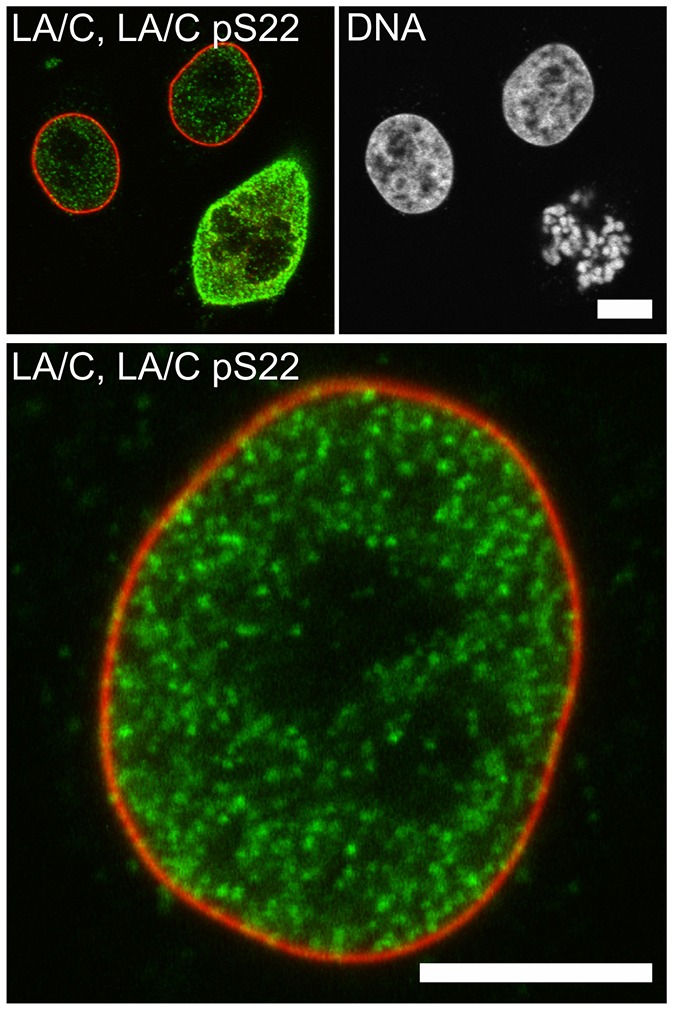
A phospho-specific antibody reveals the presence of lamin A/C that is phosphorylated at Ser22 in interphase cells. Unstimulated HeLa cells were fixed and stained with antibodies specific against lamin A/C (LA/C) and lamin A/C phosphorylated at residue Ser22 (lamin A/C pS22; LA/C pS22). Cells were then imaged by using confocal microscopy (the signal was not augumented by pre-incubation with calyculin A). Interphase cells gave an obvious positive signal upon staining with the phospho-specific antibody (lamin A/C pS22 is shown in green, and total lamin A/C is shown in red, top left and bottom). DNA was labeled with Hoechst 33258 (shown in white, top right). Total lamin A/C was predominantly localized at the lamina, whereas lamin A/C pS22 was mainly localized in the nucleoplasm. As expected, and in line with previous reports, the fluorescence intensity of p22 labeling increased substantially in mitotic cells (see upper left panel). These results demonstrate that this site is not only phosphorylated in mitotic cells but is also subjected to active phosphorylation and regulation in interphase cells. Scale bars: 5 µm.
Fig. 4.
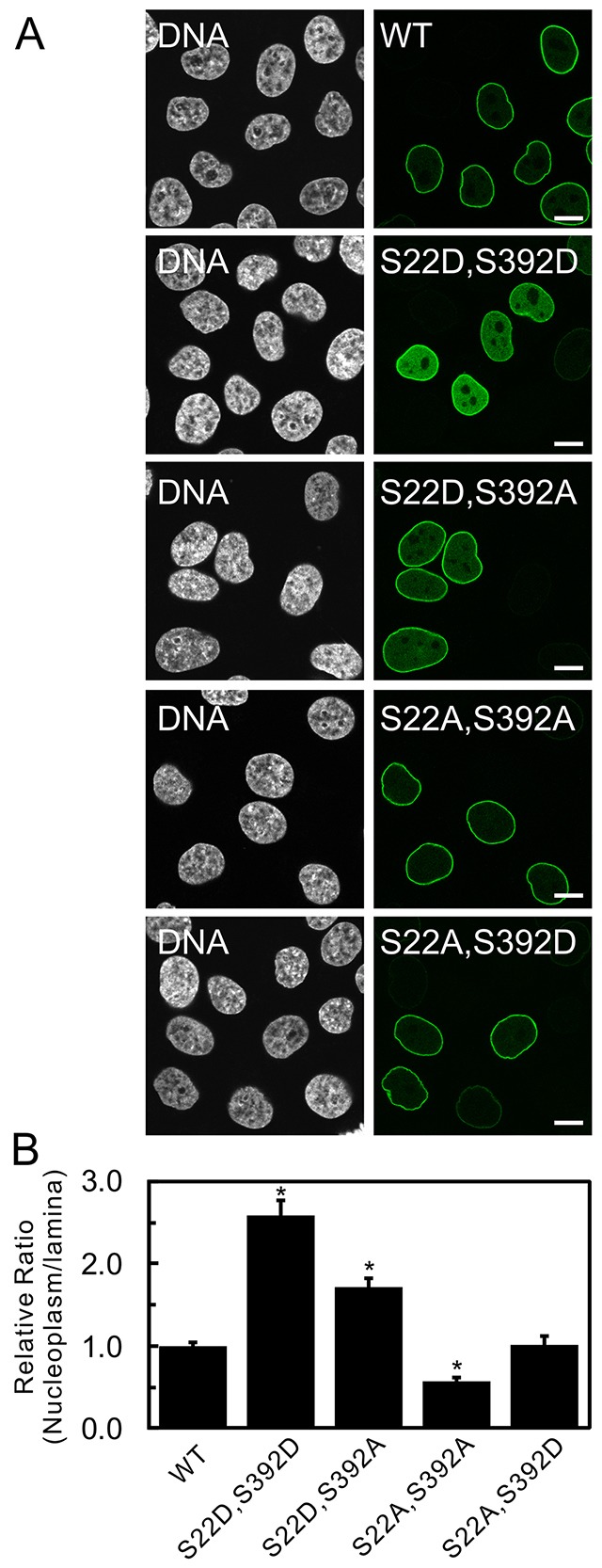
The combined effects of double mutations on the subcellular localization of lamin A. (A) The subcellular distribution of wild-type (WT) GFP–lamin A and the double mutants GFP–lamin A S22D S392D, S22D S392A, S22A S392A and S22A S392D were compared and analyzed as in Fig. 2. GFP–lamin A S22D S392D and S22D S392A accumulated in the nucleoplasm, whereas WT GFP–lamin A, S22A S392A and S22A S392D were, predominantly, localized to the lamina. DNA was stained with Hoechst 33258 and is shown in white. Scale bars: 10 µm. (B) The fluorescence intensities of the lamina and nucleoplasm were quantified, and the relative average ratios of the signals in the nucleoplasm to the signals in the lamina were plotted as in Fig. 2B (see Materials and Methods). Error bars represent the s.e.m. *P<0.005 compared with that of WT GFP–lamin A.
