

Abstract
Lung adenocarcinoma, the most common type of lung cancer, has increased in recent years. Prognosis is still poor, and pathogenesis remains unclear. This study aimed to investigate the membrane protein profile differences between lung adenocarcinoma and normal tissue. Manual microdissection was used to isolate the target cells from tumor tissue and normal tissue. iTRAQ labeling combined with 2D-LC-MS/MS yielded a differential expression profile of membrane proteins. Bioinformatic analysis was performed using Gene Ontology, WEGO, PID, and KEGG. S100A14 protein was selectively verified by Western blotting. The relationship of S100A14 expression with clinicopathological features in lung cancer patients was evaluated using immunohistochemistry. As a result, 568 differential proteins were identified; 257 proteins were upregulated and 311 were downregulated. Of these proteins, 48% were found to be membrane bound or membrane associated. These proteins enable the physiological functions of binding, catalysis, molecular transduction, transport, and molecular structure. For these differential proteins, 35 pathways were significantly enriched through the Pathway Interaction Database, whereas 19 pathways were enriched via KEGG. The overexpression and cellular distribution of S100A14 in lung cancer were confirmed. We found that upregulation of S100A14 was associated with well or moderate differentiation. The iTRAQ-coupled 2D-LC-MS/MS technique is a potential method for comparing membrane protein profiles between tumor and normal tissue. Such analysis may also help in identifying novel biomarkers and the mechanisms underlying carcinogenesis.
Keywords: Lung adenocarcinoma, iTRAQ, mass spectrometry, membrane protein, proteomic profile
Introduction
Lung cancer is one of the most frequently diagnosed cancers and the leading cause of cancer death worldwide [1]. The 5-year survival of all lung cancer patients is only approximately 16% [2]. Lung cancer is divided into two classes: non-small cell lung cancer (NSCLC) and small cell lung cancer. NSCLC includes adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and other cell types. Lung adenocarcinoma is the most common type of lung cancer and has been increasing in recent years. Although many treatments are available, its prognosis is still poor. Smoking is the most common cause of lung cancer overall, but lung adenocarcinoma is the most frequently occurring cell type in nonsmokers, and its pathogenesis remains unclear.
Proteomics, particularly quantitative proteomics, is a powerful approach developed to identify differentially expressed proteins in response to normal or tumor tissue. Proteomics has the potential to reveal underlying molecular mechanisms of disease. However, the heterogeneity of tumor tissues limits the efficacy of proteomic analysis. Laser capture microdissection (LCM) technology separates target cells from heterogeneous tumor tissue, which improves the accuracy of proteomic analyses. Many studies have established the compatibility of this method with protein extraction and analysis [3,4]. Proteomic analysis requires a large amount of cells. LCM enables a very exact selection by isolating single cells, but getting enough material for a valid study consumes considerable time [5]. Manual microdissection is an ideal, cost-efficient method for situations where a clear demarcation between tumor and non-tumor tissues is obvious, as in the lung tumor samples in our study [6].
In recent years, proteomic studies have focused on subcellular analysis. Subcellular proteomic analysis has many advantages over whole-cell proteomic analysis as the former approach has a higher probability of detecting low-abundance proteins, which are suspected to play an important role in the development of cancer [7-9].
The cell membrane is involved in many biological functions, including small molecules transport, cell-cell and cell-substrate recognition and interaction, and cell signaling transduction and communications [10,11]. Membrane proteins account for approximately 30% of the whole cell proteome and are known to be involved in cell proliferation, cell adhesion, and tumor cell invasion. They are also pivotal to the development, growth, angiogenesis, and metastasis of tumors [12-14]. Analyzing membrane proteomes may help us understand carcinogenic mechanisms and promote the discovery of new potential tumor biomarkers and therapeutic targets.
iTRAQ (isobaric tags for relative and absolute quantification) is a powerful tool in quantitative proteomic analysis that has been widely applied in many studies [15-17]. The iTRAQ labeling technology greatly increased identification sensitivity and quantitation accuracy in proteomic analysis through a multiplexed quantitation strategy. Identifying low-abundant proteins such as cell membrane proteins has become feasible via iTRAQ [18,19].
In this study, we performed iTRAQ labeling followed by LC-MS/MS to identify differential protein expression profiles of cell membranes from pooled lung adenocarcinoma and matched normal lung tissue samples. These profiles were subjected to quantitative proteomics, whereby S100A14 was found to be overexpressed in lung adenocarcinoma compared with normal lung tissue. S100A14 expression was further confirmed in clinical samples by Western blotting. We then investigated the relationship of S100A14 expression with clinicopathological features and underlying molecular mechanisms in lung cancer patients.
Materials and methods
Human tissue samples
This study was approved by the local ethnics committee. Lung adenocarcinoma and matched adjacent normal lung tissue samples were obtained from 10 patients who underwent surgery at the Second Affiliated Hospital of the Medical School of Xi’an Jiaotong University. Adjacent normal tissue was obtained at least 5 cm away from the primary tumor. All patients with lung adenocarcinoma were confirmed by pathological diagnosis. None of the patients had received chemotherapy or radiotherapy before surgery. The patients signed informed consents. All samples were immediately snap-frozen in liquid nitrogen and stored at -80°C until analyzed. The 10 pairs of lung cancer and matched normal lung tissue were used for comparative proteomic analysis and Western blotting. For immunohistochemical analysis, the formalin-fixed and paraffin-embedded 62 lung cancer specimens (41 lung adenocarcinoma, 21 lung squamous cell carcinoma) and 24 normal lung tissues from surgical resections were obtained from the Second Affiliated Hospital, Xi’an Jiaotong University and Shaanxi Cancer Hospital for this retrospective study.
Manual microdissection
We performed manual microdissection to collect cells of interest from lung adenocarcinoma and matched adjacent normal lung tissue, as previously described by Nowak [6] and Chen [20]. Frozen sections (5 μm each) from lung adenocarcinoma and matched normal lung tissues were cut in a Microm HM500 Cryostat at -25°C and identified by routine H&E staining. Under the guidance of an H&E slide, the adjacent unstained 10 to 14 μm thick continuous frozen sections were dissected with a syringe needle and/or scalpel from the area identified by a pathologist, transferred to an Eppendorf tube, and stored at -80°C until ready for use.
Purification of cell membrane proteins
Membrane proteins were extracted as previously described by Li [21], with minor modifications. Briefly, microdissected tissue was minced on ice and manually homogenized with a glass homogenizer containing precool homogenization buffer (200 mM mannitol, 70 mM sucrose, 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.5 mM EGTA, 1 mM PMSF, 0.2 mM Na3VO4, and 1 mM NaF). The homogenate was centrifuged at 10,000 g at 4°C for 15 min. The supernatant was collected and ultracentrifuged at 100,000 g for 1 hour at 4°C to purify cell membrane. The supernatant was collected as a control and used to assess the purity of the extracted membrane proteins by Western blotting. The pellets were resuspended in lysis buffer (8 Murea, 30 mM HEPES, 0.5% SDS, 1 mM PMSF, 2 mM EDTA, and 10 mM DTT). The solution was dispersed by sonication for 5 min (power 180 W, pulse 2 s on and 3 s off), then centrifuged at 20,000 g for 30 min. The supernatant was collected and proteins assayed by the Bradford method. Purity was validated by Western blotting using a plasma membrane marker enzyme (Na+/K+ATPase) and a mitochondrial marker (prohibitin) [21].
