

Abstract
Rheumatoid arthritis (RA) is an inflammatory autoimmune disorder; abnormal T cell immunity plays a critical role in the development of RA. Recently, A20 was identified as a key negative regulator for T cell activation and inflammatory signaling and may be involved in RA pathogenesis. In this study, we analyzed the expression level of A20, NF-κB, and A20 regulatory factor mucosa-associated lymphoid tissue lymphoma translocation gene 1 (MALT1) in patients with RA. Real-time PCR was used to determine the expression level of MALT1, MALT-V1, A20, and NF-κB genes in RA and healthy individuals (HI). Significantly lower A20 expression was found in RA patients compared with those in the healthy group, while NF-κB overexpression could be detected in patients with RA. Moreover, the MALT1 and MALT1-V1 expression level was downregulated in RA patients. A positive correlation between MALT1 and A20 and MALT1-V1 and A20 was found in patients with RA, and a tendency towards a negative correlation was found between MALT1 and NF-κB, MALT1-V1 and NF-κB, and A20 and NF-κB. In conclusion, we first characterized the alternative expression pattern of MALT1, A20, and NF-κB in RA, which may be related to abnormal T cell activation.
1. Introduction
Rheumatoid arthritis (RA) is a common, chronic, systemic, and inflammatory autoimmune disorder that primarily affects the small diarthrodial joints of the hands and feet, affecting approximately 1% of the world's population [1–3]. The main characteristics of this disease are synovium hyperplasia, lymphocyte infiltration, and the abnormal proliferation of fibroblast-like synoviocytes (FLS) that can lead to the destruction of bone and cartilage and eventual disability [4]. Abnormal T cell immunity plays a critical role in the development of RA. Numerous factors that are involved in alternative T cell activation have been characterized, including the activation of inflammatory cells and expression of various cytokines. Inflammatory mediators, such as interleukin-6 (IL-6), interleukin-1 (IL-1), and tumor necrosis factor-α (TNF-α), are abundant in synovial tissues and fluid from patients with RA, and the overexpression of these cytokines promotes chronic inflammation and joint destruction. Many of the inflammatory mediators involved in the pathology of RA are regulated by nuclear factor kappa B (NF-κB) transcription factors [5, 6]. Abnormal NF-κB activation occurs during many pathological conditions including allergic and autoinflammatory diseases and malignancies [7].
Recently, A20, a negative regulator of NF-κB, was identified as a key regulator for inflammation signaling and may be involved in RA pathogenesis [8]. A20 has been reported to be ubiquitin-editing enzyme with several functions. A20 is also known as tumor necrosis factor-α- (TNF-α-) induced protein 3 (TNFAIP3), which was first discovered in 1990 by Dixit and colleagues as a cytokine-induced gene in human umbilical vein endothelial cells [9, 10]. Subsequent studies demonstrated that A20 overexpression inhibits NF-κB activation in response to different stimuli [11–13]. The cloning and characterization of the A20 promoter revealed two NF-κB DNA binding elements, which are recognition sequences for NF-κB transcription factors. It was also found that multiple NF-κB activating stimuli induce A20 expression via NF-κB sites in the A20 promoter [14]. Therefore, A20 has been demonstrated to downregulate its own expression, and it has been proposed that A20 participates in a negative feedback loop to attenuate TNF-α-induced inflammatory responses. A20 overexpression was subsequently demonstrated to block the NF-κB activation mediated by TNF-α, IL-1, LPS, phorbol esters, and hydrogen peroxide in different cell types [11, 12, 15–18]. This inhibition is most likely due to the inhibition of NF-κB activation in endothelial cells in response to proinflammatory stimuli, and an antiproliferative effect on smooth muscle cells has been observed upon A20 overexpression in vitro. All of these findings suggest that A20 attenuates the activity of proximal signaling complexes at proinflammatory receptors [19–21].
A20 is regulated by the CARMA1-Bcl-10-MALT1 (mucosa-associated lymphoid tissue lymphoma translocation gene 1) upstream signaling pathway complex, which bridges T cell antigen receptor (TCR) signaling with the canonical IκB kinase (IKK)/NF-κB pathway [20, 22–25]. TCR stimulation induces the recruitment of A20 and the Bcl-10 adaptor protein into the MALT1 complex, leading to MALT1-mediated A20 processing. Similarly, API2-MALT1 expression results in A20 cleavage. MALT1 cleaves A20 at arginine 439 and impairs its NF-κB inhibitory function. Therefore, A20 was identified as a MALT1 substrate, emphasizing the importance of the MALT1 proteolytic activity in “fine-tuning” T cell antigen receptor signaling [26].