It has been previously shown that A- and B-type lamins form separate, but interacting, stable meshworks in the lamina (Shimi et al., 2008). Therefore, we decided to determine whether the phosphorylation-deficient or phosphomimetic mutations of the sites that gave the most prominent effects in the fluorescence distribution analysis – i.e. lamin A Ser22 and lamin A Ser392 – would affect the structural organization of lamin B1 in the nucleus. Endogenous lamin A, lamin C, lamin B1 and lamin B2 in these cells were localized by immunofluorescence. Neither phosphomimetic nor phosphorylation-deficient substitutions at Ser22 or Ser392 had any effect on B-type lamin distribution (supplementary material Fig. S3).
The dynamic properties of lamin A with phosphomimetic or phosphorylation-deficient amino acid replacements of Ser22 and Ser392
Of the eight identified high-turnover phosphorylation sites, our analyses suggested that Ser22 and Ser392 were the major sites that regulated the nuclear distribution and assembly state of lamin A in interphase cells. To determine how these sites affect the dynamic properties of lamin A, we performed live-cell imaging experiments by using GFP-fused wild-type lamin A, lamin A S22D S392D, lamin A S22A S392A, lamin A S22A S392D and lamin A S22D S392A. Fluorescence recovery after photobleaching (FRAP) analyses of the lamina region (at the bottom of the nucleus in order to visualize a maximally flat surface) in wild-type lamin A-expressing cells showed slow recovery rates (Fig. 5A,B), in agreement with previous findings (Broers et al., 1999; Moir et al., 2000). The double mutant lamin A constructs S22D S392D and lamin A S22D S392A were also relatively slow to recover (Fig. 5A,B). However, they did show significant increases in the percentages of their mobile fractions when compared with that of wild-type lamin A (Fig. 5C). By contrast, lamin A S22A S392A and lamin A S22A S392D showed decreased mobility when compared with wild-type lamin A (Fig. 5C). Lamin A S22D S392D showed no difference in mobility within the lamina compared with those proteins with the single Ser22 substitutions (not shown). These results showed that only the phosphomimetic substitutions of Ser22 displayed increased mobility in the lamina fraction. Correspondingly, the phosphorylation-deficient protein lamin A S22A showed decreased mobility. Taken together, these results further support the dominant effect that Ser22 has on the mobility of lamin A.
Fig. 5.
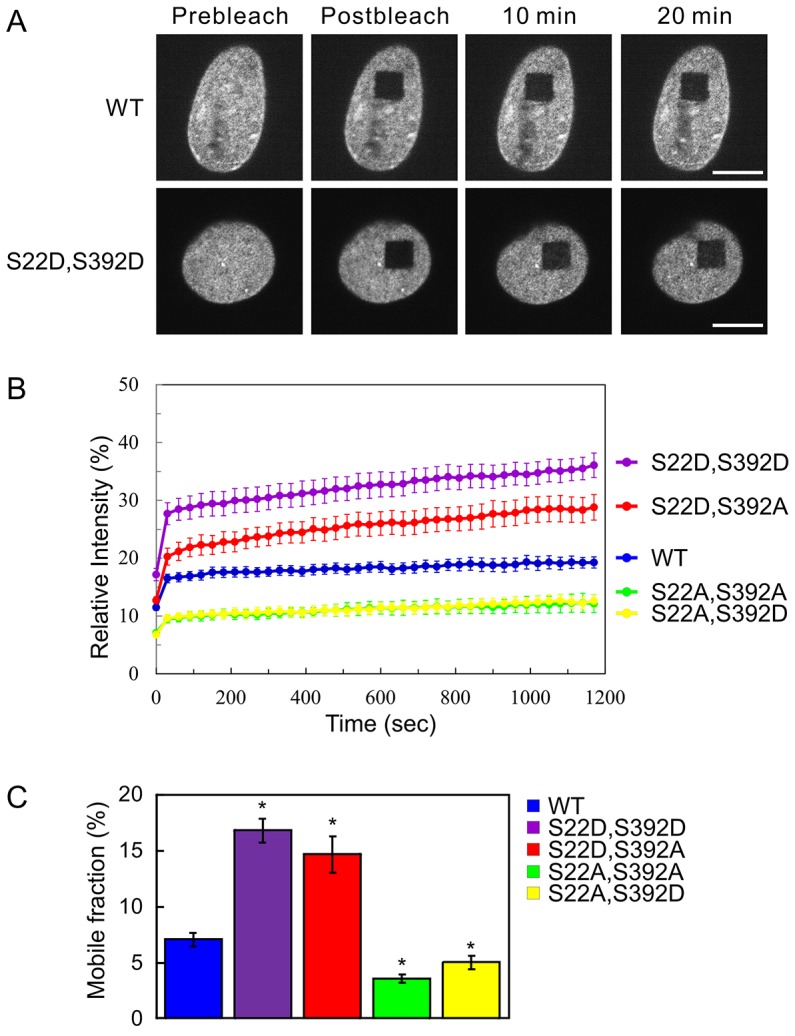
FRAP of the double mutants identifies sites that determine the rate of lamin A subunit exchange in the nuclear lamina. The subunit exchange of wild-type (WT) GFP–lamin A, or the GFP–lamin A double mutants S22D S392D, S22D S392A, S22A S392A and S22A S392D in the lamina of HeLa cell nuclei were determined by FRAP analyses. FRAP analyses were performed at either the upper or lower nuclear surfaces. (A) Representative fluorescence images of WT GFP–lamin A and GFP–lamin A S22D S392D taken at the nuclear surface immediately before and 10 min or 20 min after a square region was photobleached. Scale bars: 10 µm. (B) The mean normalized FRAP data that was measured in the bleached region (see Materials and Methods). Error bars represent the s.e.m. (C) The average mobile fraction of the various mutated lamin A proteins were calculated from the FRAP data in B (see Materials and Methods). The fractions of mobile GFP–lamin A S22D S392D and S22D S392A were significantly larger than that of WT GFP–lamin A. The fractions of mobile S22A S392A and S22A S392D were significantly smaller than those of WT GFP–lamin A. Error bars represent the s.e.m. *P<0.005 compared with that of WT GFP–lamin A.
As a comparison with the results above, we employed FRAP analysis at the midsection of the nucleus, allowing us to examine the nucleoplasmic fraction, while at the same time observing the lamina. To allow for using the lamina fraction as reference, in this analysis we followed a simplified qualitative scheme, taking one image right after photobleaching and one at plateau timepoints longer than 30 min (supplementary material Fig. S4). This permitted us to observe the contribution of single sites within the nucleoplasmic fraction (the lamina fractions showed similar results as those described in the analysis above and will not be presented here). Compared with wild-type lamin A, lamin A S22D showed a faster recovery of the nucleoplasmic fraction (supplementary material Fig. S4A). Lamin A S22A was similar to wild-type lamin A (supplementary material Fig. S4B). Lamin A S392D also showed a faster fluorescence recovery compared to wild-type lamin A, but was slower than lamin A S22D (supplementary material Fig. S4C). The fluorescence recovery of lamin A S392A was similar to wild-type lamin A (supplementary material Fig. S4D). The double mutant lamin A S22D S392D yielded the fastest recovery of nucleoplasmic fluorescence (supplementary material Fig. S4E), whereas lamin A S22A S392A showed almost no fluorescence recovery (supplementary material Fig. S4F). These results indicate that the dynamic properties of the nucleoplasmic fraction are strongly affected by modification of Ser22 and that Ser392 also markedly contributes to nucleoplasmic lamin dynamics.