Protein digestion and iTRAQ labeling
Protein digestion and iTRAQ labeling were performed according to the iTRAQ kit protocol (Applied Biosystems). The extracted membrane proteins were reduced with 10 mM DTT and alkylated with 55 mM IAM. They were then precipitated by cold acetone, stored at -20°C for 3 h, and concentrated by centrifuging at 20,000 g for 30 min. The precipitates were resuspended in solution buffer (50% TEAB, 0.1% SDS). Then 100 ug protein solutions were digested with 1 ug/ul trypsin solution at 37°C overnight and labeled with iTRAQ tags. The lung cancer and normal lung tissue samples were labeled with iTRAQ117 and iTRAQ118, respectively.
Strong cation exchange chromatography
The iTRAQ labeled peptides were mixed and then fractionated by Strong Cation Exchange (SCX) chromatography on an HPLC system (Agilent) using a Phenomenex Luna SCX Column (250 × 4.60 mm, 5 um 100A (Phenomenex). Buffer A (25% ACN, 10 mM KH2PO4, pH 3.0) and buffer B (25% ACN, 10 mM KH2PO4, 2 M KCl, pH 3.0) were used as mobile phases for gradient separation at a flow rate of 1 ml/min. The gradients were 0% B for 25 min, 0% to 5% B for 1 min, 5% to 30% B for 20 min, 30% to 50% B for 5 min, 50% B for 5 min, and 50% to 100% B for 5 min. After 100% B for 10 min, the process was stopped. Fractions collected at one-minute intervals were lyophilized in a vacuum concentrator and subsequently desalted using a strata-X C18 column.
NanoLC-MS/MS analysis
NanoLC-MS/MS analysis was performed on a QExactive Mass Spectrometer (Thermo Fisher Scientific) equipped with an UltiMate 3000 nanoHPLC system (Dionex). The desalted fractions were loaded onto a homemade analytical column [Venusil XBP, C18 (L), 75 um * 150 mm 5 μm, 150A, Agela Technologies] and separated using a mobile phase containing buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in acetonitrile). The flow rate used for separation was 400 nl/min. The gradient separation was 5% B for 10 min, 5% to 30% B for 30 min, 30% to 60% B for 5 min, 60% to 80% B for 3 min, 80% B for 7 min, 80% to 5% B for 3 min, and 5% B for 7 min. A full mass scan was performed in data-dependent mode using a QExactive Mass Spectrometer, with an acquired range of 350-2000 m/z at 70,000 resolution (m/z 200). The automated gain control (AGC) target value was 3.00 E+06 and max ion injection time (IT) was 50 ms. In MS scan, the top 20 most abundant ions in a charged state (+2-+7) were selected for tandem mass spectrometry by HCD fragmentation with normalized collision energy of 28% and an isolation width of 2.0 m/z. For subsequent MS2 scans, a resolving power of 17,500 at m/z 200 was used with an AGC target of 1 E+05 and a max ion IT of 100 ms. For each scan, dynamic exclusion was set to 15 s.
Identification and quantitation of membrane proteins
All raw spectra files were searched against UniProtKB/Swiss-Prot Homo sapiens using Proteome Discoverer (Thermo Fisher Scientific, Version 1.3) and Mascot Version 2. 3. Trypsin was used as the required enzyme, and one missed cleavage was allowed. Carbamidomethylation of cysteine was set as a fixed modification while for oxidation of methionine, Gln→Pyro-Glu (N-term Q), iTRAQ 8 plex labeling at N-terminal, K, and Y were used as variable modifications. The maximum mass deviation allowed for precursor mass was set to 15 ppm, and fragment ion tolerance was 0.02 Da. Protein quantification required at least one unique peptide. FDR less than 1% was deemed acceptable for both the peptide and protein level.
Bioinformatic analysis
The theoretical pI and molecular weight (MW) of the identified proteins were obtained for Swiss-Prot protein sequence data bank. Functional enrichment analysis was performed using Gene Ontology (http://www.geneontology.org/) and WEGO (http://wego.genomics.org.cn/) as described by Ye et al. [22]. Pathway analysis was performed by PID and KEGG; both assays proved statistically significant with p-values less than .01 and .05, respectively.
Western blotting analysis
Western blotting analysis was performed for 10 pairs of fresh lung adenocarcinoma and normal lung tissue. A measured amount (60 μg) of protein was separated by SDS-PAGE and transferred to membranes. The membranes were blocked with 5% nonfat dry milk in TBST buffer for 2 h at room temperature, incubated with anti-S100A14 antibody (1:400) overnight at 4°C, washed in TBST, and incubated again with horseradish peroxidase-conjugated secondary antibody (1:4000) for 2 h at room temperature. The immunoreactive protein bands were visualized by enhanced chemiluminescence and evaluated by densitometry using Image J software. β-actin was used as a loading control.
Immunohistochemistry and staining evaluation
Expression levels of S100A14 were determined using the standard SP immunohistochemical technique. Paraffin embedded specimens (each 4 μm) were dewaxed, rehydrated in a series of ethanol solutions, and treated with an antigen retrieval solution with a microwave. Endogenous peroxidase activity was blocked using 3% H2O2 for 10 min. Unspecific staining was blocked for 15 min using normal goat serum. The sections were then incubated with anti-S100A14 antibody (1:200) overnight at 4°C. Immunohistochemistry (IHC) was performed via the SP9001 Rabbit kit (Zhongshan Jinqiao Biotech Company, Beijing, China) according to manufacturer’s instructions. The immunoreaction was visualized using 3, 3’-diaminobenzidine (DAB) staining. Sections were counterstained with hematoxylin, dehydrated, and then mounted with coverslips. The primary antibody was replaced with PBS as the negative control. All sections were examined microscopically and blindly evaluated by two independent pathologists according to a scoring method described previously by Zhang [23]. At least 5 high-power fields were selected randomly, with >200 cells counted per field. Each specimen was assessed with reference to staining intensity and positively stained area. Staining intensity was graded on the following scale: 0, no staining; 1, light yellow; 2, yellowish brown; 3, brown. The positively stained area was evaluated as follows: 0, no staining; 1, <10% stained positive; 2, 11%-50% stained positive; 3, 51%-80% stained positive, and 4, >80% stained positive. The combined staining score (staining intensity times staining area) was then graded as 0, negative immunoreactivity; 1-4, low immunoreactivity; and >4, high immunoreactivity.
Statistical analysis
Statistical analysis was carried out using SPSS (Version 16.0; Chicago, IL, USA). The relationship between S100A14 expression levels and clinicopathological parameters was analyzed using the Mann-Whitney U or Kruskal-Wallis test. S100A14 protein expression levels between lung cancer and normal tissue were also analyzed using Mann-Whitney U. In all tests, two-sided p-values <0.05 were considered statistically significant.
