

Abstract
This study examined the bacterial and archaeal diversity from a worldwide range of wetlands soils and sediments using a meta-analysis approach. All available 16S rRNA gene sequences recovered from wetlands in public databases were retrieved. In November 2012, a total of 12677 bacterial and 1747 archaeal sequences were collected in GenBank. All the bacterial sequences were assigned into 6383 operational taxonomic units (OTUs 0.03), representing 31 known bacterial phyla, predominant with Proteobacteria (2791 OTUs), Bacteroidetes (868 OTUs), Acidobacteria (731 OTUs), Firmicutes (540 OTUs), and Actinobacteria (418 OTUs). The genus Flavobacterium (11.6% of bacterial sequences) was the dominate bacteria in wetlands, followed by Gp1, Nitrosospira, and Nitrosomonas. Archaeal sequences were assigned to 521 OTUs from phyla Euryarchaeota and Crenarchaeota. The dominating archaeal genera were Fervidicoccus and Methanosaeta. Rarefaction analysis indicated that approximately 40% of bacterial and 83% of archaeal diversity in wetland soils and sediments have been presented. Our results should be significant for well-understanding the microbial diversity involved in worldwide wetlands.
1. Introduction
Wetlands, which were estimated to be 45% of the total value of global natural ecosystems [1], are one of the most important terrestrial ecosystems and distribute in all regions throughout the world including Antarctica [2]. Microbiomes in wetlands play an important role in biogeochemical processes and microbial activities are crucial to the functions of wetland systems [3–8]. Moreover, microbial diversity is essential for exploiting potential of microbial resources from the wetland ecosystems [9–13]. It is crucial and necessary to understand the overall survival microorganisms in wetlands. Bacteria and archaea have been widely studied with respect to their biodiversity in natural and constructed wetlands [14–17]. Initial studies employed traditional culture-dependent methods and resulted in the discovery of plenty of new bacterial and archaeal taxa [18]. Employing kinds of molecular biology methods, increasing evidences have suggested that the structures of microbial communities are related to soil processes, such as cloning and sequencing of 16S rRNA genes, denaturing gradient gel electrophoresis (DGGE), terminal restriction fragment length polymorphism (T-RFLP), and quantitative PCR [4, 8, 19–23]. Cloning and sequencing of 16S rRNA genes have been widely used for their identification of potential known and unknown microbes [24]. Plenty of studies have examined the microbial diversity in wetlands using relatively large (>200 sequences) 16S rRNA clone libraries [4, 20, 25]. However, most studies to date have focused on individual wetland ecosystem [16, 26–28]. Many of the datasets published contain a small number of cloned sequences (generally >100), thus revealing only a small portion of the full diversity present in wetlands [10, 11, 29, 30]. The focus of some studies is limited to particular microbial group [31, 32]. In addition, there are many sequences recovered from wetlands with no additional information which were deposited into GenBank without being reported yet. High-throughput sequencing technologies, such as 454-pyrosequencing and ion torrent, were used to analyze the microbiomes in wetlands [30, 33–35]. These methods can produce huge datasets of short sequence reads. However, the length of these reads is too short to classify. Currently, there is no consensus on the size or nature of the microbial diversity generally found in wetlands. As a result, the understanding of the microbiomes in wetlands is fragmented and likely biased. This knowledge gap of microbiomes in wetlands will hamper the efficiency and stability of wetlands ecosystems. Few of the collective overviews of the microbial diversity in global wetlands are found up to date. The purposes of the study are to (1) perform a meta-analysis of all publicly available 16S rRNA gene sequences identified from various wetlands to provide a collective appraisal of the microbial diversity in wetland ecosystem, (2) make an effort to estimate the current coverage of the microbial diversity in wetlands, and (3) identify particular gaps in the knowledge and understanding of the microbial populations involved in wetlands.
2. Methods
2.1. Sequence Data Collection
Initial sequence sets were obtained from the GenBank (http://www.ncbi.nlm.nih.gov) and RDP (Release 10, http://rdp.cme.msu.edu) databases using the search terms (“wetland” OR “marsh” OR “fen”) AND “soil” AND “16S” on November 11, 2012. Non-16S rRNA sequences from GenBank were removed by checking the name of sequences. All 16S rRNA gene sequences from two databases were merged. Duplicate sequences identified based on accession numbers were removed. Mallared was used for checking sequences with vector nucleotides or chimera (http://www.softsea.com/review/Mallard.html). The 16S rRNA gene sequences of Escherichia coli (accession number: U00096) and Methanothermobacter thermoautotrophicus (accession number: AE000666) were selected as reference sequences for bacteria and archaea, respectively. In order to avoid uncertainties in comparing and classifying short sequences, sequences shorter than 250 bp were removed from the dataset which have few or no sequence overlap. The remaining sequences comprised the redacted composite dataset used in this work.
2.2. Phylogenetic Analysis
Sequences were aligned with Kalign [36] and classified into taxonomic ranks using the RDP Classifier with default settings [37]. Based on the output classifications from the RDP Classier, treemaps were constructed using the treemap packages in R. The dataset was divided into the following groups based on the classifications: Archaea, Bacteria, Proteobacteria, Actinobacteria, Firmicutes, Acidobacteria, Bacteroidetes, Chloroflexi, and the collected “minor phyla” of bacteria that comprised sequences not assigned to any of the aforementioned phyla. Distances matrices of aligned sequences were computed within ARB using Jukes-Cantor correction [38]. Individual distance matrices were analyzed using Mothur [39] to cluster OTUs, generated rarefaction curves, and estimated the expected maximum species richness complementary to the ACE and Chao1 richness. Unless otherwise stated, the genetic distance ≤0.03 was used to define species-level OTUs. The distance cut-off for other taxonomic ranks was set as follows: 0.05, genus; 0.10, family; 0.15, class/order; and 0.2, phylum. All the estimated asymptotes of the rarefaction curve were determined through R package monomol (https://github.com/binma/monomol) [40]. The coverage percentages were calculated as described by Nelson et al. [41].
2.3. Accession Numbers
The accession numbers for all sequences analyzed in this study were available from the corresponding author. The sequences were currently maintained in an in-house ARB database of 16S rRNA gene sequences for wetlands. A copy of this database and the sequence alignment were also available by request from the corresponding author.
3. Results and Discussion
This study was conducted as a meta-analysis ground on publicly available 16S rRNA gene sequences recovered from wetland soils worldwide. The sequences dataset collected from Genbank and RDP database was analyzed no matter their previously assigned taxonomic information or other analyses.
To address the long-term question of understanding microorganisms from wetland soil habitats, this study first aimed at characterizing prokaryotic communities inhabiting wetland soils. The prokaryotic microorganisms from wetland soil habitats drive the biogeochemical cycles of elements and may be a source of novel halophilic enzymes. Thus, we studied the diversity of prokaryotic microorganisms from wetland soils with meta-analysis approach.