A20 dysfunction by deletion or mutation was identified in numerous lymphocytic malignancies [27]. Recently, polymorphisms in the A20 region were reported in autoimmune diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Crohn's disease, and psoriasis. Single nucleotide polymorphisms in the A20 region, including rs13192841, rs2230926, and rs6922466, have been independently associated with increased susceptibility for SLE [28, 29], and this finding provides a critical link between A20 and the etiology of SLE. More recently, it was shown that A20 deficiency in myeloid cells triggers erosive polyarthritis, resembling RA in a myeloid-specific, A20-deficient mice model [30]. There are three strongly associated genetic variants, including rs6920220, rs6927127, and rs6933404, which result in an A20 functional decrease in RA [8].
The etiology of RA remains to be understood; however, A20 deficiency may be a pivotal regulator for inflammation in RA. To characterize the role of A20 in RA, we analyzed the expression level of A20, NF-κB, and the A20 regulatory factor MALT1 in samples from Chinese patients with RA in this study.
2. Materials and Methods
2.1. Samples
This study included 16 patients with untreated RA (age: 26–71 years) and 20 healthy individuals (age: 22–70 years) who served as controls. The diagnosis of RA was based on the American College of Rheumatology criteria and expert opinion (1987 ACR criteria). All patients with RA were assessed for clinical disease activity by a trained rheumatologist using the disease activity score (DAS) [28]. The most recent erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and rheumatoid factor (RF) were collected (Table 1) [31].
Table 1.
Characteristics of RA samples.
| Patient number | Gender | Age | Disease duration (mo) | RF (IU/mL) | ESR (mm/h) | CRP (mg/L) | DAS28 scores | CCP status |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 17 | 48 | 252.00 | 73 | 38.90 | 6.00 | + |
| 2 | F | 56 | 24 | 1940.00 | 111 | 7.40 | 6.78 | + |
| 3 | F | 51 | 2 | 17.50 | 77 | 37.70 | 7.09 | + |
| 4 | F | 43 | 60 | 74.30 | 37 | 5.34 | 5.53 | + |
| 5 | F | 62 | 72 | 38.30 | 69 | 17.00 | 6.88 | + |
| 6 | F | 32 | 120 | 19.90 | 85 | 9.06 | 7.51 | ND |
| 7 | F | 60 | 6 | 150.00 | 90 | 103.00 | 6.09 | ND |
| 8 | F | 26 | 9 | 31.00 | 32 | 1.84 | 4.31 | + |
| 9 | F | 53 | 6 | 368.00 | 89 | 31.90 | 7.41 | − |
| 10 | F | 54 | 9 | 9.19 | 64 | 33.25 | 7.39 | − |
| 11 | F | 45 | 12 | 102.00 | 82 | 68.98 | 6.26 | + |
| 12 | F | 53 | 240 | 65.30 | 76 | 27.08 | 7.91 | + |
| 13 | F | 71 | 12 | 58.90 | 110 | 71.35 | 6.79 | + |
| 14 | F | 63 | 12 | 153.00 | 32 | 4.84 | 5.23 | + |
| 15 | F | 33 | 84 | 299.00 | 42 | 18.70 | 5.61 | + |
| 16 | F | 60 | 6 | 10.10 | 41 | 0.57 | 6.63 | − |
Note: mo: months; F: female; +: positive; −: negative; ND: no detection.
All of the procedures were conducted according to the guidelines of the Medical Ethics Committee of the Health Bureau of Guangdong Province of China. Peripheral blood samples were collected by heparin anticoagulation, and peripheral blood mononuclear cells (PBMCs) were isolated using the Ficoll-Hypaque gradient centrifugation method. RNA extraction and cDNA synthesis were performed according to the manufacturer's instructions.