Results
Validation of membrane protein purification
Western blotting analysis determined purity of the extracted membrane protein. Signal strength analysis by ImageJ software ascertained the plasma membrane marker (Na+/K+ATPase) level was 6.5-fold and 5.3-fold higher in the solution of membrane proteins than the cytoplasm proteins for lung cancer and normal lung tissue, respectively. In contrast, the mitochondrial marker (prohibitin) level in the solution of membrane proteins was 3.2-fold and 4.6-fold lower than the cytoplasm proteins for lung cancer and normal lung tissue, respectively (Figure 1). These results indicated membrane proteins were successfully purified with few cell organelle proteins.
Figure 1.
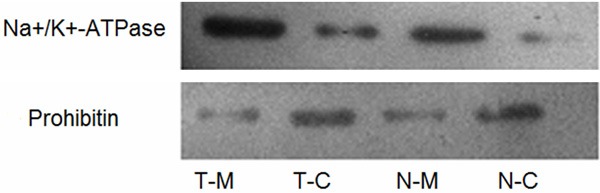
Verification of membrane protein purification using Western blotting analysis. Blots were probed with anti-Na+/K+-ATPase for the plasma membrane and anti-prohibitin for mitochondria. T-M and T-C indicate the solution of membrane proteins and the solution of cytoplasm proteins in tumor tissue, respectively. N-M and N-C indicate the solution of membrane proteins and the solution of cytoplasm proteins in normal lung tissue, respectively.
Identification of differentially expressed proteins
In this study, we used iTRAQ labeling and 2D-LC-MS/MS to compare protein expression between pooled lung adenocarcinoma and matched normal lung tissue samples. A total of 2486 proteins from both tumor and normal tissue were respectively identified using at least one peptide with ≥95% confidence. Among the 2486 proteins, 568 proteins were considered differentially expressed between lung adenocarcinoma and normal lung tissue according to ratios of fold-change (≥1.5 or ≤0.66). Two hundred fifty-seven proteins were upregulated and 311 were downregulated (Table 1, Supplementary Data). Of the differentially expressed proteins, 234 (41%) were identified by more than five unique peptides, 42 (7.4%) by four unique peptides, 48 (8.4%) by three unique peptides, 101 (14.7%) by two unique peptides, and 143 (25.2%) by one peptide. As a result, S100A14 was significantly upregulated in lung adenocarcinoma (2.10-fold) compared with normal lung tissues. MS/MS spectra of the four peptides used for identification of S100A14 are shown in Figure 2.
Table 1.
Top 50 PM or PM-related proteins highly differentially expressed in lung adenocarcinoma compared to normal lung tissue
| Accession | Description | Score | Coverage | Unique Peptides | PSMs | 117/118 |
|---|---|---|---|---|---|---|
| P51674 | Neuronal membrane glycoprotein M6-a | 23.12 | 3.96% | 1 | 1 | 0.13 |
| P01914 | HLA class II histocompatibility antigen, DR-1 beta chain | 140.22 | 15.41% | 1 | 22 | 0.14 |
| P04439 | HLA class I histocompatibility antigen, A-3 alpha chain | 571.74 | 34.52% | 1 | 172 | 0.152 |
| P28906 | Hematopoietic progenitor cell antigen CD34 | 24.64 | 2.08% | 1 | 3 | 0.187 |
| P01911 | HLA class II histocompatibility antigen, DRB1-15 beta chain | 347.41 | 44.36% | 2 | 55 | 0.205 |
| P57087 | Junctional adhesion molecule B | 51.04 | 3.69% | 1 | 2 | 0.209 |
| Q15109 | Advanced glycosylation end product-specific receptor | 283.25 | 23.76% | 6 | 47 | 0.21 |
| Q16602 | Calcitonin gene-related peptide type 1 receptor | 102.78 | 4.56% | 2 | 11 | 0.235 |
| P55087 | Aquaporin-4 | 106.77 | 6.19% | 2 | 16 | 0.238 |
| P29972 | Aquaporin-1 | 165.48 | 19.70% | 2 | 16 | 0.247 |
| Q9NY47 | Voltage-dependent calcium channel subunit alpha-2/delta-2 | 88.52 | 2.09% | 2 | 4 | 0.26 |
| Q03135 | Caveolin-1 | 404.38 | 61.80% | 10 | 129 | 0.281 |
| P37023 | Serine/threonine-protein kinase receptor R3 | 22.19 | 2.78% | 1 | 1 | 0.286 |
| Q96AP7 | Endothelial cell-selective adhesion molecule | 139.99 | 12.05% | 3 | 13 | 0.304 |
| P31645 | Sodium-dependent serotonin transporter | 52.21 | 2.54% | 1 | 4 | 0.313 |
| P56856 | Claudin-18 | 23.79 | 4.21% | 1 | 6 | 0.315 |
| P12429 | Annexin A3 | 881.28 | 58.51% | 19 | 259 | 0.322 |
| Q8NFJ5 | Retinoic acid-induced protein 3 | 97.17 | 5.32% | 2 | 9 | 0.329 |
| Q30154 | HLA class II histocompatibility antigen, DR beta 5 chain | 248.8 | 31.20% | 2 | 42 | 0.335 |
| P98172 | Ephrin-B1 | 50.37 | 5.20% | 1 | 1 | 0.343 |
| P02730 | Band 3 anion transport protein | 948.51 | 27.44% | 20 | 162 | 0.344 |
| Q96AM1 | Mas-related G-protein coupled receptor member F | 116.38 | 10.20% | 2 | 9 | 0.344 |
| P30486 | HLA class I histocompatibility antigen, B-48 alpha chain | 484.17 | 35.36% | 1 | 110 | 0.351 |
| P16671 | Platelet glycoprotein 4 | 325.47 | 19.92% | 8 | 60 | 0.355 |
| P50895 | Basal cell adhesion molecule | 468.48 | 27.07% | 12 | 40 | 0.358 |
| P07237 | Protein disulfide-isomerase | 1055.3 | 56.89% | 24 | 171 | 1.651 |
| P01597 | Ig kappa chain V-I region DEE | 104.49 | 22.22% | 1 | 28 | 1.662 |
| Q9Y666 | Solute carrier family 12 member 7 | 77.8 | 2.12% | 1 | 2 | 1.681 |
| O15427 | Monocarboxylate transporter 4 | 117.91 | 9.03% | 4 | 9 | 1.682 |
| P63241 | Eukaryotic translation initiation factor 5A-1 | 263.8 | 36.36% | 5 | 19 | 1.718 |
| Q96HE7 | ERO1-like protein alpha | 196.14 | 14.96% | 7 | 8 | 1.731 |
| Q96AG4 | Leucine-rich repeat-containing protein 59 | 206.06 | 28.66% | 7 | 10 | 1.784 |
| Q13509 | Tubulin beta-3 chain | 732.61 | 40.44% | 1 | 248 | 1.809 |
| P30453 | HLA class I histocompatibility antigen, A-34 alpha chain | 623.3 | 37.81% | 3 | 192 | 1.812 |
| P63244 | Guanine nucleotide-binding protein subunit beta-2-like 1 | 727.82 | 59.31% | 16 | 118 | 1.815 |
| P04433 | Ig kappa chain V-III region VG (Fragment) | 34.87 | 13.91% | 2 | 4 | 1.85 |
| P01877 | Ig alpha-2 chain C region | 498.16 | 55.59% | 3 | 82 | 1.874 |
| P17213 | Bactericidal permeability-increasing protein | 108.86 | 6.57% | 2 | 3 | 1.875 |
| Q14435 | Polypeptide N-acetylglucosaminyltransferase 3 | 25.84 | 1.58% | 1 | 1 | 1.881 |
| P11413 | Glucose-6-phosphate 1-dehydrogenase | 391.44 | 27.18% | 12 | 30 | 1.882 |
| P80723 | Brain acid soluble protein 1 | 372.69 | 70.93% | 10 | 32 | 1.91 |
| Q9BY50 | Signal peptidase complex catalytic subunit SEC11C | 140.79 | 27.60% | 5 | 7 | 1.933 |
| Q01650 | Large neutral amino acids transporter small subunit 1 | 37.08 | 3.55% | 1 | 2 | 1.948 |
| P01605 | Ig kappa chain V-I region Lay | 53.42 | 25.00% | 1 | 5 | 1.969 |
| Q9BYC5 | Alpha-(1,6)-fucosyltransferase | 83.78 | 4.52% | 2 | 5 | 2.063 |
| Q9HCY8 | Protein S100-A14 | 142.71 | 42.31% | 4 | 12 | 2.099 |
| O75976 | Carboxypeptidase D | 325.77 | 9.42% | 11 | 15 | 2.234 |
| Q99541 | Perilipin-2 | 28.53 | 2.52% | 1 | 1 | 2.46 |
| P09496 | Clathrin light chain A | 91.04 | 12.50% | 4 | 12 | 2.577 |
| P25815 | Protein S100-P | 83.62 | 24.21% | 2 | 15 | 2.932 |
PSMs: peptide spectrum matches.