As indicated above (supplementary material Fig. S4), the nucleoplasmic forms of some of the phosphorylation-deficient mutated lamin A proteins recovered rapidly in FRAP, making quantitative analysis difficult. Therefore, we analyzed the mobility of the nucleoplasmic fractions of lamins by fluorescence correlation spectroscopy, as we have described previously (Shimi et al., 2008) (Fig. 6). The results of these analyses showed that wild-type lamin A, lamin A S22D S392D, lamin A S22D S392A, lamin A S22A S392A and lamin A S22A S392D all displayed rapid nucleoplasmic mobility (Fig. 6B). However, the diffusion coefficients for lamin A S22D S392D and lamin A S22D S392A showed significant increases compared with those of wild-type lamin A (Fig. 6C,D). Interestingly, although Ser22 had a dominant effect on lamin A mobility in the lamina (supplementary material Fig. S4), lamin A S22A S392D also diffused faster than wild-type lamin A, whereas the diffusion coefficient of lamin A S22A S392A was indistinguishable from that of wild-type lamin A (Fig. 6C,D). These findings further corroborate that the phosphorylation of both Ser22 and Ser392 contributes to the nucleoplasmic mobility of lamin A.
Fig. 6.

Analyses of the nucleoplasmic mobility of wild-type, phosphomimetic and phosphorylation-deficient lamin A by fluorescence correlation spectroscopy. The mobilities of wild-type (WT) GFP–lamin A and the double mutants S22D S392D, S22D S392A, S22A S392A and S22A S392D in the nucleoplasm of HeLa cells were determined by fluorescence correlation spectroscopy analyses. (A) A representative fluorescence image of WT GFP-lamin A during fluorescence correlation spectroscopy measurements. The white crosses indicated the points where fluorescence correlation spectroscopy measurements were obtained. Scale bar: 10 µm. (B) The mobility of each lamin A protein compared with that of the WT, measured by using fluorescence correlation spectroscopy. The fluctuating fluorescence signals for all of these proteins were plotted. (C) Autocorrelation curves were calculated from the fluorescence intensities and then plotted (see Materials and Methods). Normalized G(τ) means that for comparison of the mobility all curves were normalized to the amplitude G = 2 at 0 µs. Pack: all of which was normalized to the same amplitude of G (0) = 2 for a comparison of the mobility. (D) Diffusion coefficients in slow (Dslow) and fast (Dfast) fractions were obtained from the fluorescence correlation spectroscopy data (see Materials and Methods). The mobilities of GFP–lamin A S22D S392D, S22D S392A and S22A S392D were significantly faster than that of WT GFP–lamin A. Error bars represent the s.e.m. *P<0.005 compared with that of WT GFP–lamin A.
Our fluorescence correlation spectroscopy analyses clearly indicate that, even though all of the nucleoplasmic fractions are highly mobile, there are differences upon expression of the mutant protein. We postulated that these differences might be reflected in protein solubility. To examine this possibility, we analyzed the detergent extractability of GFP-fused wild-type lamin A and the Ser22 and Ser392 phosphorylation site mutants in HeLa cells. Using quantitative fluorescence microscopy, we determined the ratio of extractable to non-extractable lamin A (E∶N) in the nucleoplasm (Fig. 7B upper panel) and the lamina (Fig. 7B lower panel) (see Materials and Methods). Under these conditions, more lamin A S22D S392D and lamin A S22D S392A than wild-type lamin A was extracted from both the nucleoplasm and lamina regions. lamin A S22A S392A and lamin A S22A S392D in the lamina did not show any reduction in fluorescence intensity compared with wild-type lamin A controls following extraction; however, they did show significant increases in the E∶N. These results show that the differences observed in our FRAP and fluorescence correlation spectroscopy analyses are reflections of differences in the solubility of the proteins.
Fig. 7.

The phosphorylation sites that determine nucleoplasmic distribution also affect the solubility of lamin A. The fluorescence intensities of wild-type (WT) GFP–lamin A and the double mutants S22D S392D, S22D S392A, S22A S392A and S22A S392D, which were expressed in HeLa cell nuclei, were measured quantifiably by fluorescence microscopy, with and without detergent extraction (see Materials and Methods). (A) Fluorescence images of cells without (−) and with (+) detergent extraction before fixation. Pairs of images, of the same field, are shown – the left-hand image of the pair shows DNA (stained with Hoechst 33258) and the right-hand image of the pair shows the fluorescence of the indicated lamin A protein. Scale bars: 10 µm. (B) Fluorescence intensities were determined in the nucleoplasm (top panel) and the lamina (bottom panel). The data were quantified, and the relative ratios of the fluorescence intensities (extracted/non-extracted) were calculated and normalized to that of the WT (set as 1, see Materials and Methods). Compared with WT GFP–lamin A, the solubilities of the double mutants GFP–lamin A S22D S392D and S22D S392A were significantly higher. Error bars represent the s.e.m. *P<0.005 compared with that of WT GFP–lamin A.
The contribution of the third region
The far C-terminal region contained several phosphorylation sites, of which, Ser628 was identified as being the most prominent high-turnover site. Mutation of this residue alone had no effects on the nuclear distribution of lamin A (Fig. 2); therefore, we investigated whether it affected the organization and distribution of lamin A when assessed in combination with the mutation of Ser22 and Ser392. Interestingly, phospho-mimetic substitutions at all three sites (S22D S392D S628D) had a profound effect on lamin distribution in the cytoplasm, nucleoplasm and lamina regions (Fig. 8). The effects of the triple mutation were dependent on the expression levels of GFP–lamin A, where high-expressing cells displayed higher cytoplasmic fluorescence and low-expressing cells showed more nucleoplasmic and lamina fluorescence (Fig. 8). By contrast, the distribution of lamin A S22D S392D S628A was similar to that of lamin A S22D S392D. Taken together, these data indicate that the phosphorylation of all three sites plays crucial roles in determining the distribution and organization of lamin A in the cytoplasm, nucleoplasm and nuclear lamina, as depicted in the model that summarizes our results (Fig. 9).
Fig. 8.

A possible role of the far C-terminal region of lamin A in determining subcellular localization is revealed by analysis of triple mutations. Phospho-mimetic substitutions of residues S22, S392 and S628 show that all three sites combined have a substantial effect on the distribution of GFP–lamin A. The distributions of the triple mutants GFP–lamin A S22D S392D S628D and S22D S392D S628A were determined by using confocal fluorescence microscopy (lamin A distribution is shown in green). DNA was labeled with Hoechst 33258 [shown in white, or blue in the overlay (OL)]. The distribution of the S22D S392D S628A triple mutant was indistinguishable from that of the S22D S392D double mutant, whereas the GFP–lamin A S22D S392D S628D triple mutant showed variable degrees of localization to the cytoplasm (arrow heads), as well as a prominent nucleoplasmic distribution. Scale bars: 10 µm.