3.1. Data Summary
Totally 14318 sequences longer than 250 bp were retrieved from GenBank and RDP databases. The sequences were mostly about 800 bp long, followed by approximately 600 bp (Figure 1). Interestingly, there is a small submit of sequence length between 1400 bp and 1600 bp. The 12583 bacterial sequences were assigned to 6383 OTUs, while the 1735 archaeal sequences were assigned to 521 OTUs (Table 1 and Figure 1). The most abundant bacterial and archaeal OTU contained 143 sequences and 113 sequences, respectively. Over 90% bacterial sequences were classified within five phyla, namely, Proteobacteria, Bacteroidetes, Acidobacteria, Firmicutes, and Actinobacteria (Figure 2). The remaining sequences were classified within 26 “minor” phyla, of which Chloroflexi, Planctomycetes, Cyanobacteria, and Verrucomicrobia were the only “minor” phyla with representation 1% of all bacterial sequences.
Figure 1.
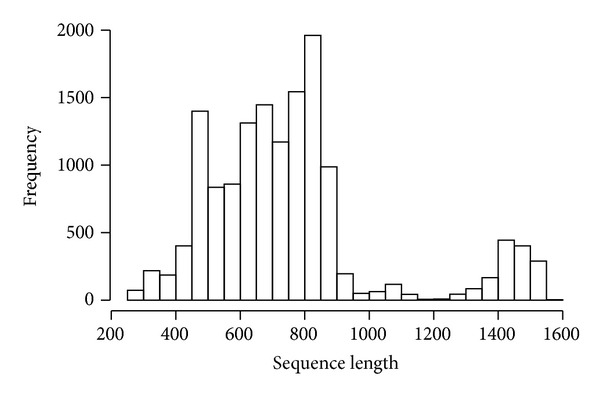
Distribution of the length of retrieved 16S rRNA sequences.
Table 1.
Diversity statistics for Archaea, Bacteria, and “Major” phylum groups. Coverage = #OTUs/rarefaction estimate; OTU and abundance were calculated using a 0.03 dissimilarity cut-off.
| Group | Total sequences | Unclassified to phylum | Number of OTUs | ACE | Chao1 | Rarefaction estimation | Current coverage (%) |
|---|---|---|---|---|---|---|---|
| Bacteria | 12583 | none | 6383 | 30581 | 17176 | 15768 | 41 |
| Pro | 5763 | 2791 | 12472 | 7245 | 6811 | 40 | |
| Act | 783 | 418 | 2280 | 1088 | 1033 | 46 | |
| Aci | 1345 | 731 | 2972 | 1693 | 1602 | 28 | |
| Fir | 973 | 540 | 3595 | 1856 | 1915 | 54 | |
| Bact | 2244 | 868 | 2700 | 1887 | 1601 | 59 | |
| Archaea | 1735 | none | 521 | 1131 | 884 | 883 | 83 |
| Eur | 925 | 418 | 681 | 505 | 504 | 62 | |
| Cre | 810 | 197 | 442 | 311 | 320 | 41 |
Figure 2.
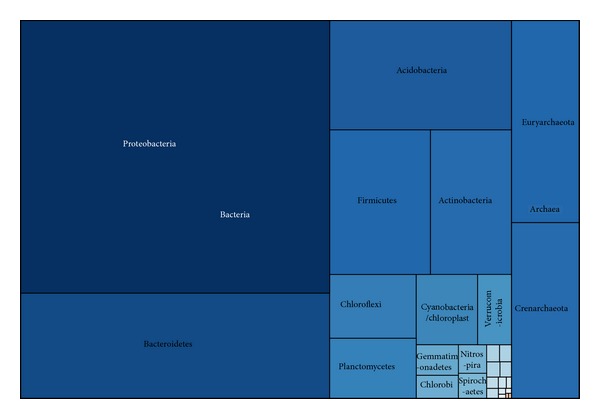
Treemap of observed prokaryotic taxons shown in their hierarchical order. Treemap showing taxonomic ranking of all taxa for all retrieved sequences. The size of each box is proportional to the number of sequences assigned to that taxon with respect to the entire dataset. The placement of boxes is arbitrary with respect to boxes within the same taxonomic rank and does not correspond to any form of phylogeny or relatedness.
Of the archaeal sequences analyzed, all of them were classified within two phyla: Euryarchaeota and Crenarchaeota, representing 925 and 810 sequences, respectively.
3.2. Bacteria
3.2.1. Proteobacteria
The Proteobacteria was the largest and most diverse phylum in the present dataset. It comprised a total of 5637 sequences, approximately 44.8% of the bacterial sequences, assigned to 466 known genera. There are 2791 OTUs generated, with a Simpson diversity index of 0.0020. All six classes within the Proteobacteria were represented, but the Delta-, Gamma-, Beta-, and Alphaproteobacteria together represented over 99% of the proteobacterial sequences (Figure 3). The classes Epsilonproteobacteria and Zetaproteobacteria were extremely rare, represented by 43 and 1 sequences, respectively, indicating a low recovery rate in most of wetlands.
Figure 3.
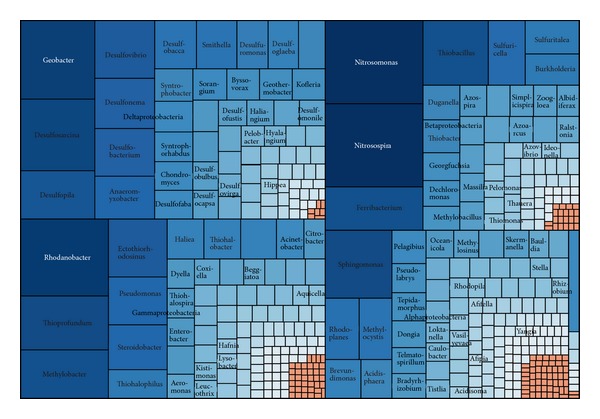
Treemap of observed Proteobacteria taxons shown in their hierarchical order.
Classes in Proteobacteria showed various tendencies in different wetlands. The wide distribution of Gammaproteobacteria and Deltaproteobacteria in marine sediment has been documented, and most of them were involved in sulfur reduction under anaerobic conditions [4]. In comparison, a high abundance of Alphaproteobacteria and Betaproteobacteria appeared in freshwater sediment, and it is significantly correlated with pH and nutrients [34]. Some genera of Betaproteobacteria were confirmed to inhabit extremely alkaline wetland filled with historic steel slag [42]. The Epsilonproteobacteria is relatively abundant at oxic-anoxic interfaces such as intertidal wetland [43].
Deltaproteobacteria was the largest class in the phylum, with 1627 sequences (28.9% of the proteobacteria). Geobacter of family Geobacteraceae was the most abundant genus (9.8% of the Deltaproteobacteria) in Deltaproteobacteria. It was abundant in the rhizosphere and has been widely known as a kind of Fe (III)-reducing bacterium [44]. The followed abundant genera were Deltaproteobacteria, Desulfosarcina, Desulfopila, Desulfovibrio, Desulfonema, and Desulfobacterium, which represented greater than 1.0% of proteobacterial sequences. All of them were sulfate-reducing bacteria, and their distributions were influenced by salinity and plant nutrient [45]. They played important roles in the metabolism of nitrogen, phosphorus, sulfur, and some organic compounds in wetland systems [18, 46]. Anaeromyxobacter was also the genus owning more than 1.0% proteobacterial sequences. As a kind of facultative bacteria, its unique respiratory reduction of nitrate and nitrite to ammonia was not linked to its ability to reduce nitrous oxide to nitrogen gas [47].