Figure 2.

MS/MS spectra of four identified peptides: (A) The MS spectra of sequence MSPDEGQEELEEVQAELK with m/z of 895.78375 Da (z = +3), MH+: 2685.33670 Da; (B) The MS spectra of sequence NKEPPAPAQQLQPQPVAVQGPEPAR with m/z of 984.53918 Da (z = +3), MH+: 2951.60300 Da; (C) The MS spectra of sequence MSPDEGQEELEEVQAELK with m/z of 890.45135 Da (z = +3), MH+: 2669.33951 Da; and (D) the MS/MS spectra of MSPDEGQEELEEVQAELK, with m/z of 794.38354 Da (z = +3), MH+: 2381.13608 Da.
Bioinformatic analysis of differentially expressed proteins
The molecular weights of the identified differentially expressed proteins ranged from 6.6 kDa to 628.7 kDa; 481 proteins (84.7%) were between 10 kDa and 100 kDa. Moreover, the isoelectric points ranged from 3.78 to 12.15; 501 proteins (88.2%) were between 4 and 10. Gene Ontology (GO) annotation was applied to describe functions of the identified differentially expressed proteins, which were classified into three major categories: cellular component, molecular function, and biological process [24]. To visualize the annotation of gene sets, WEGO was performed to plot the distribution of GO annotation [22] (Figure 3).
Figure 3.
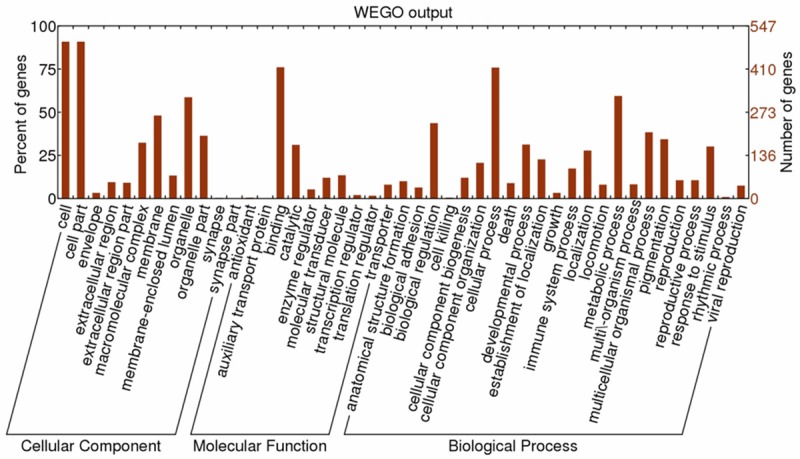
Distribution of the GO terms for differential proteins. The identified differential proteins were classified into cellular component, molecular function, and biological process by WEGO.
The cellular component category showed that differential proteins are mainly involved in the cell, cell parts, membrane, and organelles. GO annotation shows that approximately 48% of the differential proteins were found to be membrane bound or membrane associated, with PM or PM-related proteins accounting for approximately 38%. In the molecular function category, differentially expressed proteins are mainly associated with binding, catalysis, molecular transduction, transport, and molecular structure. The biological process category indicated that the differential proteins are mainly related to cellular processes, metabolic processes, biological regulation, cellular component organization, response to stimulus and pigmentation.
For more insight into the pathways and biological processes of the 568 differential proteins, pathway enrichment analysis was performed through the Pathway Interaction Database (PID), which provided batch query options. For the differential proteins, 169 pathways were detected via PID and 35 pathways were significantly enriched with association signals at the p<0.01 level (Figure 4). Among these, there were many remarkable signaling pathways involved in cell surface interactions and cell signaling events, including integrin beta1 cell surface interactions, integrin family cell surface interactions, CXCR4-mediated signaling events, Syndecan-2-mediated signaling events, Par-4-mediated thrombin signaling events, plasma membrane estrogen receptor signaling, etc. These results indicated that the differential proteins might affect carcinogenesis of lung adenocarcinoma through these pathways.
Figure 4.
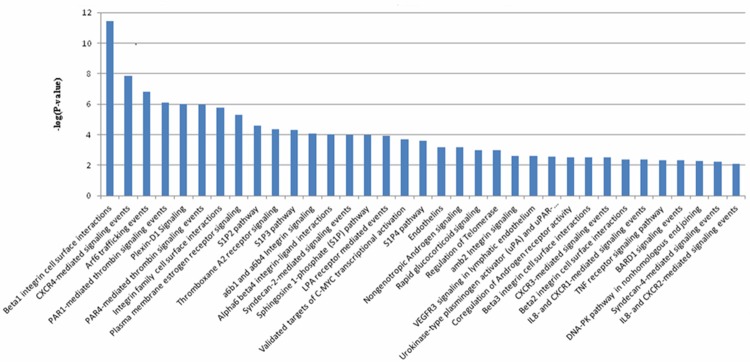
Significant pathways (p<0.01) were involved in differential proteins according to PID enrichment analysis.