Fig. 9.

Summary of the effects of three major phosphorylation sites that affect the distribution and assembly of lamin A. Phosphorylation of different combinations of three phosphorylation sites has substantially different effects on the distribution and assembly of lamin A (LA). The schematics of the lamin A protein correspond to that shown in Fig. 1A.
DISCUSSION
Soon after the identification of the nuclear lamins as the major proteins of the nuclear lamina (Gerace et al., 1978), the question of how they disassembled and reassembled during the process of nuclear envelope breakdown and reformation in mitosis came to the forefront. One of the early studies on the mitotic disassembly of nuclear lamins identified reversible phosphorylation as a key factor (Gerace and Blobel, 1980). Subsequent searches for mitosis-specific phosphorylation sites revealed the presence of Cdk1-specific sites, which are conserved in each of the lamins, as well as sites that are phosphorylated by other kinases, such as PKC (Haas and Jost, 1993; Hennekes et al., 1993; Mall et al., 2012). Each of the lamin phosphorylation sites could be involved in regulating the assembly state of the lamina during mitosis.
In contrast with mitosis, very little is known about the regulation of lamin structure and assembly states in interphase nuclei. However, in recent years, studies of the laminopathies have shown that mutations in LMNA can have profound effects on nuclear structure and function. Alterations in lamin assembly, the structural organization of the nucleus and the interactions of mutant lamins with tissue-specific signaling molecules and/or transcription factors have been reported (Dechat et al., 2008). Still, there is very scarce information about the underlying mechanisms that determine the structural organization of lamins during interphase. Although studies of cytoskeletal IFs indicate that reversible phosphorylation is a key determinant in regulating IF structure (Eriksson et al., 2009; Pallari and Eriksson, 2006), this aspect of interphase lamin dynamics remains unexplored.
Databases that are devoted to post-translational modification proteomics contain a large collection of positive results for lamin phosphorylation sites (Simon and Wilson, 2013), but there are inherent problems with these data as a certain proportion of large-scale proteomic screening can be mis-identifications, particularly when it comes to localizing phosphorylation sites. The large studies also include events in mitosis; hence, some of the data is not relevant to interphase cells. Only a few of the identified sites have been verified as bona fide in vivo phosphorylation sites, and nothing is known about the roles of these sites in regulating interphase assembly and organization, or any other functions for that matter.
This study identifies 20 interphase phosphorylation sites in lamin A/C in vivo, of which eight are high phosphate-turnover sites. All of the identified Ser and Thr sites (no Tyr sites were identified) are restricted to three regions, and each of these regions contain at least one of the high phosphate-turnover sites. The importance of the identified sites is supported by their extremely high degree of evolutionary conservation (see Fig. 1B). Furthermore, there is a rather broad mix of kinase motifs, and, interestingly, a significant portion of these are either established targets of the Cdk group (Ser22, Ser390, Ser392) or TP/SP motifs that could potentially be Cdk targets and/or targets of other proline-directed kinases (Thr3, Thr19, Ser22, Ser390, Ser392, Ser652). A number of the targets have also been shown to be, or are predicted to be, PKC targets (Ser403, Ser404, Ser406, Ser407), and it has been shown that Ser404 is phosphorylated by Akt (Bertacchini et al., 2013). The residues surrounding the other phosphorylation sites identified in our study do not reveal any distinguishable kinase motifs. One notable feature is the absence of protein kinase A (also known as cAMP-dependent protein kinase) or Rho-associated protein kinase (ROCK) consensus sequences in lamin A/C. This is in contrast with the N-termini of many other cytoskeletal IF proteins, which typically contain these motifs (Eriksson et al., 2004; Schofield and Bernard, 2013).
The finding that the two major mitotic phosphorylation sites Ser22 and Ser392 are also two of the predominant interphase phosphorylation sites was somewhat surprising. However, in light of what is known about the profound effects of substitutions in Ser22 and Ser392 on mitosis (Heald and McKeon, 1990; Peter et al., 1990), it is unsurprising to find that these mutations also had profound effects on lamin organization and dynamics in interphase cells. This was especially the case in lamin A S22D, which caused a dramatic shift towards a nucleoplasmic distribution. The phosphomimetic substitution at Ser392 also increased the nucleoplasmic signal, but to a lesser extent than S22D. Interestingly, when these two sites were substituted with phosphorylation-deficient alanine residues, only S392A showed less nucleoplasmic fluorescence, whereas S22A localization was indistinguishable from that of wild-type lamin A. In addition, there was an apparent decrease in the nucleoplasmic distribution of the S390A mutant, whereas S390D had no obvious effect on the nucleoplasmic-to-lamina localization ratio. No other substitution of a single serine residue had substantial effects on lamin A distribution.
When combinations of mutated serine residues were analyzed, the double mutant lamin A S22D S392D was strikingly nucleoplasmic, whereas, in the phosphorylation-deficient lamin A S22A S392A double mutant, the nucleoplasmic fluorescence was significantly lower than that of wild-type lamin A. The nucleoplasmic fluorescence of lamin A S22D S392A was indistinguishable from that of lamin A S22D, and the nucleoplasmic fluorescence lamin A S22A S392D was indistinguishable from that of wild-type lamin A. These results indicate that Ser22 has a dominant effect on the nucleoplasmic distribution of lamin A, but that Ser392 clearly participates in determining the nucleoplasmic-to-lamina localization ratio. These findings suggest that site-specific phosphorylation of these serine residues regulates the steady-state distribution between the nucleoplasm and lamina of non-assembled and assembled lamin A. In this steady-state, if Ser22 is available for phosphorylation, then phosphorylated Ser392 is, in fact, quite efficient at inducing a shift towards a nucleoplasmic distribution, as demonstrated by the effect of the lamin A S392D mutation. If, however, Ser22 is substituted with alanine and is, therefore, unavailable for phosphorylation, then a shift from the lamina to the nucleoplasm is less efficient.
Taken together, the immunofluorescence analyses of single and double mutants suggest that phosphorylation of Ser22 is dominant in terms of shifting the equilibrium towards the nucleoplasm, whereas phosphorylation of Ser392, and most likely Ser390, contributes to this equilibrium by facilitating the nucleoplasmic localization of lamin A. It should also be noted that lamin A S404D S407D yielded a significant increase in nucleoplasmic fluorescence and that corresponding sites have been identified as relevant for the disassembly of lamin B1 during mitosis (Mall et al., 2012).