For the class Gammaproteobacteria, 1456 sequences were identified. It was the second largest class in Proteobacteria. Approximately 12.6% of gammaproteobacterial sequences (184 sequences) were assigned to the genus Rhodanobacter of family Xanthomonadaceae. This genus might be engaged in acidic denitrification in wetland soils [3]. The following abundant genera were Thioprofundum and Methylobacter, accounting for 8.9% (129 sequences) and 8.0% (108 sequences) of gammaproteobacterial sequences, respectively. Thioprofundum was recently considered as a mesophilic, facultatively anaerobic, sulfur-oxidizing bacterial strain [48]. Methylobacter was reported as dominating in the Zoige wetland where the centers of methane emission were [24]. However, it was not affected by nitrogen leached from the catchment area in boreal littoral wetlands [9]. The other genera representing more than 1.0% proteobacteria sequences were Ectothiorhodosinus, Pseudomonas, and Steroidobacter. Pseudomonas was one of the widely studied PAH-degrading bacteria; it spread widely in contaminated wetlands environment [29] and was predominant microbial populations in the constructed wetland for nitrobenzene wastewater [32].
The 1420 betaproteobacterial sequences were identified in Proteobacteria. The genus Nitrosomonas was the predominant genera with 222 assigned sequences, while the genus Nitrosospira was the second abundant genus with 217 sequences. They were also the first and second most abundant proteobacterial genera, and both of them belonged to the family Nitrosomonadaceae which were well known as the main ammonia-oxidizing microorganisms contributing to N2O production in wetlands and sediments [31, 49, 50]. The genera Ferribacterium, Thiobacillus, and Sulfuricella owned more than 1.0% of proteobacterial sequences.
The fourth largest proteobacterial class was Alphaproteobacteria, with 1090 sequences (over 19.3%). The dominating genus Sphingomonas in class Alphaproteobacteria was widely distributed in wetland and sediments, due to its ability to survive in low concentrations of nutrients, as well as to metabolize a wide variety of carbon sources [7, 51]. Except for Sphingomonas which contains over 2.0% of the proteobacterial sequences (122 sequences), other genera of Alphaproteobacteria represented less than 1.0% proteobacteria sequences.
3.2.2. Bacteroidetes
Bacteroidetes was the second abundant phylum in the present dataset, including 2244 sequences (nearly 17.8% of all bacterial sequences), which were assigned to 109 known genera, with 868 OTUs and a Simpson diversity index of 0.0007 (Figure 4). A plenty of Bacteroidetes strains isolated from wetland soils and sediments were reported to be anaerobic and saprophytic representative bacteria [52, 53]. Highlighting the unevenness of the phylum, over 70% of all the Bacteroidetes sequences (12.8% of all bacterial sequences) were assigned to class Flavobacteria. As a common heterotrophic obligate aerobe, Flavobacteria was the second largest class in the dataset. It is widespread in various wetlands, even in swine wastewater lagoon and constructed wetlands [54, 55]. The class Sphingobacteria was represented by only 491 sequences, while the class Bacteroidia was represented by only 75 sequences. “Undefined Bacteroidetes” comprised 65 sequences.
Figure 4.
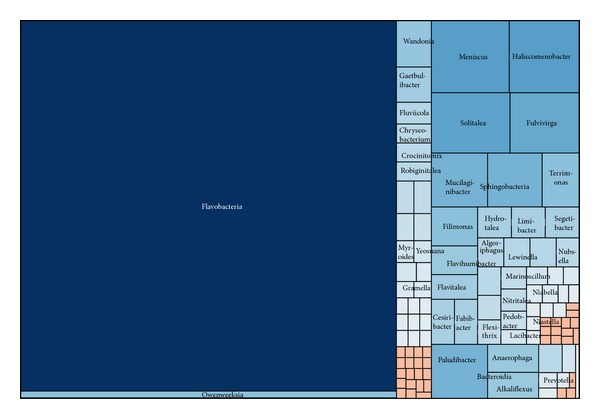
Treemap of observed Bacteroidetes taxons shown in their hierarchical order.
The most frequently observed genus in Flavobacteria was Flavobacterium (1459 sequences), which was also the most abundant bacterial genus in this dataset. A number of species of Flavobacterium have been isolated from rhizosphere of wetland [52, 53].
3.2.3. Acidobacteria
Acidobacteria was the third largest phylum in our dataset, including 1345 sequences assigned to 29 genera. Acidobacteria is a new phylum, whose members are physiologically diverse and ubiquitous in soils, but are underrepresented in culture at present. There were 731 OTUs identified, with a Simpson diversity index of 0.0031 (Figure 5). Just over 90% of all the acidobacterial sequences (9.7% of all bacterial sequences) were assigned to 21 unclassified groups, only 130 sequences represented to class Holophagae. In total, nearly 40% of the Acidobacteria sequences were able to be classified to Gp1, which was the second largest class of bacteria. The following classes were group Gp3 and then group Gp6, with 196 and 116 sequences, respectively. As the reports, Acidobacteria group was more abundant in natural wetlands than in created wetlands [10, 34], especially in freshwater sediment [34]. Acidobacteria has been reported as the largest division in the active layer and the associated permafrost of a moderately acidic wetland in Canada [11]. Future studies are needed to examine the interrelations of environmental parameters with Acidobacteria and individual populations within subgroups [56].
Figure 5.
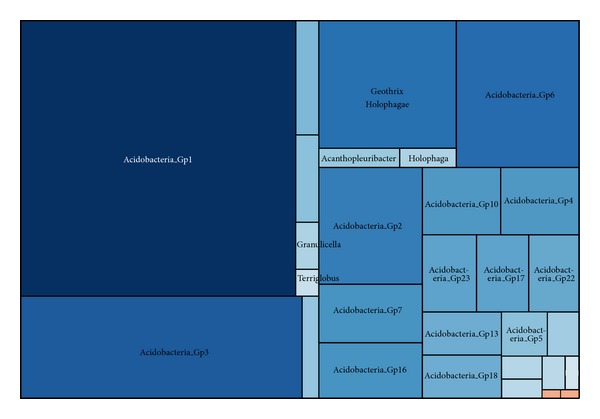
Treemap of observed Acidobacteria taxons shown in their hierarchical order.
3.2.4. Firmicutes
The fourth largest phylum was the Firmicutes, assigned into 973 sequences and 540 OTUs with a Simpson diversity index of 0.0041 (Figure 6). As saprophytic microbes, some members of Firmicutes are known to produce endospores under stressful environmental conditions such as in intertidal sediment [34], extremely alkaline (pH > 12) constructed wetland [42].
Figure 6.
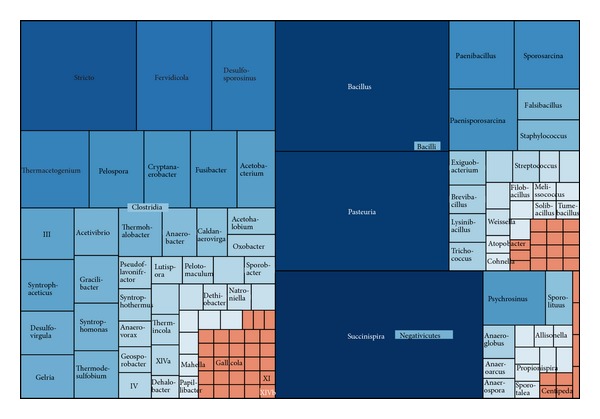
Treemap of observed Firmicutes taxons shown in their hierarchical order.