An additional pathway enrichment analysis was conducted by KEGG for 568 differential proteins. By this means, we identified 19 pathways significantly enriched with association signals at the p<0.05 level. These pathways encompassed metabolism, environmental information processing, organismal systems, human diseases, and genetic information processing. Among the 19 pathways (Table 2), 6 are related to metabolism (nicotinate and nicotinamide metabolism, nitrogen metabolism, isoquinoline alkaloid biosynthesis, phenylalanine metabolism, alanine, aspartate and glutamate metabolism, tyrosine metabolism). Two pathways are related to signaling molecules and interaction (ECM-receptor interaction, cell adhesion molecules (CAMs). These findings suggested that amino acid metabolism is clearly different between tumor and normal cells, and membrane proteins may be involved at the metabolic level in lung cancer.
Table 2.
19 pathways significantly enriched by KEGG enrichment analysis for differential proteins
| No | Pathway | Proteins with pathway annotation (333) | P value | Pathway ID |
|---|---|---|---|---|
| 1 | Ribosome | 73 (21.92%) | <0.001 | ko03010 |
| 2 | Non-homologous end-joining | 5 (1.5%) | 0.001 | ko03450 |
| 3 | Hematopoietic cell lineage | 14 (4.2%) | 0.002 | ko04640 |
| 4 | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 15 (4.5%) | 0.003 | ko05412 |
| 5 | Nicotinate and nicotinamide metabolism | 6 (1.8%) | 0.004 | ko00760 |
| 6 | Nitrogen metabolism | 4 (1.2%) | 0.015 | ko00910 |
| 7 | Isoquinoline alkaloid biosynthesis | 3 (0.9%) | 0.015 | ko00950 |
| 8 | Dilated cardiomyopathy | 16 (4.8%) | 0.017 | ko05414 |
| 9 | Toxoplasmosis | 17 (5.11%) | 0.021 | ko05145 |
| 10 | Cell adhesion molecules (CAMs) | 27 (8.11%) | 0.024 | ko04514 |
| 11 | Malaria | 8 (2.4%) | 0.025 | ko05144 |
| 12 | Phenylalanine metabolism | 5 (1.5%) | 0.026 | ko00360 |
| 13 | Alanine, aspartate and glutamate metabolism | 5 (1.5%) | 0.026 | ko00250 |
| 14 | ECM-receptor interaction | 17 (5.11%) | 0.027 | ko04512 |
| 15 | Hypertrophic cardiomyopathy (HCM) | 14 (4.2%) | 0.032 | ko05410 |
| 16 | Tyrosine metabolism | 6 (1.8%) | 0.032 | ko00350 |
| 17 | Intestinal immune network for IgA production | 10 (3%) | 0.037 | ko04672 |
| 18 | Bile secretion | 8 (2.4%) | 0.037 | ko04976 |
| 19 | Viral myocarditis | 24 (7.21%) | 0.043 | ko05416 |
Validation of S100A14 expression by Western blotting
To further validate the differential expression of S100A14 identified by iTRAQ labeling and LC-MS/MS, we examined the expression levels of S100A14 in 10 lung adenocarcinoma tissue samples and paired normal lung tissues using Western blotting. As shown in Figure 5, S100A14 was significantly upregulated in lung adenocarcinoma compared with matched normal lung tissue, which confirmed the LC-MS/MS result.
Figure 5.
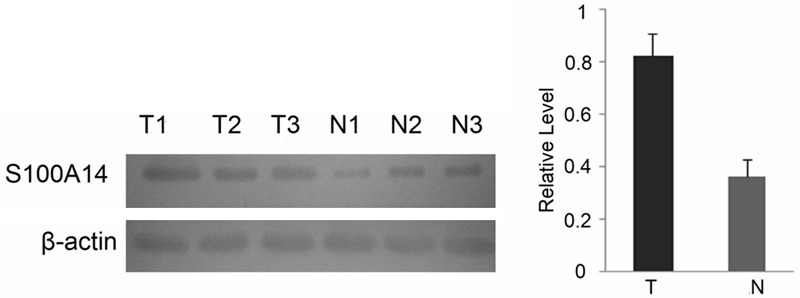
Expressional levels of S100A14 in lung adenocarcinoma and paired normal lung tissues. β-actin was used as a loading control. T = tumor tissues; N = normal tissues.
S100A14 expression in lung cancer and normal lung tissues
To further verify the results obtained from quantitative proteomics, we performed IHC to detect the expression and cellular distribution of S100A14 in a series of 62 specimens of lung cancer, including 41 adenocarcinomas and 21 lung squamous cell carcinomas, along with a series of 24 specimens of normal lung tissues. Increased expression of S100A14 was observed at the membranes of some of the tumor cells in most of the lung adenocarcinoma specimens and squamous cell carcinomas (Figure 6A-C). However, negative or low S100A14 expression levels were observed in a large majority of normal lung specimens (Figure 6D). Compared with normal lung specimens, the expression level of S100A14 in adenocarcinoma significantly increased (p<0.01). Similarly, S100A14 expression was significantly higher in lung squamous cell carcinoma than normal lung specimens (p<0.01) (Table 3). Furthermore, we evaluated the relationship between clinicopathological characteristics and S100A14 expression in lung cancer. We found that S100A14 expression is higher in well or moderately differentiated lung cancer than in poorly differentiated lung cancer. There were no significant correlations between S100A14 expression and other clinicopathologic characteristics, including pathological type, lymph node metastasis, patient age, gender, tumor size, or TNM stage (Table 4).
Figure 6.
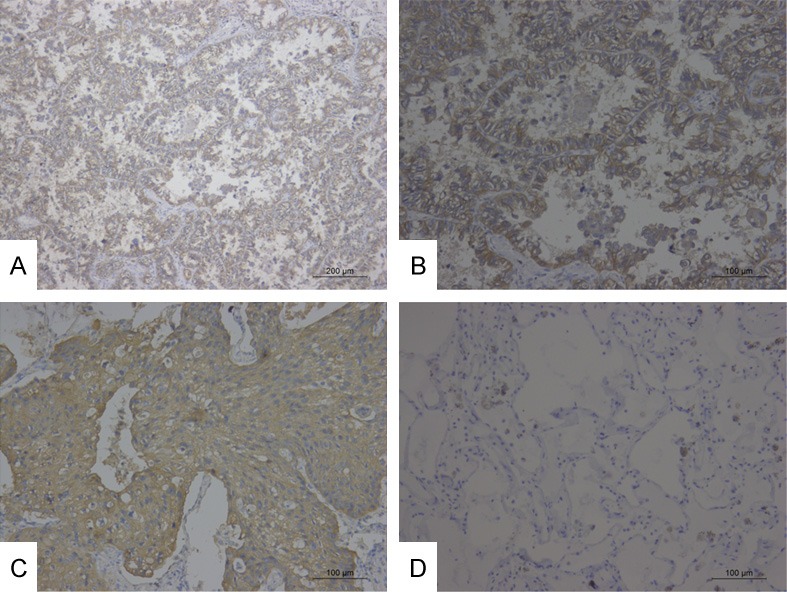
(A, B) High staining of S100A14 in LAC. (C) High staining of S100A14 in LSCC. (D) Negative staining of S100A14 in normal lung tissue. (A ×100; B-D ×200).
Table 3.