2.2. Quantitative Real-Time RT-PCR (qRT-PCR)
The sequences of the primers for MALT1, A20, and NF-κB gene amplification are listed in Table 2. According to the structure of the MALT1 gene, there are two variants, that is, MALT1-V1 and MALT1-V2, and the latter contains a 33 bp deletion located between exons 6 and 8. To amplify the two MALT1 transcript variants, the MALT-V1-for and MALT-V1-rev primer pair was designed for MALT1-V1 amplification to cover the region that is missing in MALT1-V2, and the MALT1-for and MALT1-rev primer pair was designed to amplify the conserved region, which is contained by both variants.
Table 2.
List of primer information.
| Primer | Sequences | Accession number | PCR products |
|---|---|---|---|
| A20-for | 5′-CTGGGACCATGGCACAACTC-3′ | NM_006290 | 182 bp |
| A20-rev | 5′-CGGAAGGTTCCATGGGATTC-3′ | ||
| MALT1-V1-for | 5′-AAGCCCTATTCCTCACTACCAG-3′ | NM_006785.2 | 195 bp |
| MALT1-V1-rev | 5′-CACTCCACTGCCTCATCTGTTC-3′ | ||
| MALT1-for | 5′-TCTTGGCTGGACAGTTTGTGA-3′ | NM_006785.2 | 230 bp |
| MALT1-rev | 5′-GCTCTCTGGGATGTCGCAA-3′ | ||
| NF-κB-for | 5′-CCACAAGACAGAAGCTGAAG-3′ | NM_003998 | 149 bp |
| NF-κB-rev | 5′-AGATACTATCTGTAAGTGAACC-3′ | ||
| β2M-for | 5′-TACACTGAATTCACCCCCAC-3′ | J00105 | 145 bp |
| β2M-rev | 5′-CATCCAATCCAAATGCGGCA-3′ |
The expression level of the A20, MALT1, MALT1-V1, NF-κB, and β2-microglobulin (β2M) genes was determined by SYBR Green I real-time PCR. Briefly, PCR in a 20 μL total volume was performed with approximately 1 μL of cDNA, 0.5 μM of each primer pair, 9 μL of 2.5 × Real Master Mix (Tiangen Biotech (Beijing) Co., Ltd., Beijing, China), and 9 μL of dH2O. After initial denaturation at 95°C for 15 minutes, 45 cycles of the following procedure were performed: 30 seconds at 95°C and 40 seconds at 60°C for the β2M, MALT1-V1, MALT1, A20, and NF-κB genes. The plate was read immediately after the 60°C step using an MJ Research DNA Engine Opticon 2 PCR cycler (Bio-Rad, Hercules, CA, USA) [32]. The relative amount of the genes of interest and β2M reference gene was measured in two independent assays. The specific, amplified PCR products were analyzed by melting curve analysis. The data are presented as the relative expression of the genes of interest compared with the internal control gene as determined by the 2(−ΔCT) method [33]. In addition, to analyze the MALT1-V1 expression characteristics, we calculated the MALT1-V1 expression ratio as MALT1-V1/MALT1 × 100%.
2.3. Statistical Analysis
Two independent-samples Wilcoxon tests were performed to compare the median expression level for each gene between patients with RA and the control group. Pearson correlation and linear regression analyses were used to determine the association between different genes in different groups. A P < 0.05 was considered statistically significant [34].
3. Results and Discussions
Abnormal T cell activation is a common feature of RA [35], and the upregulation of some positive regulating factors such as TNF-α, IL-6, and IL-2 was identified during the initiation of RA. In contrast, the downregulation of negative regulatory factors has the same effect on initiation of RA [16]. For example, T cell activation leads to the downregulation of A20 expression in mature thymocytes and peripheral T cells [9, 10]. Recently, abnormal A20 expression was described in patients with RA [8], and decreased A20 results in increased NF-κB expression and enhanced inflammation [25]. In this study, we analyzed the expression of A20 in 16 patients with RA in the active phase, and a significantly lower expression of A20 (median: 6.530) was found compared with those in the healthy group (median: 44.614, P < 0.001) (Figure 1(a)). These results are similar to findings of different reports examining mouse models or patients with RA [8, 36]. A20-deficient mice develop severe multiorgan inflammation. Moreover, it is well accepted that the activation of NF-κB-dependent gene expression plays a key role in the development of RA [30]; thus, decreased A20 expression may be a key reason for NF-κB overexpression in RA. Our study also demonstrated that NF-κB overexpression could be detected in patients with RA (median: 0.798) in comparison with healthy controls (median: 0.605, P = 0.042) (Figure 1(b)), indicating that decreased A20 resulting in NF-κB overexpression is also a common feature for Chinese patients with RA.