In total, about 45% of the Firmicutes sequences were classified to the class Clostridia, and nearly 36% were classified into the class Bacilli. The Clostridia (sulfite-reducing bacteria) is an anaerobic and highly polyphyletic bacterium, while Bacilli can be obligate aerobes or facultative anaerobes. There was a long record of evidence to suggest that both of them were the abundant taxa in sewage sludge [57]. Some species of them exhibit great ability to degrade hydrocarbons in crude oil contaminated wetland ultisol [6]. Within the class Bacilli, two primary genera were Bacillus and Pasteuria, representing 107 and 98 sequences, respectively. While in Clostridia, genus Stricto was the most abundant genus, with 56 sequences.
The class Negativicutes represented 178 sequences. The genus Succinispira represented over 70% of sequences in Negativicutes. The genus Succinispira, the most abundant genus in Firmicutes, was capable of decarboxylating succinate in anaerobic conditions. The class Erysipelotrichia represented only three sequences.
3.2.5. Actinobacteria
As the fifth abundant phylum, Actinobacteria represented 783 sequences, clustered into 418 OTUs, with a Simpson diversity index of 0.0054. All of Acidobacteria sequences were classified to the class Actinobacteria and over 66% of them belonged to order Actinomycetales (Figure 7). Actinobacteria can be terrestrial or aquatic, playing an important role in the decomposition of organic materials. Although understood primarily as soil bacteria, they might be more abundant in freshwaters [10, 57].
Figure 7.
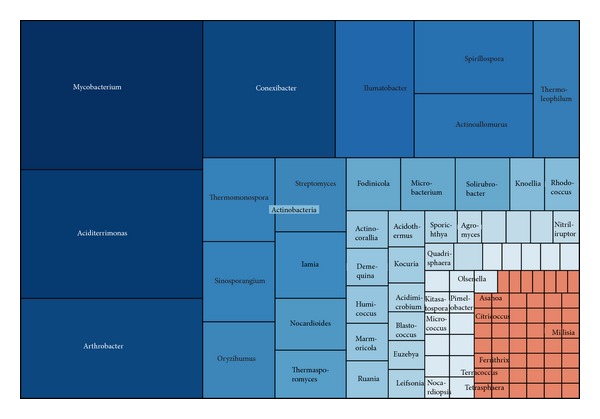
Treemap of observed Actinobacteria taxons shown in their hierarchical order.
Mycobacterium (103 sequences) was the most frequently observed genus in Actinobacteria. It has been widely detected from contaminated soil or sediments [51]. Some species of Mycobacterium were the dominant PAH-degraders and played an important role in degrading PAHs in contaminated mangrove sediments [7]. The following abundant genera were Aciditerrimonas, Conexibacter, Arthrobacter, and Ilumatobacter. The rest of genera were less than 5% of actinobacterial sequences.
3.2.6. Minor Phyla
In addition to the five phyla described above, 26 minor phyla with 1601 sequences were also observed based on the dataset. Of these minor phyla, only the phyla Chloroflexi (2.96%), Planctomycetes (2.77%), Cyanobacteria (2.28%), and Verrucomicrobia (1.28%) represented more than 1% of all the bacterial sequences and accounted for over 73% of all minor phyla sequences (Figure 1).
Some known genera were represented in these “minor phyla.” The most abundant of the minor phyla, Chloroflexi, comprised 372 sequences. Members of the Chloroflexi are generally found in intertidal sediment and moderately acidic wetland [11, 13, 34, 58]. Planctomycetes was the second most abundant of the minor phyla, to which 349 sequences were assigned. A number of genera of the Planctomycetes, which were once thought to occur primarily in aquatic environments, have been discovered in wetlands [12, 29]. As the third most abundant minor phyla, Cyanobacteria occupy a broad range of habitats across all latitudes. They are widespread in freshwater, marine, and even in the most extreme niches such as hot springs and hypersaline bays [12, 59, 60]. Evidence suggests that Verrucomicrobia are abundant within the environment and important. The species of Verrucomicrobia have been identified and isolated from fresh water and soil environments [61].
3.3. Archaea
3.3.1. Euryarchaeota
Euryarchaeota comprised 925 sequences, approximately 53.3% archaeal sequences. They were clustered into 418 OTUs with a Simpson diversity index of 0.0054 (Figure 8). The majority (70.9%) of Euryarchaeota sequences were assigned to the methanogenic class Methanomicrobia (656 sequences). The class Thermoplasmata comprised 132 sequences, while the class Methanobacteria comprised 75 sequences. Only 59 sequences were classified into class Halobacteria. Classes Archaeoglobi and Methanopyri represented only 2 and 1 sequences, respectively.
Figure 8.
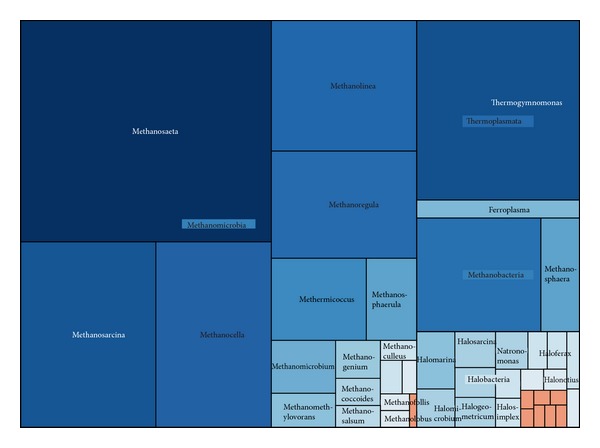
Treemap of observed Euryarchaeota taxons shown in their hierarchical order.
Methanomicrobia contributes a large proportion of methane emission in wetlands, no matter in cold area or in subtropical places [19, 62]. As seen in Figure 6, the most predominate Methanomicrobia genus (223 sequences) was Methanosaeta (formerly Methanothrix), which was also the second most abundant archaeal genus. It was reported precisely as the dominant acetoclastic methanogen in the high arctic wetlands [63]. The methanogens genera Methanosarcina, Methanocella, Methanolinea, and Methanoregula each represented nearly 10% of Euryarchaeota sequences. The other 12 genera were only represented by a small number of sequences in the dataset.
The largest genus in class Thermoplasmata was Thermogymnomonas (120 sequences), which was detected widely even at low pH wetlands. It was known as a kind of iron-oxidizing microorganisms [64]. The rest 12 sequence of Euryarchaeota were assigned to genus Ferroplasma, an anaerobic and acidophilic archaea, which coupled to the reduction of ferric iron [5]. Of the class Methanobacteria, there were two genera, Methanobacterium and Methanosphaera, with 57 and 18 sequences, respectively. Both of these genera were detected from water and sediments of a high-altitude athalassohaline wetland [25]. As a facultative anaerobic archaea, Halobacteria was common in most environments where large amounts of salt, moisture, and organic material are available [25].
3.3.2. Crenarchaeota
Crenarchaeota owned less abundant sequences than Euryarchaeota in the dataset, with 810 sequences. Crenarchaeota diversity was lower, with only 197 OTUs generated and a Simpson diversity index of 0.0443. It suggested that Crenarchaeota was more related to aerobic metabolisms in the water and surface sediment [65].