S100A14 expression in lung cancer and normal lung tissues
| Groups | N | Expressional levels | P* | ||
|---|---|---|---|---|---|
|
| |||||
| Neg | Low | High | |||
| LAC | 41 | 3 | 13 | 25 | <0.01a |
| LSCC | 21 | 1 | 7 | 13 | <0.01b |
| N | 24 | 8 | 15 | 1 | <0.01c |
LAC versus normal lung tissue;
LSCC versus normal lung tissue;
Lung cancer versus normal lung tissue;
P<0.05 by Mann-Whitney U test;
LAC, lung adenocarcinoma; LSCC, lung squamous cell carcinoma; Lung cancer, LAC+LSCC; N, normal lung tissue.
Table 4.
Correlation between clinicopathological characteristics and S100A14 expression in lung cancer
| Parameter | N | Expressional levels | P* | ||
|---|---|---|---|---|---|
|
| |||||
| Neg | 1+ | 2+ | |||
| Age | |||||
| <60 | 32 | 2 | 13 | 17 | 0.218 |
| ≥60 | 30 | 2 | 7 | 21 | |
| Gender | |||||
| Male | 41 | 3 | 14 | 24 | 0.521 |
| Female | 21 | 1 | 6 | 14 | |
| Grade | |||||
| G1+G2 | 34 | 1 | 8 | 25 | 0.026 |
| G3 | 28 | 3 | 12 | 13 | |
| Histology | |||||
| LAC | 41 | 3 | 13 | 25 | 0.890 |
| LSCC | 21 | 1 | 7 | 13 | |
| Lymphatic invasion | |||||
| N0 | 33 | 2 | 12 | 19 | 0.576 |
| N+ | 29 | 2 | 8 | 19 | |
| Tumor size | |||||
| ≤3 | 22 | 0 | 6 | 16 | 0.183 |
| 3<T≤7 | 31 | 4 | 11 | 16 | |
| >7 | 9 | 0 | 3 | 6 | |
| TNM stage | |||||
| I+II | 35 | 2 | 13 | 20 | 0.519 |
| III+IV | 27 | 2 | 7 | 18 | |
P<0.05 by Mann-Whitney U test or Kruskal-Wallis test.
Discussion
By delineating the differences in protein expression profiles between tumor and normal tissue, comparative quantitative proteomics offers a powerful method to investigate molecular mechanisms and identify potential tumor markers and therapeutic targets in cancer. To date, a number of studies have identified differential proteins in normal and cancer cells using traditional 2DGE, followed by mass spectrometry [25-27]. Although 2DGE remains an important tool in comparative proteomics, it is less than satisfactory when a large amount of sample handling is required. The procedure is also deficient in identifying low abundance proteins, proteins with extreme molecular weights and pI value, and hydrophobic proteins such as membrane proteins [28]. iTRAQ technology can overcome these limitations and provide a better solution to identify cell membrane proteins. Subcellular proteomic analysis has many advantages over whole-cell proteome analysis. Most significantly, the enhanced technology increases the probability of detecting low-abundance proteins that may play a role in the development of cancer. Manual microdissection from frozen sections provides a rapid and inexpensive means to substantially enrich tumor cells for downstream analysis. This technique yielded findings consistent with LCM data [6].
In this study, we isolated tumor cells from adjacent non-tumor cells and used iTRAQ labeling 2D-LC-MS/MS to identify the differential membrane proteins between lung adenocarcinoma tissue and matched normal lung tissue. As a result, a total of 568 differential expression proteins were identified between the two types of tissues, including well-known membrane markers Protein S100-P, Ig kappa chain V-I region Lay, Caveolin-1, Voltage-dependent calcium channel subunit alpha-2/delta-2, Annexin A3, Claudin-18, etc. To better understand the basic biological information of differential expression proteins identified in our study, we further analyzed the proteins using WEGO. WEGO has been applied in several genomics studies, including those of the rice genome [29], Arabidopsis genome [30], and the silkworm genome [31]. WEGO provides a visualization of the annotation sets of genes, comparing the provided gene datasets by plotting a histogram of the distribution of GO annotation [22]. In addition, the differential proteins involved with binding, catalysis, molecular transduction, and transport were detected. These results indicated that proteins engaged in these functions may play important roles in carcinogenesis.
The cell membrane plays a critical role in cell signaling transduction and signal pathways closely associated with carcinogenesis and tumor development. Therefore, we performed pathway enrichment analysis using PID and KEGG. PID is a growing collection of human cellular signaling pathways. The database focuses on signaling and regulatory pathways, particularly those impacting cancer research and treatment. KEGG, however, focuses on metabolic processes and generic mechanisms such as transcription and translation [32]. In this study, the enrichment analysis results derived from PID indicated that the enriched pathways were involved in many membrane proteins, which shows the importance of these proteins in carcinogenesis. The results obtained from KEGG enrichment analysis revealed pathways associated with nicotinate and nicotinamide metabolism, which have been implicated in lung adenocarcinoma carcinogenesis. Several studies have already shown that nicotinamide is involved in tumor formation and tumor cell apoptosis [33,34]. Our study offered valuable information for further exploring the underlying mechanism in carcinogenesis of lung adenocarcinoma.
S100A14 is a novel member of the S100 protein family [35]. S100 is a subfamily of proteins related by Ca2+-binding to the EF-hand superfamily that appear to be involved in the regulation of many cellular processes (e.g., cell cycle progression, differentiation, cell-cell communication, intracellular signaling, energy metabolism) [35,36]. The study by Lukanidin [37] showed that the S100 family is pivotal in cell migration, invasion, and cancer metastasis.
In our study, the higher expression levels of S100A14 were identified in lung adenocarcinoma versus normal lung tissues by MS/MS analysis. We then confirmed this result via Western blotting analysis. Findings were consistent with the results from quantitative proteomics: they showed that S100A14 was significantly increased in lung adenocarcinoma compared with normal lung tissue.
Recently, some studies reported S100A14 expression in various cancers, but the results were inconsistent. Some data have indicated S100A14 is upregulated in several cancers, including ovarian, breast, and hepatocellular cancer [35,38]. On the other hand, some data suggest that S100A14 is downregulated in kidney, colon, rectal, esophageal, and oral carcinoma [35,39]. These apparent discrepancies suggest that S100A14 plays different roles at different tumor and development stages, although the underlying mechanism remains unclear. Although northern blot hybridization has shown that S100A14 mRNA expression is upregulated in lung tumors [35], S100A14 protein expression in lung cancer still remains unclear. Therefore, using IHC we further detected S100A14 expression in paraffin-embedded archival tissue specimens and evaluated the relationship between S100A14 and clinicopathological characteristics in patients with lung cancer. We found that S100A14 expression was increased in lung adenocarcinoma and squamous cell carcinoma compared with that in normal lung tissue. Furthermore, our results showed that S100A14 was expressed at higher levels in well or moderately differentiated lung cancer than in poorly differentiated lung cancer, which was consistent with a previous report [40]. The IHC data indicated that S100A14 may play a potential role in cell differentiation.