Figure 1.
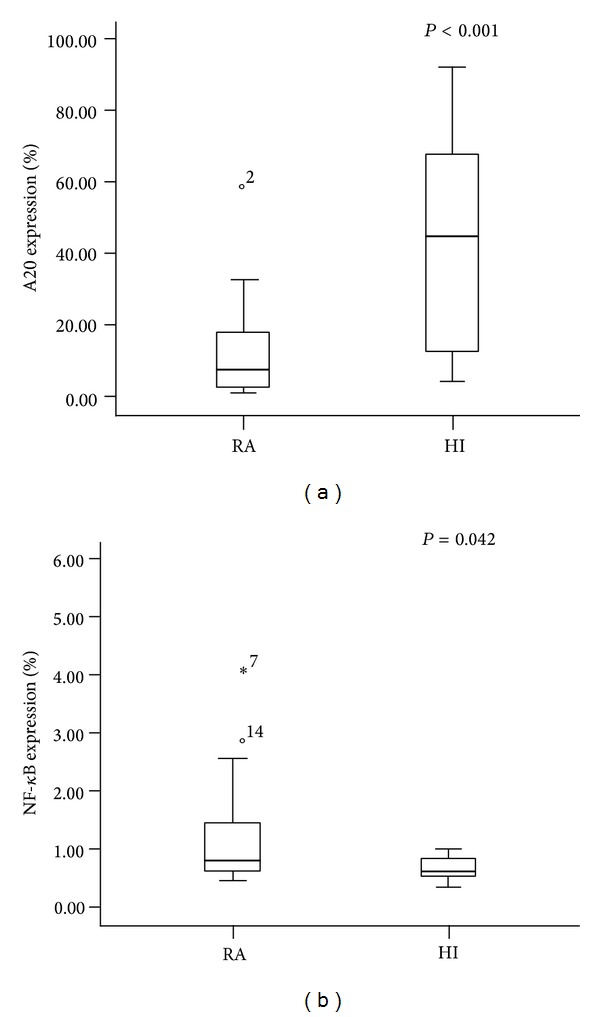
The expression level of A20 and NF-κB in patients with RA and healthy individuals.
Lower A20 expression is associated with polymorphisms in the A20 genomic locus [29]; however, whether there is any dysregulation in A20 by upstream pathway factors is unknown. MALT1 is an upstream A20 pathway factor that cleaves A20 at arginine 439 and impairs its NF-κB inhibitory function. To characterize the relationship between MALT1 and A20 expression, we also examined the expression level of MALT1. Interestingly, the MALT1 expression level is downregulated in patients with RA (median: 0.541) compared with those in the healthy group (median: 1.638, P < 0.001) (Figure 2(a)). This result appears to be inconsistent with the lower A20 and higher NF-κB expression level results because MALT1 is also a positive regulatory factor of NF-κB. It is known that there are two MALT1 variants, MALT1-V1 and MATL1-V2, according to data in GenBank, and our previous study has found that these MALT1 variants could be identified by RT-PCR and sequencing (data not shown). In addition, we analyzed the expression level of the different variants, and similar results were found including the fact that a significantly lower MALT1-V1 expression level was detected in patients with RA (median: 0.062) compared with healthy controls (median: 0.140, P < 0.001) (Figure 2(b)).
Figure 2.