The results of our immunofluorescence analyses were comparable to those obtained by using live-cell imaging of FRAP, fluorescence correlation spectroscopy and nuclear extraction techniques. Each of these approaches demonstrated that the key sites that were identified within the three phosphorylation regions play important roles in the assembly, subunit exchange and mobility of lamin A between the lamina and the nucleoplasm of interphase cells. The advantage of fluorescence correlation spectroscopy is that it allowed us to specifically address the effects of phosphorylation on the mobility of lamin A within the nucleoplasm. The fluorescence correlation spectroscopy data revealed that lamin A S22A S392D did have a significant effect on lamin A mobility in the nucleoplasm, although our other data showed that the mutation did not alter the nucleoplasmic-to-lamina fluorescence ratio. These results suggest that, in addition to facilitating the nucleoplasmic localization of lamin A, Ser392 plays a substantial role in regulating the nucleoplasmic mobility of lamin A. This is further supported by the inhibition of fluorescence recovery by FRAP analysis of phosphorylation-deficient alanine substitutions of Ser392 (S392A).
The sites that we identified as being responsible for regulating the nucleoplasmic lamin organization, assembly and dynamics all have motifs that indicate that they could be regulated by either cell cycle or growth factor signaling. The phoshorylation-mediated release, or restructuring, of lamin subunits provides excellent opportunities for both negative- and positive-feedback loops for the signaling pathways that regulate lamins at these phosphorylation sites. In this way, the kinases responsible for modifying these sites could promote a broad spectrum of effects on nuclear functions by changing the assembly-state and lamin A/C dynamics. For example, there are a number of specific chromatin-associated factors – such as SREBP1, Mok2 and PCNA – whose activity and organization is directly affected by binding to lamin proteins, and some of the identified lamin A/C phosphorylation sites are located within the reported binding regions (Lloyd et al., 2002; Dreuillet et al., 2002; Shumaker et al., 2008). Furthermore, there are lamin A/C-interacting proteins whose organization could have secondary effects on cell signaling and/or transcription regulation. For example, lamin A is known to be associated with lamina-associated polypeptide 2 α (TMPO; hereafter referred to as LAP2α) in the nucleoplasm, which in turn regulates pRb, an important regulator of the cell cycle (Dorner et al., 2006; Pekovic et al., 2007). Our results indicate that phosphorylation of lamin A/C is a very good candidate mechanism for regulating the LAP2α–lamin A/C interaction and, consequently, the interaction of LAP2α with pRb as the identified phosphorylation sites fall into the LAP2α-binding region that is located between amino acid residues 319 and 566 of lamin A/C (Dechat et al., 2000). Another example of this kind of interaction is derived from LMNA mutations that have been found to alter developmental signaling through their effects on emerin, which in turn affects the dynamics of actin transport. Actin dynamics, and especially G-actin levels, are crucial for the functions of the mechanosensitive transcription factor megakaryoblastic leukaemia 1 (MKL1), which is important for cardiac development and function (Ho et al., 2013). These examples illustrate that lamin localization, as regulated by post-translational modifications, could provide a platform for the integration of growth factor and cell cycle signaling. In this way, a structural protein could be employed as a signal processing unit and feedback mechanism, which would also integrate mechanical and architectural cues, a highly timely topic in the context of lamins (Swift et al., 2013). In this respect, the numerous other phosphorylation sites that are identified here, even though they do not display effects on lamin distribution or morphology, provide further possibilities for the regulation of interactions with signaling proteins, transcription factors and structural proteins, etc.
To date, studies of the premature aging disease HGPS have been focused on perturbations of the nuclear architecture and function that are attributable to the permanently farnesylated and truncated dominant-negative form of lamin A, termed progerin (Eriksson et al., 2003; Goldman et al., 2004). Importantly, the third phosphorylation region that we have identified lies within the 50-amino-acid sequence that is lacking in progerin. This deleted region contains three of our identified interphase phosphorylation sites, of which Ser628 is a high-turnover site. Because the triple phosphomimetic lamin A (S22 S392 S628) is distributed in the lamina, the nucleoplasm and, surprisingly, the cytoplasm, it is possible that the loss of Ser628 in HGPS, and the other two phosphorylated serine residues in progerin, might have effects on the activity of the protein, in addition to the effects of aberrant permanent farnesylation. It is also possible that other specific lamin A/C phosphorylation sites are involved in the physiological impact of laminopathy mutations. However, very few of the known laminopathy mutation sites lie within close proximity to the phosphorylation sites that we have identified in this study. Alternatively, even laminopathy mutations that are located outside of the phosphorylation domains could affect the access or efficacy of phosphorylation sites within lamin A/C by creating defects in lamin polymer structure. These relationships require further investigation, nevertheless, our study clearly shows that site-specific phosphorylation in different regions of the protein have marked consequences for the distribution and dynamics of lamin A/C.
MATERIALS AND METHODS
Cell culture and transfection
HeLa cervical carcinoma cells were grown under a humidified 5% CO2 atmosphere at 37°C in Dulbecco's modified Eagle's medium (Sigma-Aldrich) that was supplemented with 10% fetal calf serum, antibiotics (penicillin, streptomycin) and 2 mM l-glutamine. The GFP–lamin A fusion constructs were introduced into cells by electroporation. Cells at 80–90% confluence, grown on a 10-cm cell culture plastic dish, were detached by using 0.25% trypsin with 0.02% K-EDTA solution and then centrifuged at 200 g for 5 min and resuspended in 400–450 µl Opti-MEM I (GIBCO). The cells that had been collected from one 10-cm cell culture dish were then mixed with 10 µg of the corresponding GFP–lamin A plasmid in a 4-mm electroporation cuvette (BTX, Holliston, MA) and electroporated at 220–240 V and 975 µF. The cells were then plated onto 22×22-mm no. 1.5 coverslips (Menzel GmbH), Lab-Tek Chamber Slides (NalgeNunc, Rochester, NY) or 35-mm no. 1.5 glass-bottomed culture dishes (MatTek, Ashland, MA). The transfection efficiency varied from 60–80%. In some experiments, mitotic cells were removed from the cultures, as described previously (Chou et al., 1990; Eriksson et al., 1992).
Phosphorylation analysis by mass spectrometry
TiO2 affinity chromatography and LC-MS/MS were performed as described previously (Imanishi et al., 2007) with some modifications. Briefly, lamin A/C was immunoprecipitated from the calyculin-A-treated Hela cells that remained after the removal of mitotic cells by mechanical shake-off, and then in-gel digested by using trypsin. The digests with and without phosphopeptide enrichment by TiO2 affinity chromatography were analyzed by LC-MS/MS using an EASY-nLC 1000 nanoflow liquid chromatograph coupled to a Q Exactive quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific). A database search was performed against the SwissProt database (version 2011_05; Homo sapiens) using Mascot 2.4 (Matrix Science) via Proteome Discoverer 1.3 (Thermo Fisher Scientific). MS/MS spectra of lamin phosphopeptides that were identified with a high probability (Mascot expectation value <0.05) were manually inspected. Label-free peptide quantification was performed in two technical replicates using Progenesis LC-MS 4.1 (Nonlinear Dynamics). Other methods used for phosphorylation site identification and the assignment of major high-turnover sites are described in detail in conjunction with the proteomics data that is presented in the supplementary figures.