In conclusion, a large number of differential proteins were identified in the membrane fraction from lung adenocarcinoma and normal lung tissue samples using the iTRAQ-coupled 2D-LC-MS/MS technique. Furthermore, we verified the differential expression of S100A14 and found the protein may be a suitable biomarker potentially involved in tumor cell differentiation. However, a larger group of lung cancer samples is needed to confirm the results. Our findings in this study helped elucidate the underlying carcinogenesis of lung cancer by providing a potential novel biomarker and new therapeutic targets.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (no. 81350032).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3.Neubauer H, Clare SE, Kurek R, Fehm T, Wallwiener D, Sotlar K, Nordheim A, Wozny W, Schwall GP, Poznanovic S, Sastri C, Hunzinger C, Stegmann W, Schrattenholz A, Cahill MA. Breast cancer proteomics by laser capture microdissection, sample pooling, 54-cm IPG IEF, and differential iodine radioisotope detection. Electrophoresis. 2006;27:1840–1852. doi: 10.1002/elps.200500739. [DOI] [PubMed] [Google Scholar]
- 4.Bu LN, Yang SY, Li FT, Shang WL, Zhang W, Huo SF, Nan YD, Tian YX, Du J, Lin XL, Liu YF, Lin YR, Rong BX. Study of differential proteins in lung adenocarcinoma using laser capture microdissection combined with liquid chip-mass spectrometry technology. Chin Med J (Engl) 2010;123:3309–3313. [PubMed] [Google Scholar]
- 5.Rabien A. Laser microdissection. Methods Mol Biol. 2010;576:39–47. doi: 10.1007/978-1-59745-545-9_3. [DOI] [PubMed] [Google Scholar]
- 6.Nowak NJ, Miecznikowski J, Moore SR, Gaile D, Bobadilla D, Smith DD, Kernstine K, Forman SJ, Mhawech-Fauceglia P, Reid M, Stoler D, Loree T, Rigual N, Sullivan M, Weiss LM, Hicks D, Slovak ML. Challenges in array comparative genomic hybridization for the analysis of cancer samples. Genet Med. 2007;9:585–595. doi: 10.1097/gim.0b013e3181461c4a. [DOI] [PubMed] [Google Scholar]
- 7.Taylor RS, Wu CC, Hays LG, Eng JK, Yates JR 3rd, Howell KE. Proteomics of rat liver Golgi complex: minor proteins are identified through sequential fractionation. Electrophoresis. 2000;21:3441–3459. doi: 10.1002/1522-2683(20001001)21:16<3441::AID-ELPS3441>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 8.Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT, Matunis MJ. Proteomic analysis of the mammalian nuclear pore complex. J Cell Biol. 2002;158:915–927. doi: 10.1083/jcb.200206106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Zhan X, Li M, Li G, Zhang P, Xiao Z, Shao M, Peng F, Hu R, Chen Z. Mitochondrial proteomics of nasopharyngeal carcinoma metastasis. BMC Med Genomics. 2012;5:62. doi: 10.1186/1755-8794-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prior MJ, Larance M, Lawrence RT, Soul J, Humphrey S, Burchfield J, Kistler C, Davey JR, La-Borde PJ, Buckley M, Kanazawa H, Parton RG, Guilhaus M, James DE. Quantitative proteomic analysis of the adipocyte plasma membrane. J Proteome Res. 2011;10:4970–4982. doi: 10.1021/pr200446r. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Y, Zhang W, Kho Y, Zhao Y. Proteomic analysis of integral plasma membrane proteins. Anal Chem. 2004;76:1817–1823. doi: 10.1021/ac0354037. [DOI] [PubMed] [Google Scholar]
- 12.Stockwin LH, Blonder J, Bumke MA, Lucas DA, Chan KC, Conrads TP, Issaq HJ, Veenstra TD, Newton DL, Rybak SM. Proteomic analysis of plasma membrane from hypoxia-adapted malignant melanoma. J Proteome Res. 2006;5:2996–3007. doi: 10.1021/pr0601739. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Zhang M, Go VL, Hu S. Membrane proteomic analysis of pancreatic cancer cells. J Biomed Sci. 2010;17:74. doi: 10.1186/1423-0127-17-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dowling P, Walsh N, Clynes M. Membrane and membrane-associated proteins involved in the aggressive phenotype displayed by highly invasive cancer cells. Proteomics. 2008;8:4054–4065. doi: 10.1002/pmic.200800098. [DOI] [PubMed] [Google Scholar]
- 15.Seshi B. An integrated approach to mapping the proteome of the human bone marrow stromal cell. Proteomics. 2006;6:5169–5182. doi: 10.1002/pmic.200600209. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Lv L, Liu H, Cui L, Chen G, Bi P, Li Z. Profiling the potential biomarkers for cell differentiation of pancreatic cancer using iTRAQ and 2-D LC-MS/MS. Proteomics Clin Appl. 2009;3:862–871. doi: 10.1002/prca.200800029. [DOI] [PubMed] [Google Scholar]
- 17.Muraoka S, Kume H, Watanabe S, Adachi J, Kuwano M, Sato M, Kawasaki N, Kodera Y, Ishitobi M, Inaji H, Miyamoto Y, Kato K, Tomonaga T. Strategy for SRM-based verification of biomarker candidates discovered by iTRAQ method in limited breast cancer tissue samples. J Proteome Res. 2012;11:4201–4210. doi: 10.1021/pr300322q. [DOI] [PubMed] [Google Scholar]
- 18.Chen JS, Chen KT, Fan CW, Han CL, Chen YJ, Yu JS, Chang YS, Chien CW, Wu CP, Hung RP, Chan EC. Comparison of membrane fraction proteomic profiles of normal and cancerous human colorectal tissues with gel-assisted digestion and iTRAQ labeling mass spectrometry. FEBS J. 2010;277:3028–3038. doi: 10.1111/j.1742-4658.2010.07712.x. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z, Zhang L, Hua Y, Jia X, Li J, Hu S, Peng X, Yang P, Sun M, Ma F, Cai Z. Comparative proteomic analysis of plasma membrane proteins between human osteosarcoma and normal osteoblastic cell lines. BMC Cancer. 2010;10:206. doi: 10.1186/1471-2407-10-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen XL, Zhou L, Yang J, Shen FK, Zhao SP, Wang YL. Hepatocellular carcinoma-associated protein markers investigated by MALDI-TOF MS. Mol Med Rep. 2010;3:589–596. doi: 10.3892/mmr_00000302. [DOI] [PubMed] [Google Scholar]
- 21.Li B, Chang J, Chu Y, Kang H, Yang J, Jiang J, Ma H. Membrane proteomic analysis comparing squamous cell lung cancer tissue and tumour-adjacent normal tissue. Cancer Lett. 2012;319:118–124. doi: 10.1016/j.canlet.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 22.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Liu J, Yue D, Gao L, Wang D, Zhang H, Wang C. Clinical significance of E-cadherin, beta-catenin, vimentin and S100A4 expression in completely resected squamous cell lung carcinoma. J Clin Pathol. 2013;66:937–945. doi: 10.1136/jclinpath-2013-201467. [DOI] [PubMed] [Google Scholar]