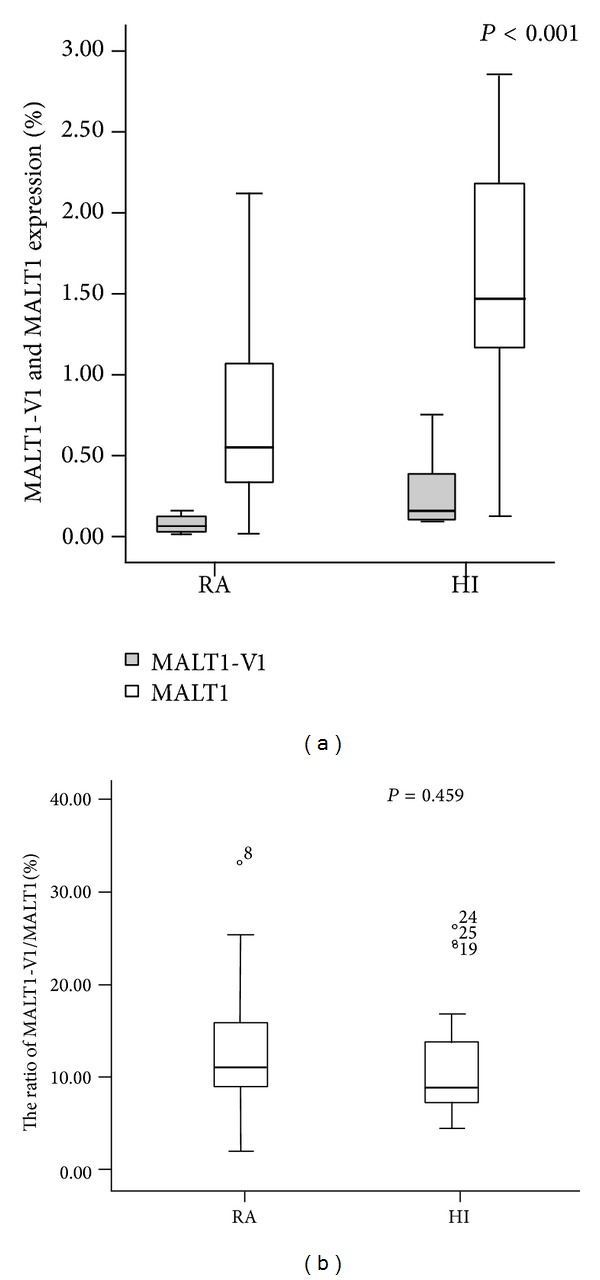
The expression level of MALT1-V1 and total MALT1 in patients with RA and healthy individuals.
Because we could not directly amplify MALT1-V2, which contains a 33 bp deletion, the expression level of MALT1-V2 could only be indirectly calculated by the relative expression of MALT1-V1/total MALT1 [37], and there are no significant differences in the ratio of MALT1-V1/total MALT1 between patients with RA and healthy controls (13.43 ± 7.98% versus 11.76 ± 6.66%), implying that the MALT1-V2 expression level was also downregulated in RA. Overall, either MALI1-V1 or MALT1-V2 was decreased in RA, unlike the finding in T cells from acute myeloid leukemia (AML), in which we found that the MALT1-V1 expression level was significantly higher in T cells from AML patients compared with healthy controls, while the MALT1-V2 expression level was downregulated [37]; this may indicate different expression pattern of these two MALT1 variants in RA. Little is known about the functional difference between the variants, and whether this is a feedback response from the expression pattern of A20 and NF-κB in RA is unknown because the impact of A20 cleavage by MALT1 on its capacity to regulate NF-κB has only been partially elucidated [24]. Further investigation is needed to characterize the upstream pathway regulators of A20 in addition to MALT1.
The role of MALT1 in the development of inflammation is largely unknown for RA and other autoimmune diseases, and only one study has reported that patients who had MALT-type lymphomas may also suffer from rheumatoid arthritis due to MALT1 dysfunction and continuous NF-κB activation [38]. A study has reported that MALT1 rearrangement in gastric MALT lymphoma is frequently associated with Sjögren's syndrome [39]. More recently, Brüstle A and coworkers indicated that MALT1 is a central cell intrinsic factor that determines the experimental autoimmune encephalitogenic potential of inflammatory Th17 cells in vivo [40]. It appears that MALT1 may play a role in the development of autoimmune diseases, and it may interfere with specific T cell subsets. Thus, our finding of lower MALT1 expression may imply a loss of the control of T cell activation in some T cell subsets in RA, which remains an open question. Further studies are needed to investigate the pathways upstream of MALT1 and TCR-CD3 signaling in different T cell subsets in RA.
In general, A20 is cleaved by MALT1; thus, the expression level of both genes should be negatively correlated with the expression pattern of A20 and MALT1 [24]. However, we found a positive correlation between MALT1 and A20 (r = 0.520, P = 0.039) (Figure 3(a)) and MALT1-V1 and A20 (r = 0.511, P = 0.043) (Figure 3(b)) in RA, and a tendency towards a negative correlation was found between MALT1 and NF-κB (r = −0.098, P = 0.718), MALT1-V1 and NF-κB (r = −0.204, P = 0.448), and A20 and NF-κB (r = −0.264, P = 0.322), indicating that the MALT1-A20-NF-κB expression pattern may be more complex in RA.