All of the Crenarchaeota sequences were assigned to the class Thermoprotei (Figure 9). As the reports, class Thermoprotei dominated in archaeal phyla in Pacific influenced sediments, while Methanomicrobia inhabited in methane-containing Atlantic influenced sediments [58]. Within the class, 258 sequences were classified to the genera Fervidicoccus. Fervidicoccus was the most abundant genera in archaeal and has been cultivated and characterized widely. The following abundant genera in this phylum were Thermofilum (19.5%), Caldisphaera (13.3%), and Stetteria (11.5%). The other genera sequences were less than 10%.
Figure 9.
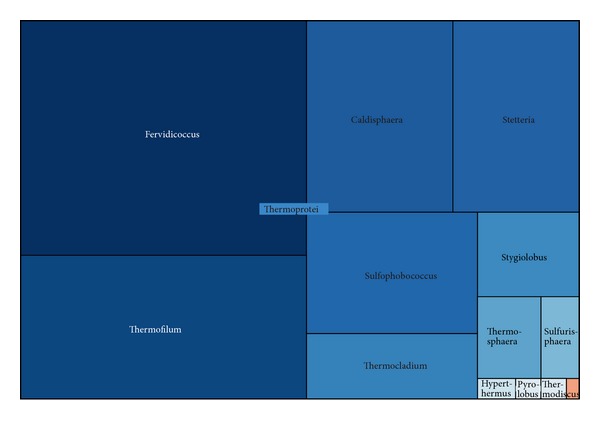
Treemap of observed Crenarchaeota taxons shown in their hierarchical order.
3.4. Diversity Estimates
For all of the bacterial groups, the ACE value of richness was the greatest, while the majority of corresponding rarefaction estimates were the lowest (Table 1). Similar with rarefaction estimates, the ACE and Chao1 estimate the maximum species richness for an OUT definition. However, the richness estimates derived from the rarefaction curves differed less from the Chao1 estimate, comparing with those from ACE estimates. The richness estimates derived from ACE differed greatly (72~120%) for Bacteria, Proteobacteria, Acidobacteria, Firmicutes, and Actinobacteria, while the corresponding estimates for the Bacteroidetes, Archaea, Euryarchaeota, and Crenarchaeota were less than 70% different.
The present results showed that the coverage of microbial diversity in wetlands was remaining rather low. Rarefaction analysis of Bacteria showed that only sampling at the phylum (0.20 phylogenetic distance) level has begun to reach a horizontal plateau. The other sampling at the taxonomic ranks was still projecting upward (Figure 10 and Table 2). At the species (0.03 phylogenetic distance) level, only 41% of the expected diversity has been revealed. The estimates of current coverage suggest that of Bacteria was less than that of Actinobacteria, Bacteroidetes, and Firmicutes, greater than that of Acidobacteria. Coverage rate of Proteobacteria was similar to that of the Bacteria. For the archaea, the coverage of diversity was greater than bacteria, but still low compared to estimated richness. There was about 59% of the expected diversity revealed at the species level. The estimates of current coverage of Euryarchaeota and Crenarchaeota were much greater than that of Archaea. As the results of rarefaction analysis and diversity statistics, it was obvious that the known bacterial and archaeal diversity in wetlands were incomplete below the phylum level. Nevertheless, the global microbial diversity in wetlands revealed in this study had ability to serve as a framework for future studies of alpha and beta diversity. More specifically, the collected sequence dataset could give a hand on detecting and quantifying specific groups of either bacteria or archaea at the nucleotide level. Additionally, these studies will greatly advance the ecology of individual microbia collected in the dataset.
Figure 10.
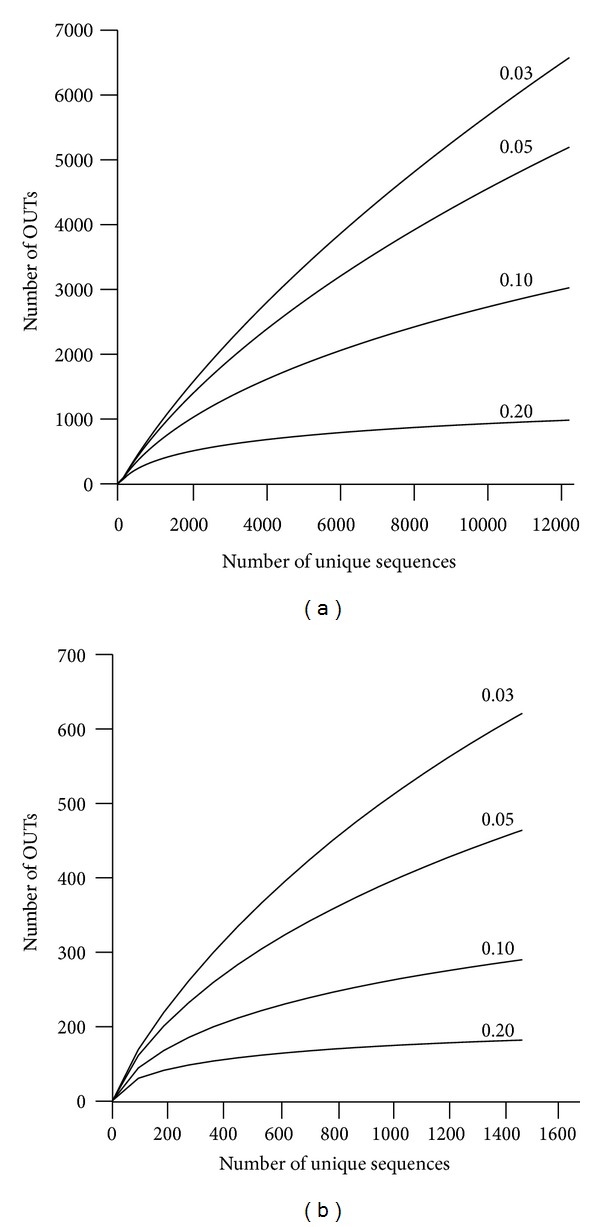
Rarefaction curve for the Archaea (a) and Bacteria (b) with different dissimilarity cut-off.
Table 2.
Estimates of current taxonomic coverage for Archaea and Bacteria.
| Distance | Number of Current OTUs | Rarefaction estimation | Coveragea (%) |
|---|---|---|---|
| Archaea | |||
| 0.03 | 521 | 883 | 59 |
| 0.05 | 364 | 587 | 62 |
| 0.10 | 190 | 278 | 68 |
| 0.20 | 82 | 91 | 90 |
| Bacteria | |||
| 0.03 | 6383 | 15768 | 40 |
| 0.05 | 5042 | 9854 | 51 |
| 0.10 | 2937 | 4617 | 63 |
| 0.20 | 954 | 1118 | 85 |
aCoverage = number of OTUs/rarefaction estimate.