- 24.Zhong XW, Zou Y, Liu SP, Yi QY, Hu CM, Wang C, Xia QY, Zhao P. Proteomic-based insight into malpighian tubules of silkworm Bombyx mori. PLoS One. 2013;8:e75731. doi: 10.1371/journal.pone.0075731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalra RS, Bapat SA. Expression proteomics predicts loss of RXR-gamma during progression of epithelial ovarian cancer. PLoS One. 2013;8:e70398. doi: 10.1371/journal.pone.0070398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yousefi Z, Sarvari J, Nakamura K, Kuramitsu Y, Ghaderi A, Mojtahedi Z. Secretomic analysis of large cell lung cancer cell lines using two-dimensional gel electrophoresis coupled to mass spectrometry. Folia Histochem Cytobiol. 2012;50:368–374. doi: 10.5603/18762. [DOI] [PubMed] [Google Scholar]
- 27.Yonglitthipagon P, Pairojkul C, Bhudhisawasdi V, Mulvenna J, Loukas A, Sripa B. Proteomics-based identification of alpha-enolase as a potential prognostic marker in cholangiocarcinoma. Clin Biochem. 2012;45:827–834. doi: 10.1016/j.clinbiochem.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Gilmore JM, Washburn MP. Advances in shotgun proteomics and the analysis of membrane proteomes. J Proteomics. 2010;73:2078–2091. doi: 10.1016/j.jprot.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Huang X, Li W, Li J, Liu Z, Li L, Liu J, Qi Q, Liu J, Li L, Li T, Wang X, Lu H, Wu T, Zhu M, Ni P, Han H, Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Zhang J, Xu J, Zhang K, Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Ren X, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Wang J, Zhao W, Li P, Chen W, Wang X, Zhang Y, Hu J, Wang J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B, Zheng W, Chen S, Guo W, Li G, Liu S, Tao M, Wang J, Zhu L, Yuan L, Yang H. A draft sequence of the rice genome (Oryza sativa L. ssp. indica) Science. 2002;296:79–92. doi: 10.1126/science.1068037. [DOI] [PubMed] [Google Scholar]
- 30.Yu J, Wang J, Lin W, Li S, Li H, Zhou J, Ni P, Dong W, Hu S, Zeng C, Zhang J, Zhang Y, Li R, Xu Z, Li S, Li X, Zheng H, Cong L, Lin L, Yin J, Geng J, Li G, Shi J, Liu J, Lv H, Li J, Wang J, Deng Y, Ran L, Shi X, Wang X, Wu Q, Li C, Ren X, Wang J, Wang X, Li D, Liu D, Zhang X, Ji Z, Zhao W, Sun Y, Zhang Z, Bao J, Han Y, Dong L, Ji J, Chen P, Wu S, Liu J, Xiao Y, Bu D, Tan J, Yang L, Ye C, Zhang J, Xu J, Zhou Y, Yu Y, Zhang B, Zhuang S, Wei H, Liu B, Lei M, Yu H, Li Y, Xu H, Wei S, He X, Fang L, Zhang Z, Zhang Y, Huang X, Su Z, Tong W, Li J, Tong Z, Li S, Ye J, Wang L, Fang L, Lei T, Chen C, Chen H, Xu Z, Li H, Huang H, Zhang F, Xu H, Li N, Zhao C, Li S, Dong L, Huang Y, Li L, Xi Y, Qi Q, Li W, Zhang B, Hu W, Zhang Y, Tian X, Jiao Y, Liang X, Jin J, Gao L, Zheng W, Hao B, Liu S, Wang W, Yuan L, Cao M, McDermott J, Samudrala R, Wang J, Wong GK, Yang H. The Genomes of Oryza sativa: a history of duplications. PLoS Biol. 2005;3:e38. doi: 10.1371/journal.pbio.0030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia Q, Zhou Z, Lu C, Cheng D, Dai F, Li B, Zhao P, Zha X, Cheng T, Chai C, Pan G, Xu J, Liu C, Lin Y, Qian J, Hou Y, Wu Z, Li G, Pan M, Li C, Shen Y, Lan X, Yuan L, Li T, Xu H, Yang G, Wan Y, Zhu Y, Yu M, Shen W, Wu D, Xiang Z, Yu J, Wang J, Li R, Shi J, Li H, Li G, Su J, Wang X, Li G, Zhang Z, Wu Q, Li J, Zhang Q, Wei N, Xu J, Sun H, Dong L, Liu D, Zhao S, Zhao X, Meng Q, Lan F, Huang X, Li Y, Fang L, Li C, Li D, Sun Y, Zhang Z, Yang Z, Huang Y, Xi Y, Qi Q, He D, Huang H, Zhang X, Wang Z, Li W, Cao Y, Yu Y, Yu H, Li J, Ye J, Chen H, Zhou Y, Liu B, Wang J, Ye J, Ji H, Li S, Ni P, Zhang J, Zhang Y, Zheng H, Mao B, Wang W, Ye C, Li S, Wang J, Wong GK, Yang H. A draft sequence for the genome of the domesticated silkworm (Bombyx mori) Science. 2004;306:1937–1940. doi: 10.1126/science.1102210. [DOI] [PubMed] [Google Scholar]
- 32.Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37:D674–679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato F, Mitaka T, Mizuguchi T, Mochizuki Y, Hirata K. Effects of nicotinamide-related agents on the growth of primary rat hepatocytes and formation of small hepatocyte colonies. Liver. 1999;19:481–488. doi: 10.1111/j.1478-3231.1999.tb00080.x. [DOI] [PubMed] [Google Scholar]
- 34.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- 35.Pietas A, Schluns K, Marenholz I, Schafer BW, Heizmann CW, Petersen I. Molecular cloning and characterization of the human S100A14 gene encoding a novel member of the S100 family. Genomics. 2002;79:513–522. doi: 10.1006/geno.2002.6744. [DOI] [PubMed] [Google Scholar]
- 36.Schafer BW, Wicki R, Engelkamp D, Mattei MG, Heizmann CW. Isolation of a YAC clone covering a cluster of nine S100 genes on human chromosome 1q21: rationale for a new nomenclature of the S100 calcium-binding protein family. Genomics. 1995;25:638–643. doi: 10.1016/0888-7543(95)80005-7. [DOI] [PubMed] [Google Scholar]
- 37.Lukanidin E, Sleeman JP. Building the niche: the role of the S100 proteins in metastatic growth. Semin Cancer Biol. 2012;22:216–225. doi: 10.1016/j.semcancer.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 38.Zhao FT, Jia ZS, Yang Q, Song L, Jiang XJ. S100A14 promotes the growth and metastasis of hepatocellular carcinoma. Asian Pac J Cancer Prev. 2013;14:3831–3836. doi: 10.7314/apjcp.2013.14.6.3831. [DOI] [PubMed] [Google Scholar]
- 39.Sapkota D, Costea DE, Blo M, Bruland O, Lorens JB, Vasstrand EN, Ibrahim SO. S100A14 inhibits proliferation of oral carcinoma derived cells through G1-arrest. Oral Oncol. 2012;48:219–225. doi: 10.1016/j.oraloncology.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Chen H, Ma J, Sunkel B, Luo A, Ding F, Li Y, He H, Zhang S, Xu C, Jin Q, Wang Q, Liu Z. S100A14: novel modulator of terminal differentiation in esophageal cancer. Mol Cancer Res. 2013;11:1542–1553. doi: 10.1158/1541-7786.MCR-13-0317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.