Figure 3.
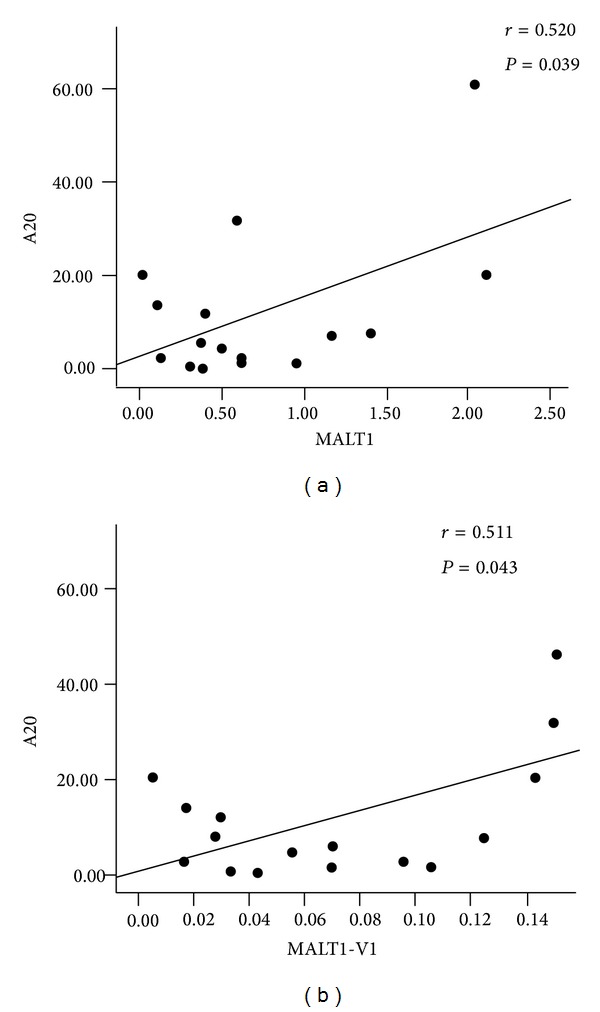
Correlation between the gene expression levels of MALT1 and A20 (a) and MALT1-V1 and A20 (b) in patients with RA.
In conclusion, we characterized, for the first time, the alternative expression pattern of MALT1, A20, and NF-κB in RA, which may be related to abnormal T cell activation. Lacking A20 and MALT1 dysfunction are common characteristics of Chinese patients with RA, and our results provide new inflammation targets to consider for RA treatment. Moreover, further investigation is needed to follow up on patients with different MALT1-A20-NF-κB expression patterns and their association with cancer development.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (no. 91129720) and a Collaborated Grant for HK-Macao-TW of the Ministry of Science and Technology (2012DFH30060).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Yangqiu Li contributed to the concept development and study design. Xu Wang, Lihua Zhu, Ziwei Liao, and Fan Zhang performed the real-time PCR. Ling Xu, Yan Xu, Shaohua Chen, and Lijian Yang performed the PBMC isolation, RNA extraction, and cDNA synthesis. Lihua Zhu and Yi Zhou were responsible for the collection of clinical data. Yangqiu Li, Xu Wang, Lihua Zhu, and Ziwei Liao coordinated the study and helped draft the paper. All authors read and approved the final paper. Xu Wang, Lihua Zhu, and Ziwei Liao contributed equally to this paper.