Sufficient coverage and depth were provided to explore an individual sample or compare multiple samples through multiplexing, with the developing of second generation sequencing technologies. Moreover, new sequences dataset could be added to the composite datasets analyzed in this study to increase our knowledge on the diversity of this ecosystem. The knowledge on the diversity may shine light on the understanding of the microbiomes of wetlands and define the significance of individual microbia. It is also suited for continuous following of the succession variation of the diversity of wetlands. However, the beta diversity was hardly determined because most of studies could not contain large sequence datasets and detailed information with same methodologies and sequence submission criteria. A “core group” was defined after analyzing seven municipal sludge digesters [66]. Although distinct microbiomes are possibly being selected under a unique environment, only a small number of “core OTUs” can be found among the large numbers of OTUs identified. Systematic studies examining multiple wetlands designs with great depth of coverage should help further define the “core microbiomes” in wetlands.
Now that analysis of 16S rRNA gene sequences can provide insight into the functional diversity of wetlands, the metabolic functions of organisms are getting more concerned. For a good comprehension of the metabolic capacities of these organisms, metagenomic studies techniques such as SIP and MAR-FISH should be used more frequently. Cultivation-based studies are also needed to define the functions of uncharacterized species of bacteria and archaea in wetlands.
4. Conclusions
The present dataset generated from GenBank and RDP databases was largely dominated by Proteobacteria. Approximately 40% of sequences and OTUs belonged to Proteobacteria. Our results showed that (1) nearly 56% of the archaeal and 45% of the bacterial species-level diversity in wetlands have been witnessed; (2) sequences from the bacterial phyla Proteobacteria, Bacteroidetes, Chloroflexi, Firmicutes, Actinobacteria, and archaeal class were well represented by the available sequences and the corresponding microorganisms were probably important participants in the wetland environments; (3) the global diversity contains numerous groups for which there was no close cultured representative, especially the majority of sequences assigned to the phyla Chloroflexi and Bacteroidetes. Therefore future studies should utilize multiple approaches to characterize the microbial diversity and its function in wetlands.
Acknowledgments
The authors would like to thank the Project of National Science & Technology Pillar Program in “12th Five Year” period (2011BAC02B01), National Natural Science Foundation for Distinguished Young Scholar of Shandong Province (no. JQ201114), the National Science Foundation of China (41301333), and the CAS/SAFEA International Partnership Program for Creative Research Teams “Representative environmental processes and resources effects in coastal zone.” They also would like to thank the Yellow River Delta Wetland Ecological Experimental Station, CAS, for providing experimental and residential place for this study.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Xiaofei Lv, Junbao Yu, and Yuqin Fu contributed equally to this work.
References
- 1.Costanza R, d'Arge R, de Groot R, et al. The value of the world's ecosystem services and natural capital. Nature. 1997;387(6630):253–260. [Google Scholar]
- 2.Jungblut AD, Wood SA, Hawes I, Webster-Brown J, Harris C. The pyramid trough wetland: environmental and biological diversity in a newly created Antarctic protected area. FEMS Microbiology Ecology. 2012;82(2):356–366. doi: 10.1111/j.1574-6941.2012.01380.x. [DOI] [PubMed] [Google Scholar]
- 3.van den Heuvel RN, van der Biezen E, Jetten MSM, Hefting MM, Kartal B. Denitrification at pH 4 by a soil-derived Rhodanobacter-dominated community. Environmental Microbiology. 2010;12(12):3264–3271. doi: 10.1111/j.1462-2920.2010.02301.x. [DOI] [PubMed] [Google Scholar]
- 4.Lenk S, Arnds J, Zerjatke K, Musat N, Amann R, Mußmann M. Novel groups of Gammaproteobacteria catalyse sulfur oxidation and carbon fixation in a coastal, intertidal sediment. Environmental Microbiology. 2011;13(3):758–774. doi: 10.1111/j.1462-2920.2010.02380.x. [DOI] [PubMed] [Google Scholar]
- 5.Justice NB, Pan C, Mueller R, et al. Heterotrophic archaea contribute to carbon cycling in low-pH, suboxic biofilm communities. Applied and Environmental Microbiology. 2012;78(23):8321–8330. doi: 10.1128/AEM.01938-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.John RC, Itah AY, Essien JP, Ikpe DI. Fate of nitrogen-fixing bacteria in crude oil contaminated wetland ultisol. Bulletin of Environmental Contamination and Toxicology. 2011;87(3):343–353. doi: 10.1007/s00128-011-0320-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo C, Ke L, Dang Z, Tam NF. Temporal changes in Sphingomonas and Mycobacterium populations in mangrove sediments contaminated with different concentrations of polycyclic aromatic hydrocarbons (PAHs) Marine Pollution Bulletin. 2011;62(1):133–139. doi: 10.1016/j.marpolbul.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 8.Hu B, Shen L, Lian X, et al. Evidence for nitrite-dependent anaerobic methane oxidation as a previously overlooked microbial methane sink in wetlands. Proceedings of the National Academy of Science. 2014;111:4495–4500. doi: 10.1073/pnas.1318393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siljanen HM, Saari A, Bodrossy L, Martikainen PJ. Effects of nitrogen load on the function and diversity of methanotrophs in the littoral wetland of a boreal lake. Frontiers in Microbiology. 2012;3:p. 39. doi: 10.3389/fmicb.2012.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peralta RM, Ahn C, Gillevet PM. Characterization of soil bacterial community structure and physicochemical properties in created and natural wetlands. Science of the Total Environment. 2012;443:725–732. doi: 10.1016/j.scitotenv.2012.11.052. [DOI] [PubMed] [Google Scholar]
- 11.Wilhelm RC, Niederberger TD, Greer C, Whyte LG. Microbial diversity of active layer and permafrost in an acidic wetland from the Canadian high arctic. Canadian Journal of Microbiology. 2011;57(4):303–315. doi: 10.1139/w11-004. [DOI] [PubMed] [Google Scholar]
- 12.Prasanna R, Nain L, Pandey AK, Saxena AK. Microbial diversity and multidimensional interactions in the rice ecosystem. Archives of Agronomy and Soil Science. 2012;58(7):723–744. [Google Scholar]
- 13.Farias ME, Contreras M, Rasuk MC, et al. Characterization of bacterial diversity associated with microbial mats, gypsum evaporites and carbonate microbialites in thalassic wetlands: tebenquiche and La Brava, Salar de Atacama, Chile. Extremophiles. 2014;18(2):311–329. doi: 10.1007/s00792-013-0617-6. [DOI] [PubMed] [Google Scholar]