Lamin A plasmid constructs
The cDNA for full-length wild-type pre-lamin A was expressed in pEGFP-C1 (Clontech lab) (Moir et al., 2000). Transfection into mammalian cells results in expression of a 664-amino-acid long human prelamin A with green fluorescent protein (GFP) fused to the lamin N-terminus. In the cell, the recombinant protein undergoes a set of post-translational modifications, such as CAAX-box isoprenylation, carboxymethylation and a series of proteolytic cleavages to produce mature GFP–lamin A. Point mutations of individual phosphorylation sites were made in the GFP–lamin A pEGFP-C1 construct using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, California) and confirmed by using DNA sequencing. We introduced either phosphomimetic (Ser to Asp) or phosphorylation-deficient (Ser to Ala) mutations into the construct as single GFP–lamin A-SxxA or GFP–lamin A-SxxD, where ‘xx’ denotes the position of the mutated serine in the human lamin A sequence – 12, 22, 390, 392, 404, 407, 423 or 628; or the double and triple mutations – GFP–lamin A-S404D S407D, GFP–lamin A-S404A S407A, GFP–lamin A-S404A S407D, GFP–lamin A-S22D S392D, GFP–lamin A-S22A S392A, GFP–lamin A-S22A S392D, GFP–lamin A-S22D S392A, GFP–lamin A-S22D S392D S628D and GFP–lamin A-S22D S392D S628A. The resulting constructs were transiently expressed in HeLa cells and analyzed by using confocal microscopy to observe possible changes in nuclear morphology, compared with control cells that expressed GFP–lamin A.
FRAP measurement and analysis
The cells were examined 48 h post-transfection. For imaging, the cells were overlaid with Leibovitz's L15 medium containing 10% serum and antibiotics (penicillin and streptomycin). Cells were maintained at 37°C and under 5% CO2 using a Tokai-HIT stage-top CO2 incubator. For FRAP measurements, all time-lapse images of cells expressing the GFP–lamin A constructs were made with the Andor spinning disk confocal microscope that was based on a Nikon Ti Perfect Focus microscope that was equipped with a Yokogawa CSU-X1 spinning disk unit and an iXon 3 14-bit EM CCD camera (Andor Technology). Square-shaped areas (4.8 µm×4.6 µm) that contained a region of the lamina in a single nucleus were bleached using a 488-nm laser, and both non-bleached and bleached areas of the nucleus were monitored for recovery at 30 s intervals using Andor iQ2 software for FRAP analyses (Andor Technology). FRAP data were analyzed using GraphPad Prism 6 software as previously described (Shimi et al., 2008). Mobile fractions of GFP–lamin A constructs were calculated by curve-fitting the fluorescence recovery data with the following formula to estimate one-phase disassociation–association kinetics:
I (t): Normalized fluorescence intensity in the bleached area at a time t
I: Normalized initial fluorescence intensity in the bleached area immediately after photobleaching
P: Normalized fluorescence at plateau
k: constant
Fluorescence correlation spectroscopy measurement and analysis
The methods used for the fluorescence correlation spectroscopy measurement of transfected cells has been described elsewhere (Pack et al., 2006; Shimi et al., 2008). Briefly, fluorescence correlation spectroscopy was performed with a ConfoCor2 spectrometer (Carl Zeiss) that was equipped with an Argon laser and a water immersion objective [C-Apochromat, 40×, 1.2 NA (Carl Zeiss)]. Fluorescence correlation spectroscopy measurements were all performed at 25°C. During fluorescence correlation spectroscopy measurements, the GFP–lamin A constructs were excited at the 488-nm laser line and the confocal pinhole diameter was adjusted to 70 µm. The cells that were used for fluorescence correlation spectroscopy analysis expressed low levels of GFP–lamin A [nuclear fluorescence less than 100 kilocounts/s (kcps)]. Data analysis by using a two-component diffusional model and the determination of mean diffusion coefficients of fast and slow diffusion have been described elsewhere (Pack et al., 2006; Shimi et al., 2008).
Indirect immunofluorescence
Cells that were grown on coverslips were fixed 48 h post-transfection by using 3.7% formaldehyde in PBS for 20 min at room temperature. The fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min at room temperature and then rinsed with PBST (0.06% Tween-20 in PBS) and PBS, and incubated with the following primary antibodies against the indicated proteins: mouse monoclonal against lamin A/C (diluted 1∶10,000, Shimi et al., 2011); rabbit polyclonal against lamin A (diluted 1∶1000, clone 323, Dechat et al., 2008), rabbit polyclonal against LC (diluted 1∶1000, clone 321, generated against a synthetic peptide with the sequence CHHVSGSRR), rabbit polyclonal against lamin B1 (diluted 1∶1000, Moir et al., 1994), rabbit polyclonal against lamin B2 (diluted 1∶1000, clone 327, generated against a synthetic peptide with the sequence CSGPSVLGTGTGGSG) and a rabbit polyclonal against lamin A/C phosphorylated Ser22 (diluted 1∶100, Thermo Scientific). Secondary antibodies were Alexa-Fluor-568-conjugated against mouse or rabbit IgG (Molecular Probes). DNA was stained with Hoechst 33258 dye (diluted 1∶10,000, Molecular Probes). The fixed cells were observed using a Zeiss AxioImager.Z1 (Carl Zeiss) that was equipped with a Plan-Neofluar Ph ×40 1.3 NA oil immersion objective (Carl Zeiss) or a Zeiss LSM 510 META (Carl Zeiss) equipped with a Plan Apochromat ×63, Ph1.40 NA oil immersion objective (Carl Zeiss). Nucleoplasmic∶lamina ratios of GFP–lamin A intensity were calculated using the LSM 510 software. For each of the substituted lamin A proteins, the nuclei of ten cells from different images were used, except for S628A (n = 9). A straight line was drawn across each one of the cells using the overlay tool, avoiding the nucleolus and regions rich in heterochromatin (based on Hoechst staining). The ratios were calculated by dividing the nucleoplasm intensity by the lamina intensity for each nucleus, and the ratios from the mutated proteins were normalized to the wild type. The mean, standard error of the mean (s.e.m.) and Student's t-test were calculated by using Excel. Asterisks indicate statistically significant differences (P<0.05) compared with cells that had been transfected with wild-type lamin A. For extraction assays, cells were grown on coverslips for 48 h post-transfection. The cells were washed with PBS, and soluble GFP–lamin A was extracted by incubation in 0.5% Triton X-100 in PBS for 5 min, followed by fixation for 20 min with 3.7% formaldehyde at room temperature. The cells were then incubated in 0.1% Triton X-100 in PBS for 15 min at room temperature, and the DNA was stained with Hoechst 33258. The images were analyzed using ImageJ software in order to determine the fluorescence intensities of GFP–lamin A within the lamina and the nucleoplasm in Triton-extracted and non-extracted cells. Briefly, 30 to 40 nuclei in cells that expressed GFP–lamin A were randomly selected, and the mean fluorescence intensities of GFP–lamin A within the lamina (L) and nucleoplasmic (N) regions were quantified. The ratio of fluorescence between the lamina and nucleoplasmic areas were calculated as follows:
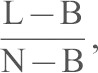 |
where B is background subtraction.