References
- 1.Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85(3):307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 2.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 3.Vinay DS, Kwon BS. Targeting TNF superfamily members for therapeutic intervention in rheumatoid arthritis. Cytokine. 2012;57(3):305–312. doi: 10.1016/j.cyto.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Cooles FA, Isaacs JD. Pathophysiology of rheumatoid arthritis. Current Opinion in Rheumatology. 2011;23(3):233–240. doi: 10.1097/BOR.0b013e32834518a3. [DOI] [PubMed] [Google Scholar]
- 5.Campbell IK, Gerondakis S, O’Donnell K, Wicks IP. Distinct roles for the NF-κB1 (p50) and c-Rel transcription factors in inflammatory arthritis. Journal of Clinical Investigation. 2000;105(12):1799–1806. doi: 10.1172/JCI8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okamoto H, Kobayashi A. Tyrosine kinases in rheumatoid arthritis. Journal of Inflammation. 2011;8:p. 21. doi: 10.1186/1476-9255-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwai K. Diverse ubiquitin signaling in NF-κB activation. Trends in Cell Biology. 2012;22(7):355–364. doi: 10.1016/j.tcb.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Elsby LM, Orozco G, Denton J, Worthington J, Ray DW, Donn RP. Functional evaluation of TNFAIP3 (A20) in rheumatoid arthritis. Clinical and Experimental Rheumatology. 2010;28(5):708–714. [PMC free article] [PubMed] [Google Scholar]
- 9.Opipari AW, Jr., Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor α encodes a novel type of zinc finger protein. Journal of Biological Chemistry. 1990;265(25):14705–14708. [PubMed] [Google Scholar]
- 10.Dixit VM, Green S, Sarma V, et al. Tumor necrosis factor-α induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. Journal of Biological Chemistry. 1990;265(5):2973–2978. [PubMed] [Google Scholar]
- 11.Jaattela M, Mouritzen H, Elling F, Bastholm L. A20 zinc finger protein inhibits TNF and IL-1 signaling. Journal of Immunology. 1996;156(3):1166–1173. [PubMed] [Google Scholar]
- 12.Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(13):6721–6725. doi: 10.1073/pnas.93.13.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tewari M, Wolf FW, Seldin MF, O’Shea KS, Dixit VM, Turka LA. Lymphoid expression and regulation of A20, an inhibitor of programmed cell death. Journal of Immunology. 1995;154(4):1699–1706. [PubMed] [Google Scholar]
- 14.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor α-inducible zinc finger protein, A20, is mediated by κB elements. Journal of Biological Chemistry. 1992;267(25):17971–17976. [PubMed] [Google Scholar]
- 15.Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKγ) upon receptor stimulation. Immunity. 2000;12(3):301–311. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 16.De Valck D, Heyninck K, Van Criekinge W, et al. A20 inhibits NF-kappaB activation independently of binding to 14-3-3 proteins. Biochemical and Biophysical Research Communications. 1997;238(2):590–594. doi: 10.1006/bbrc.1997.7343. [DOI] [PubMed] [Google Scholar]
- 17.Ferran C, Stroka DM, Badrichani AZ, et al. A20 inhibits NF-κB activation in endothelial cells without sensitizing to tumor necrosis factor-mediated apoptosis. Blood. 1998;91(7):2249–2258. [PubMed] [Google Scholar]
- 18.Heyninck K, De Valck D, Vanden Berghe W, et al. The zinc finger protein A20 inhibits TNF-induced NF-κB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-κB-inhibiting protein ABIN. Journal of Cell Biology. 1999;145(7):1471–1482. doi: 10.1083/jcb.145.7.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beyaert R, Heyninck K, Van Huffel S. A20 and A20-binding proteins as cellular inhibitors of nuclear factor- κB-dependent gene expression and apoptosis. Biochemical Pharmacology. 2000;60(8):1143–1151. doi: 10.1016/s0006-2952(00)00404-4. [DOI] [PubMed] [Google Scholar]
- 20.Hymowitz SG, Wertz IE. A20: from ubiquitin editing to tumour suppression. Nature Reviews Cancer. 2010;10(5):332–340. doi: 10.1038/nrc2775. [DOI] [PubMed] [Google Scholar]