- 14.Wang YF, Gu JD. Higher diversity of ammonia/ammonium-oxidizing prokaryotes in constructed freshwater wetland than natural coastal marine wetland. Applied Microbiology and Biotechnology. 2013;97(15):7015–7033. doi: 10.1007/s00253-012-4430-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt H, Eickhorst T. Spatio-temporal variability of microbial abundance and community structure in the puddled layer of a paddy soil cultivated with wetland rice (Oryza sativa L.) Applied Soil Ecology. 2013;72:93–102. [Google Scholar]
- 16.Decker CJ, Parker R. Analysis of double-stranded RNA from microbial communities identifies double-stranded RNA virus-like elements. Cell Reports. 2014;7(3):898–906. doi: 10.1016/j.celrep.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouali M, Pelletier E, Chaussonnerie S, le Paslier D, Bakhrouf A, Sghir A. Characterization of rhizosphere prokaryotic diversity in a horizontal subsurface flow constructed wetland using a PCR cloning-sequencing based approach. Applied Microbiology and Biotechnology. 2013;97(9):4221–4231. doi: 10.1007/s00253-012-4239-1. [DOI] [PubMed] [Google Scholar]
- 18.Vladár P, Rusznyák A, Márialigeti K, Borsodi AK. Diversity of sulfate-reducing bacteria inhabiting the rhizosphere of Phragmites australis in Lake Velencei (Hungary) revealed by a combined cultivation-based and molecular approach. Microbial Ecology. 2008;56(1):64–75. doi: 10.1007/s00248-007-9324-0. [DOI] [PubMed] [Google Scholar]
- 19.Zhang G, Tian J, Jiang N, Guo X, Wang Y, Dong X. Methanogen community in Zoige wetland of Tibetan plateau and phenotypic characterization of a dominant uncultured methanogen cluster ZC-I. Environmental Microbiology. 2008;10(7):1850–1860. doi: 10.1111/j.1462-2920.2008.01606.x. [DOI] [PubMed] [Google Scholar]
- 20.Kato S, Itoh T, Yamagishi A. Archaeal diversity in a terrestrial acidic spring field revealed by a novel PCR primer targeting archaeal 16S rRNA genes. FEMS Microbiology Letters. 2011;319(1):34–43. doi: 10.1111/j.1574-6968.2011.02267.x. [DOI] [PubMed] [Google Scholar]
- 21.Tang J, Ding X, Wang L, et al. Effects of wetland degradation on bacterial community in the Zoige Wetland of Qinghai-Tibetan Plateau (China) World Journal of Microbiology and Biotechnology. 2012;28(2):649–657. doi: 10.1007/s11274-011-0858-4. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z-Y, Xin Y-Z, Gao D-M, Li F-M, Morgan J, Xing B-S. Microbial community characteristics in a degraded wetland of the yellow river delta. Pedosphere. 2010;20(4):466–478. [Google Scholar]
- 23.Ma B, Lv X, Warren A, Gong J. Shifts in diversity and community structure of endophytic bacteria and archaea across root, stem and leaf tissues in the common reed, Phragmites australis, along a salinity gradient in a marine tidal wetland of northern China. Antonie van Leeuwenhoek International Journal of General and Molecular Microbiology. 2013;104:759–768. doi: 10.1007/s10482-013-9984-3. [DOI] [PubMed] [Google Scholar]
- 24.Yun J, Zhuang G, Ma A, Guo H, Wang Y, Zhang H. Community structure, abundance, and activity of methanotrophs in the Zoige wetland of the Tibetan Plateau. Microbial Ecology. 2012;63(4):835–843. doi: 10.1007/s00248-011-9981-x. [DOI] [PubMed] [Google Scholar]
- 25.Dorador C, Vila I, Remonsellez F, Imhoff JF, Witzel K-P. Unique clusters of Archaea in Salar de Huasco, an athalassohaline evaporitic basin of the Chilean Altiplano. FEMS Microbiology Ecology. 2010;73(2):291–302. doi: 10.1111/j.1574-6941.2010.00891.x. [DOI] [PubMed] [Google Scholar]
- 26.Wang YF, Feng YY, Ma XJ, Gu JD. Seasonal dynamics of ammonia/ammonium-oxidizing prokaryotes in oxic and anoxic wetland sediments of subtropical coastal mangrove. Applied Microbiology and Biotechnology. 2013;97(17):7919–7934. doi: 10.1007/s00253-012-4510-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scavino AF, Ji Y, Pump J, Klose M, Claus P, Conrad R. Structure and function of the methanogenic microbial communities in Uruguayan soils shifted between pasture and irrigated rice fields. Environmental Microbiology. 2013;15(9):2588–2602. doi: 10.1111/1462-2920.12161. [DOI] [PubMed] [Google Scholar]
- 28.Dorador C, Vila I, Witzel KP, Imhoff JF. Bacterial and archaeal diversity in high altitude wetlands of the Chilean Altiplano. Fundamental and Applied Limnology. 2013;182(2):135–159. [Google Scholar]
- 29.Khan MA. Environmental contamination of Hokersar Wetland Waters in Kashmir Himalayan Valley, India. Journal of Environmental Science and Engineering. 2010;52(2):157–162. [PubMed] [Google Scholar]
- 30.Frank-Fahle BA, Yergeau E, Greer CW, Lantuit H, Wagner D. Microbial functional potential and community composition in permafrost-affected soils of the NW Canadian Arctic. Plos ONE. 2014;9(1) doi: 10.1371/journal.pone.0084761.e84761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domingos SS, Dallas S, Skillman L, Felstead S, Ho G. Nitrogen removal and ammonia-oxidising bacteria in a vertical flow constructed wetland treating inorganic wastewater. Water Science and Technology. 2011;64(3):587–594. doi: 10.2166/wst.2011.565. [DOI] [PubMed] [Google Scholar]
- 32.Lin Y, Yin J, Wang J, Tian W. Performance and microbial community in hybrid Anaerobic Baffled Reactor-constructed wetland for nitrobenzene wastewater. Bioresource Technology. 2012;118:128–135. doi: 10.1016/j.biortech.2012.05.056. [DOI] [PubMed] [Google Scholar]
- 33.Yu Y, Wang H, Liu J, et al. Shifts in microbial community function and structure along the successional gradient of coastal wetlands in Yellow River Estuary. European Journal of Soil Biology. 2012;49:12–21. [Google Scholar]
- 34.Wang H Sheng Y, He Y, Wu J, Jiang Y, Tam N, Zhou H. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Applied and Environmental Microbiology. 2012;78(23):8264–8271. doi: 10.1128/AEM.01821-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yergeau E, Lawrence JR, Sanschagrin S, Waiser MJ, Korber DR, Greer CW. Next-generation sequencing of microbial communities in the Athabasca River and its tributaries in relation to oil sands mining activities. Applied and Environmental Microbiology. 2012;78(21):7626–7637. doi: 10.1128/AEM.02036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lassmann T, Sonnhammer ELL. Kalign—an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics. 2005;6, article 298 doi: 10.1186/1471-2105-6-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cole JR, Chai B, Farris RJ, et al. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Research. 2005;33:D294–D296. doi: 10.1093/nar/gki038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen DL, Townsend RR. Is there added value to adding ARB to ACE inhibitors in the management of CKD? Journal of the American Society of Nephrology. 2009;20(8):1666–1668. doi: 10.1681/ASN.2008040381. [DOI] [PubMed] [Google Scholar]
- 39.Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma B, Gong J. A meta-analysis of the publicly available bacterial and archaeal sequence diversity in saline soils. World Journal of Microbiology & Biotechnology. 2013;29(12):2325–2334. doi: 10.1007/s11274-013-1399-9. [DOI] [PubMed] [Google Scholar]