Supplementary Material
Acknowledgments
This study represents a continuing collaboration between the J.E.E.'s and R.D.G.'s laboratories. We thank all colleagues and technical personnel who have in some way contributed to the finalization of this study. Northwestern University Cell Imaging Facility and the Cell Imaging Core of Turku Centre for Biotechnology and Turku Bioimaging contributed to the microscopic imaging and analyses.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
V.K. and S.Y.I. conducted the phosphoproteomic analysis. V.K. generated and validated the constructs expressing mutant lamins. V.T and T.S. conducted most of the imaging-based analysis. FCS analysis was done by T.S. and C.-G.P. E.T., S.A.A., A.G. and J.M.-C. participated in various aspects of the imaging-based analysis. E.T. performed the analyses combining immunoprecipitation and western blotting. The experiments were planned and executed under the guidance of R.D.G and J.E.E. V.K., T.S., S.A.A., and S.Y.I. wrote the article together with R.D.G. and J.E.E.
Funding
J.E.E. is supported by grants from the Academy of Finland, the Sigrid Jusélius Foundation, the Finnish Cancer Foundations and a Center of Excellence grant from the Endowment of the Åbo Akademi University. R.D.G. is supported by National Institutes of Health grants Super resolution microscopy of nuclear lamin and spindle envelope/matrix function [GM106023] and Interactions between intermediate filaments and nucleus [CA03176], the Progeria Research Foundation grant, and grants from the National Cancer Institute. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.141820/-/DC1
References
- Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villén J., Li J., Cohn M. A., Cantley L. C., Gygi S. P. (2004). Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 101, 12130–12135 10.1073/pnas.0404720101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertacchini J., Beretti F., Cenni V., Guida M., Gibellini F., Mediani L., Marin O., Maraldi N. M., de Pol A., Lattanzi G. et al. (2013). The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J. 27, 2145–2155 10.1096/fj.12--218214 [DOI] [PubMed] [Google Scholar]
- Broers J. L., Machiels B. M., van Eys G. J., Kuijpers H. J., Manders E. M., van Driel R., Ramaekers F. C. (1999). Dynamics of the nuclear lamina as monitored by GFP-tagged A-type lamins. J. Cell Sci. 112, 3463–3475 [DOI] [PubMed] [Google Scholar]
- Burke B., Stewart C. L. (2002). Life at the edge: the nuclear envelope and human disease. Nat. Rev. Mol. Cell Biol. 3, 575–585 10.1038/nrm879 [DOI] [PubMed] [Google Scholar]
- Butin-Israeli V., Adam S. A., Goldman A. E., Goldman R. D. (2012). Nuclear lamin functions and disease. Trends Genet. 28, 464–471 10.1016/j.tig.2012.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell B. C., Collins F. S. (2006). Human laminopathies: nuclei gone genetically awry. Nat. Rev. Genet. 7, 940–952 10.1038/nrg1906 [DOI] [PubMed] [Google Scholar]
- Chou Y. H., Bischoff J. R., Beach D., Goldman R. D. (1990). Intermediate filament reorganization during mitosis is mediated by p34cdc2 phosphorylation of vimentin. Cell 62, 1063–1071 10.1016/0092--8674(90)90384--Q [DOI] [PubMed] [Google Scholar]
- Dechat T., Korbei B., Vaughan O. A., Vlcek S., Hutchison C. J., Foisner R. (2000). Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J. Cell Sci. 113, 3473–3484 [DOI] [PubMed] [Google Scholar]
- Dechat T., Pfleghaar K., Sengupta K., Shimi T., Shumaker D. K., Solimando L., Goldman R. D. (2008). Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832–853 10.1101/gad.1652708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessev G. N., Iovcheva-Dessev C., Goldman R. D. (1990). Lamin dimers. Presence in the nuclear lamina of surf clam oocytes and release during nuclear envelope breakdown. J. Biol. Chem. 265, 12636–12641 [PubMed] [Google Scholar]
- Dessev G., Iovcheva-Dessev C., Bischoff J. R., Beach D., Goldman R. (1991). A complex containing p34cdc2 and cyclin B phosphorylates the nuclear lamin and disassembles nuclei of clam oocytes in vitro. J. Cell Biol. 112, 523–533 10.1083/jcb.112.4.523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner D., Vlcek S., Foeger N., Gajewski A., Makolm C., Gotzmann J., Hutchison C. J., Foisner R. (2006). Lamina-associated polypeptide 2alpha regulates cell cycle progression and differentiation via the retinoblastoma-E2F pathway. J. Cell Biol. 173, 83–93 10.1083/jcb.200511149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreuillet C., Tillit J., Kress M., Ernoult-Lange M. (2002). In vivo and in vitro interaction between human transcription factor MOK2 and nuclear lamin A/C. Nucleic Acids Res. 30, 4634–4642 10.1093/nar/gkf587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckersley-Maslin M. A., Bergmann J. H., Lazar Z., Spector D. L. (2013). Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 4, 53–60 10.4161/nucl.23384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch T., Peter M., Nurse P., Nigg E. A. (1991). p34cdc2 acts as a lamin kinase in fission yeast. J. Cell Biol. 112, 797–807 10.1083/jcb.112.5.797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson J. E., Brautigan D. L., Vallee R., Olmsted J., Fujiki H., Goldman R. D. (1992). Cytoskeletal integrity in interphase cells requires protein phosphatase activity. Proc. Natl. Acad. Sci. USA 89, 11093–11097 10.1073/pnas.89.22.11093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson J. E., Toivola D. M., Sahlgren C., Mikhailov A., Härmälä-Braskén A. S. (1998). Strategies to assess phosphoprotein phosphatase and protein kinase-mediated regulation of the cytoskeleton. Methods Enzymol. 298, 542–569 10.1016/S0076--6879(98)98044--2 [DOI] [PubMed] [Google Scholar]
- Eriksson M., Brown W. T., Gordon L. B., Glynn M. W., Singer J., Scott L., Erdos M. R., Robbins C. M., Moses T. Y., Berglund P. et al. (2003). Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson J. E., He T., Trejo-Skalli A. V., Härmälä-Braskén A-S., Hellman J., Chou Y-H., Goldman R. D. (2004). Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 117, 919–932 10.1242/jcs.00906 [DOI] [PubMed] [Google Scholar]
- Eriksson J. E., Dechat T., Grin B., Helfand B., Mendez M., Pallari H-M., Goldman R. D. (2009). Introducing intermediate filaments: from discovery to disease. J. Clin. Invest. 119, 1763–1771 10.1172/JCI38339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerace L., Blobel G. (1980). The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 19, 277–287 10.1016/0092--8674(80)90409--2 [DOI] [PubMed] [Google Scholar]
- Gerace L., Blum A., Blobel G. (1978). Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction. Interphase and mitotic distribution. J. Cell Biol. 79, 546–566 10.1083/jcb.79.2.546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman R. D., Shumaker D. K., Erdos M. R., Eriksson M., Goldman A. E., Gordon L. B., Gruenbaum Y., Khuon S., Mendez M., Varga R. et al. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 101, 8963–8968 10.1073/pnas.0402943101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss V. L., Hocevar B. A., Thompson L. J., Stratton C. A., Burns D. J., Fields A. P. (1994). Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J. Biol. Chem. 269, 19074–19080 [PubMed] [Google Scholar]