- 21.Zhang F, Yang L, Li Y. The role of A20 in the pathogenesis of lymphocytic malignancy. Cancer Cell International. 2012;12(1):p. 44. doi: 10.1186/1475-2867-12-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu YC, Penninger J, Karin M. Immunity by ubiquitylation: a reversible process of modification. Nature Reviews Immunology. 2005;5(12):941–952. doi: 10.1038/nri1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Düwel M, Welteke V, Oeckinghaus A, et al. A20 negatively regulates T cell receptor signaling to NF-κB by cleaving Malt1 ubiquitin chains. Journal of Immunology. 2009;182(12):7718–7728. doi: 10.4049/jimmunol.0803313. [DOI] [PubMed] [Google Scholar]
- 24.Malinverni C, Unterreiner A, Staal J, et al. Cleavage by MALT1 induces cytosolic release of A20. Biochemical and Biophysical Research Communications. 2010;400(4):543–547. doi: 10.1016/j.bbrc.2010.08.091. [DOI] [PubMed] [Google Scholar]
- 25.Verstrepen L, Verhelst K, van Loo G, Carpentier I, Ley SC, Beyaert R. Expression, biological activities and mechanisms of action of A20 (TNFAIP3) Biochemical Pharmacology. 2010;80(12):2009–2020. doi: 10.1016/j.bcp.2010.06.044. [DOI] [PubMed] [Google Scholar]
- 26.Coornaert B, Baens M, Heyninck K, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-κB inhibitor A20. Nature Immunology. 2008;9(3):263–271. doi: 10.1038/ni1561. [DOI] [PubMed] [Google Scholar]
- 27.Kato M, Sanada M, Kato I, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459(7247):712–716. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- 28.Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nature Genetics. 2008;40(9):1062–1064. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vereecke L, Beyaert R, Van Loo G. Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochemical Society Transactions. 2011;39(4):1086–1091. doi: 10.1042/BST0391086. [DOI] [PubMed] [Google Scholar]
- 30.Matmati M, Jacques P, Maelfait J, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nature Genetics. 2011;43(9):908–912. doi: 10.1038/ng.874. [DOI] [PubMed] [Google Scholar]
- 31.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis and Rheumatism. 1988;31(3):315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 32.Qin Y, Tian H, Wang G, Lin C, Li Y. A BCR/ABL-hIL-2 DNA vaccine enhances the immune responses in BALB/c mice. BioMed Research International. 2013;2013:9 pages. doi: 10.1155/2013/136492.136492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zha X, Yan X, Shen Q, et al. Alternative expression of TCRζ related genes in patients with chronic myeloid leukemia. Journal of Hematology and Oncology. 2012;5:p. 74. doi: 10.1186/1756-8722-5-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmad R, Al-Mass A, Atizado V, et al. Elevated expression of the toll like receptors 2 and 4 in obese individuals: its significance for obesity-induced inflammation. Journal of Inflammation. 2012;9(1):p. 48. doi: 10.1186/1476-9255-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aggarwal A, Srivastava R, Agrawal S. T cell responses to citrullinated self-peptides in patients with rheumatoid arthritis. Rheumatology International. 2013;33(9):2359–2363. doi: 10.1007/s00296-013-2731-2. [DOI] [PubMed] [Google Scholar]
- 36.Hah YS, Lee Y, Jun J, et al. A20 suppresses inflammatory responses and bone destruction in human fibroblast-like synoviocytes and in mice with collagen-induced arthritis. Arthritis and Rheumatism. 2010;62(8):2313–2321. doi: 10.1002/art.27545. [DOI] [PubMed] [Google Scholar]
- 37.Shi L, Chen S, Lu Y, et al. Changes in the MALT1-A20-NF-κB expression pattern may be related to T cell dysfunction in AML. Cancer Cell International. 2013;13(1):p. 37. doi: 10.1186/1475-2867-13-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grønbæk K, Ralfkiaer E, Kalla J, Skovgaard GL, Guldberg P. Infrequent somatic Fas mutations but no evidence of Bcl10 mutations or t(11;18) in primary cutaneous MALT-type lymphoma. Journal of Pathology. 2003;201(1):134–140. doi: 10.1002/path.1426. [DOI] [PubMed] [Google Scholar]
- 39.Streubel B, Huber D, Wöhrer S, Chott A, Raderer M. Frequency of chromosomal aberrations involving MALT1 in mucosa-associated lymphoid tissue lymphoma in patients with Sjögren’s Syndrome. Clinical Cancer Research. 2004;10(2):476–480. doi: 10.1158/1078-0432.ccr-0873-03. [DOI] [PubMed] [Google Scholar]
- 40.Brüstle A, Brenner D, Knobbe CB, et al. The NF-κB regulator MALT1 determines the encephalitogenic potential of Th17 cells. Journal of Clinical Investigation. 2012;122(12):4698–4709. doi: 10.1172/JCI63528. [DOI] [PMC free article] [PubMed] [Google Scholar]