- 41.Nelson MC, Morrison M, Yu Z. A meta-analysis of the microbial diversity observed in anaerobic digesters. Bioresource Technology. 2011;102(4):3730–3739. doi: 10.1016/j.biortech.2010.11.119. [DOI] [PubMed] [Google Scholar]
- 42.Roadcap GS, Sanford RA, Jin Q, Pardinas JR, Bethke CM. Extremely alkaline (pH > 12) ground water hosts diverse microbial community. Ground Water. 2006;44(4):511–517. doi: 10.1111/j.1745-6584.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 43.dos Santos HF, Cury JC, do Carmo FL, et al. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: bacterial proxies for oil pollution. PLoS ONE. 2011;6(3) doi: 10.1371/journal.pone.0016943.e16943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Imfeld G, Aragonés CE, Fetzer I, et al. Characterization of microbial communities in the aqueous phase of a constructed model wetland treating 1,2-dichloroethene-contaminated groundwater. FEMS Microbiology Ecology. 2010;72(1):74–88. doi: 10.1111/j.1574-6941.2009.00825.x. [DOI] [PubMed] [Google Scholar]
- 45.Miletto M, Loy A, Antheunisse AM, Loeb R, Bodelier PLE, Laanbroek HJ. Biogeography of sulfate-reducing prokaryotes in river floodplains. FEMS Microbiology Ecology. 2008;64(3):395–406. doi: 10.1111/j.1574-6941.2008.00490.x. [DOI] [PubMed] [Google Scholar]
- 46.Li YH, Zhu JN, Zhai ZH, Zhang Q. Endophytic bacterial diversity in roots of Phragmites australis in constructed Beijing Cuihu Wetland (China) FEMS Microbiology Letters. 2010;309(1):84–93. doi: 10.1111/j.1574-6968.2010.02015.x. [DOI] [PubMed] [Google Scholar]
- 47.Sanford RA, Wagner DD, Wu Q, et al. Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(48):19709–19714. doi: 10.1073/pnas.1211238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mori K, Suzuki K-I, Urabe T, et al. Thioprofundum hispidum sp. nov., an obligately chemolithoautotrophic sulfur-oxidizing gammaproteobacterium isolated from the hydrothermal field on Suiyo Seamount, and proposal of Thioalkalispiraceae fam. nov. in the order Chromatiales . International Journal of Systematic and Evolutionary Microbiology. 2011;61, part 10:2412–2418. [Google Scholar]
- 49.Wu J, Zhang J, Jia W-L, Xie H-J, Gu RR. Nitrous oxide fluxes of constructed wetlands to treat sewage wastewater. Huan Jing Ke Xue. 2009;30(11):3146–3151. [PubMed] [Google Scholar]
- 50.Tietz A, Hornek R, Langergraber G, Kreuzinger N, Haberi R. Diversity of ammonia oxidising bacteria in a vertical flow constructed wetland. Water Science and Technology. 2007;56(3):241–247. doi: 10.2166/wst.2007.505. [DOI] [PubMed] [Google Scholar]
- 51.Li C-H, Zhou H-W, Wong Y-S, Tam NF-Y. Vertical distribution and anaerobic biodegradation of polycyclic aromatic hydrocarbons in mangrove sediments in Hong Kong, South China. Science of the Total Environment. 2009;407(21):5772–5779. doi: 10.1016/j.scitotenv.2009.07.034. [DOI] [PubMed] [Google Scholar]
- 52.Liu M, Li YH, Liu Y, et al. Flavobacterium phragmitis sp. nov., an endophyte of reed (Phragmites australis) International Journal of Systematic and Evolutionary Microbiology. 2011;61, part 11:2717–2721. doi: 10.1099/ijs.0.027417-0. [DOI] [PubMed] [Google Scholar]
- 53.Xiao Y-P, Hui W, Lee J-S, Lee KC, Quan Z-X. Flavobacterium dongtanense sp. nov., isolated from the rhizosphere of a wetland reed. International Journal of Systematic and Evolutionary Microbiology. 2011;61, part 2:343–346. doi: 10.1099/ijs.0.022301-0. [DOI] [PubMed] [Google Scholar]
- 54.Dong X, Reddy GB. Nutrient removal and bacterial communities in swine wastewater lagoon and constructed wetlands. Journal of Environmental Science and Health A: Toxic/Hazardous Substances and Environmental Engineering. 2010;45(12):1526–1535. doi: 10.1080/10934529.2010.506109. [DOI] [PubMed] [Google Scholar]
- 55.Jin G, Kelley TR. Characterization of microbial communities in a pilot-scale constructed wetland using PLFA and PCR-DGGE analyses. Journal of Environmental Science and Health A: Toxic/Hazardous Substances and Environmental Engineering. 2007;42(11):1639–1647. doi: 10.1080/10934520701518125. [DOI] [PubMed] [Google Scholar]
- 56.Naether A, Foesel BU, Naegele V, et al. Environmental factors affect Acidobacterial communities below the subgroup level in grassland and forest soils. Applied and Environmental Microbiology. 2012;78(20):7398–7406. doi: 10.1128/AEM.01325-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang G, Zhao Q, Jiao Y, Wang K, Lee D-J, Ren N. Efficient electricity generation from sewage sludge usingbiocathode microbial fuel cell. Water Research. 2012;46(1):43–52. doi: 10.1016/j.watres.2011.10.036. [DOI] [PubMed] [Google Scholar]
- 58.Hamdan LJ, Coffin RB, Sikaroodi M, Greinert J, Treude T, Gillevet PM. Ocean currents shape the microbiome of Arctic marine sediments. ISME Journal. 2012;7(4):685–696. doi: 10.1038/ismej.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi O, Das A, Yu C-P, Hu Z. Nitrifying bacterial growth inhibition in the presence of algae and cyanobacteria. Biotechnology and Bioengineering. 2010;107(6):1004–1011. doi: 10.1002/bit.22860. [DOI] [PubMed] [Google Scholar]
- 60.Barberán A, Casamayor EO. Global phylogenetic community structure and β-diversity patterns in surface bacterioplankton metacommunities. Aquatic Microbial Ecology. 2010;59(1):1–10. [Google Scholar]
- 61.Yu K, Zhang T. Metagenomic and metatranscriptomic analysis of microbial community structure and gene expression of activated sludge. Plos ONE. 2012;7(5) doi: 10.1371/journal.pone.0038183.e38183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Utsumi M, Belova SE, King GM, Uchiyama H. Phylogenetic comparison of methanogen diversity in different wetland soils. Journal of General and Applied Microbiology. 2003;49(2):75–83. doi: 10.2323/jgam.49.75. [DOI] [PubMed] [Google Scholar]
- 63.Høj L, Olsen RA, Torsvik VL. Archaeal communities in High Arctic wetlands at Spitsbergen, Norway (78°N) as characterized by 16S rRNA gene fingerprinting. FEMS Microbiology Ecology. 2005;53(1):89–101. doi: 10.1016/j.femsec.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 64.González-Toril E, Águilera Á, Souza-Egipsy V, Pamo EL, España JS, Amils R. Geomicrobiology of La Zarza-Perrunal acid mine effluent (Iberian Pyritic Belt, Spain) Applied and Environmental Microbiology. 2011;77(8):2685–2694. doi: 10.1128/AEM.02459-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Costa KC, Navarro JB, Shock EL, Zhang CL, Soukup D, Hedlund BP. Microbiology and geochemistry of great boiling and mud hot springs in the United States Great Basin. Extremophiles. 2009;13(3):447–459. doi: 10.1007/s00792-009-0230-x. [DOI] [PubMed] [Google Scholar]
- 66.Rivière D, Desvignes V, Pelletier E, et al. Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME Journal. 2009;3(6):700–714. doi: 10.1038/ismej.2009.2. [DOI] [PubMed] [Google Scholar]