- Haas M., Jost E. (1993). Functional analysis of phosphorylation sites in human lamin A controlling lamin disassembly, nuclear transport and assembly. Eur. J. Cell Biol. 62, 237–247 [PubMed] [Google Scholar]
- Heald R., McKeon F. (1990). Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 61, 579–589 10.1016/0092--8674(90)90470--Y [DOI] [PubMed] [Google Scholar]
- Hennekes H., Peter M., Weber K., Nigg E. A. (1993). Phosphorylation on protein kinase C sites inhibits nuclear import of lamin B2. J. Cell Biol. 120, 1293–1304 10.1083/jcb.120.6.1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C. Y., Jaalouk D. E., Vartiainen M. K., Lammerding J. (2013). Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497, 507–511 10.1038/nature12105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocevar B. A., Burns D. J., Fields A. P. (1993). Identification of protein kinase C (PKC) phosphorylation sites on human lamin B. Potential role of PKC in nuclear lamina structural dynamics. J. Biol. Chem. 268, 7545–7552 [PubMed] [Google Scholar]
- Hutchison C. J., Worman H. J. (2004). A-type lamins: guardians of the soma? Nat. Cell Biol. 6, 1062–1067 10.1038/ncb1104--1062 [DOI] [PubMed] [Google Scholar]
- Imanishi S. Y., Kochin V., Ferraris S. E., de Thonel A., Pallari H-M., Corthals G. L., Eriksson J. E. (2007). Reference-facilitated phosphoproteomics: fast and reliable phosphopeptide validation by microLC-ESI-Q-TOF MS/MS. Mol. Cell. Proteomics 6, 1380–1391 10.1074/mcp.M600480--MCP200 [DOI] [PubMed] [Google Scholar]
- Kim J-E., Tannenbaum S. R., White F. M. (2005). Global phosphoproteome of HT-29 human colon adenocarcinoma cells. J. Proteome Res. 4, 1339–1346 10.1021/pr050048h [DOI] [PubMed] [Google Scholar]
- Kochin V., Imanishi S. Y., Eriksson J. E. (2006). Fast track to a phosphoprotein sketch - MALDI-TOF characterization of TLC-based tryptic phosphopeptide maps at femtomolar detection sensitivity. Proteomics 6, 5676–5682 10.1002/pmic.200600457 [DOI] [PubMed] [Google Scholar]
- Leukel M., Jost E. (1995). Two conserved serines in the nuclear localization signal flanking region are involved in the nuclear targeting of human lamin A. Eur. J. Cell Biol. 68, 133–142 [PubMed] [Google Scholar]
- Lloyd D. J., Trembath R. C., Shackleton S. (2002). A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum. Mol. Genet. 11, 769–777 10.1093/hmg/11.7.769 [DOI] [PubMed] [Google Scholar]
- Lüscher B., Brizuela L., Beach D., Eisenman R. N. (1991). A role for the p34cdc2 kinase and phosphatases in the regulation of phosphorylation and disassembly of lamin B2 during the cell cycle. EMBO J. 10, 865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M., Walter T., Gorjánácz M., Davidson I. F., Nga Ly-Hartig T. B., Ellenberg J., Mattaj I. W. (2012). Mitotic lamin disassembly is triggered by lipid-mediated signaling. J. Cell Biol. 198, 981–990 10.1083/jcb.201205103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir R. D., Montag-Lowy M., Goldman R. D. (1994). Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J. Cell Biol. 125, 1201–1212 10.1083/jcb.125.6.1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir R. D., Yoon M., Khuon S., Goldman R. D. (2000). Nuclear lamins A and B1: different pathways of assembly during nuclear envelope formation in living cells. J. Cell Biol. 151, 1155–1168 10.1083/jcb.151.6.1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 10.1016/j.cell.2006.09.026 [DOI] [PubMed] [Google Scholar]
- Omary M. B. (2009). “IF-pathies”: a broad spectrum of intermediate filament-associated diseases. J. Clin. Invest. 119, 1756–1762 10.1172/JCI39894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto H., Dreger M., Bengtsson L., Hucho F. (2001). Identification of tyrosine-phosphorylated proteins associated with the nuclear envelope. Eur. J. Biochem. 268, 420–428 10.1046/j.1432--1033.2001.01901.x [DOI] [PubMed] [Google Scholar]
- Pack C., Saito K., Tamura M., Kinjo M. (2006). Microenvironment and effect of energy depletion in the nucleus analyzed by mobility of multiple oligomeric EGFPs. Biophys. J. 91, 3921–3936 10.1529/biophysj.105.079467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallari H-M., Eriksson J. E. (2006). Intermediate filaments as signaling platforms. Sci. STKE 2006, pe53 10.1126/stke.3662006pe53 [DOI] [PubMed] [Google Scholar]
- Pekovic V., Harborth J., Broers J. L. V., Ramaekers F. C. S., van Engelen B., Lammens M., von Zglinicki T., Foisner R., Hutchison C., Markiewicz E. (2007). Nucleoplasmic LAP2alpha-lamin A complexes are required to maintain a proliferative state in human fibroblasts. J. Cell Biol. 176, 163–172 10.1083/jcb.200606139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M., Nakagawa J., Dorée M., Labbé J. C., Nigg E. A. (1990). In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 61, 591–602 10.1016/0092--8674(90)90471--P [DOI] [PubMed] [Google Scholar]
- Peter M., Heitlinger E., Häner M., Aebi U., Nigg E. A. (1991). Disassembly of in vitro formed lamin head-to-tail polymers by CDC2 kinase. EMBO J. 10, 1535–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield A. V., Bernard O. (2013). Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit. Rev. Biochem. Mol. Biol. 48, 301–316 10.3109/10409238.2013.786671 [DOI] [PubMed] [Google Scholar]
- Shimi T., Pfleghaar K., Kojima S., Pack C-G., Solovei I., Goldman A. E., Adam S. A., Shumaker D. K., Kinjo M., Cremer T. et al. (2008). The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 22, 3409–3421 10.1101/gad.1735208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimi T., Butin-Israeli V., Adam S. A., Hamanaka R. B., Goldman A. E., Lucas C. A., Shumaker D. K., Kosak S. T., Chandel N. S., Goldman R. D. (2011). The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 25, 2579–2593 10.1101/gad.179515.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker D. K., Solimando L., Sengupta K., Shimi T., Adam S. A., Grunwald A., Strelkov S. V., Aebi U., Cardoso M. C., Goldman R. D. (2008). The highly conserved nuclear lamin Ig-fold binds to PCNA: its role in DNA replication. J. Cell Biol. 181, 269–280 10.1083/jcb.200708155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon D. N., Wilson K. L. (2013). Partners and post-translational modifications of nuclear lamins. Chromosoma 122, 13–31 10.1007/s00412--013--0399--8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift J., Ivanovska I. L., Buxboim A., Harada T., Dingal P. C. D. P., Pinter J., Pajerowski J. D., Spinler K. R., Shin J-W., Tewari M. et al. (2013). Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341, 1240104 10.1126/science.1240104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward G. E., Kirschner M. W. (1990). Identification of cell cycle-regulated phosphorylation sites on nuclear lamin C. Cell 61, 561–577 10.1016/0092--8674(90)90469--U [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.