

Abstract
Introduction: Although HER2 and ER pathways are predominant pathways altered in breast cancer, it is now well accepted that many other signaling pathways are also involved in the pathogenesis of breast cancer. The understanding of these additional pathways may assist in identifying new therapeutic approaches for breast cancer. Methods: 13 invasive ductal carcinoma tissues and 5 benign breast tissues were analyzed for the mRNA expression level of 1243 cancer pathway-related genes using SmartChip (WaferGen, CA), a real-time PCR-base method. In addition, the levels of 131 cancer pathway-related proteins and phosphoproteins in 33 paired breast cancers were measured using our innovative Protein Pathway Array. Results: Out of 1,243 mRNAs, 68.7% (854) were detected in breast cancer and 395 mRNAs were statistically significant (fold change >2) between benign and cancer tissues. Of these mRNAs, 105 only expressed in breast cancer tissues and 33 mRNAs only expressed in normal breast tissues. Out of 131 proteins and phosphoproteins, 68% (89) were detected in cancer tissues and 57 proteins were significantly differentiated between tumor and normal tissues. Interestingly, only 3 genes (CDK6, Vimentin and SLUG) showed decreases in both protein and mRNA. Six proteins (BCL6, CCNE1, PCNA, PDK1, SRC and XIAP) were differentially expressed between tumor and normal tissues but no differences were observed at mRNA levels. Analyses of mRNA and protein data using Ingenuity Pathway Analysis showed more than 15 pathways were altered in breast cancer and 6 of which were shared between mRNAs and proteins, including p53, IL17, HGF, NGF, PTEN and PI3K/AKT pathways. Conclusions: There is a broad dysregulation of various pathways in breast cancer both at protein levels and mRNA levels. It is important to note that mRNA expression does not correlate with protein level, suggesting different regulation mechanisms between proteins and mRNAs.
Keywords: Breast cancer, gene expression, signaling network, signal transduction, protein abundance
Introduction
Breast cancer is the most common cancer in women worldwide with approximately 1.15 million new cases diagnosed annually. Some significant risk factors for breast cancer are older age, null parity, obesity, smoking and estrogen exposure. In approximately 10% of cases of breast cancer, the patients’ have a strong genetic predisposition. For example, patients with BRCA1 and BRCA2 gene mutations as well as those with mutations in p53 (Li-Fraumeni syndrome), PTEN (Cowden syndrome), and STK11 (Peutz-Jeghers syndrome) have a high risk of breast cancer [1]. Initial treatment of breast cancer primarily involves surgical resection of the tumor. Based on the stage of the disease as well as the presence of lymph node invasion, local radiation and chemotherapy (i.e. cyclophosphamide, pacitaxol and doxorubicin) are also used. Also, many tumors express estrogen (ER+) and progesterone (PR+) receptors as well as human epidermal growth factor receptor 2 (HER2+) and, therefore, can be treated with targeted agents like tamoxifen for the ER+ and PR+ tumors, and trastuzumab,a monoclonal antibody for HER2+ tumors [2].
Heterogeneous disease with multiple molecular mechanisms involved in its pathogenesis which correlate with different clinical behaviors. This phenotypic diversity severely affects the diagnosis and prognosis of breast cancer [3]. Recently, gene expression studies using DNA microarrays have identified several distinct breast cancer subtypes based on the expression patterns of 496 genes. These various subtypes differ substantially in prognosis and in the therapeutic targets they express [4].
The intrinsic subtypes comprise 2 main subtypes of ER-negative tumors (basal-like and HER2+/ER- subtype) and at least 2 types of ER-positive tumors (luminal A and luminal B). Basal-like tumors generally show lower expression of HER2 and ER and have higher expression of genes characteristic of the basal epithelial cell layer, like expression of cytokeratins 5, 6, and 17. The HER2+ tumors make up at least 2 separate expression groups: those that are ER+ (and may also be PR+) and cluster with tumors of luminal cell origins as part of the luminal B subtype and those that are ER- and cluster near the basal-like tumors (HER2+/ER- subtype). The luminal subtype A and B tumors express ER, GATA3, and genes regulated by both ER and GATA3. In contrast to luminal B tumors, luminal A tumors express higher levels of ER and GATA3 and are associated with a better clinical prognosis,15 whereas luminal B tumors more often express HER1, HER2, and/or cyclin E1 [4].
However, all these mRNA expression patterns do not directly relate genes and their expression to a functional context and the underlying pathogenesis. In past decade, many important signaling pathways have been shown to be involved in the pathogenesis of breast cancer. Endogenous signal transduction in cancer cells is systematically disturbed to redirect the cellular decisions from differentiation and apoptosis to proliferation and, later, invasion. Among them, hormonal pathways (i.e. ER, PR), membrane kinases (i.e. HER2) [5], DNA damage repair (i.e. P53) and tumor suppressor (i.e. PTEN) [6] have demonstrated importance in breast cancer proliferation, metastasis, and response to treatment [4,7]. These pathways do not occur in isolation and there is significant and complicated interactions among these pathways [8]. However, how these pathways interact with each other in a cellular network remains unclear.
In this study, we utilized high-throughput quantitative PCR array (WaferGen’s SmartChip mRNA array) [9] and proteomic array (Protein Pathway Array) [10-12] to investigate the gene expression and protein levels between normal and tumor tissues. The unique aspect of these arrays compared to other microarrays is that the mRNAs and proteins detected by this array are those involved in cancer pathogenesis as well as signaling pathways. Therefore, the results of this study can provide more mechanistic information as compared to other studies based on global gene expression. The goal of this study is to understand signaling pathways involved in breast cancer and to compare the difference between mRNA expression and protein levels in breast cancer.
Methods
Clinical information and tissue samples
Thirty three paired normal and tumor tissues were obtained from thirty three patients with invasive ductal carcinoma who underwent lumpectomy between August, 2006 and January, 2008 at The First Hospital of Jilin University, Jilin, China. Among 33 breast cancers, 20 were PR+, 13 PR-, 25 ER+, 8 ER-, 11 HER2+, 22 HER2- and 3 triple negative (staining intensity greater than 2+). In addition, 18 paraffin embedded tissue samples (13 cancer and 5 normal) were collected from these 33 patients where enough tissue were available. The patients’ mean age was 51.7 (ranged from 37 to 73). Of these 13 cancer cases, 11 were ER+, 8 PR+, and 4+ HER2. Informed consent was obtained from each participants. This study was reviewed and approved by the Institution Ethical Review Boards of The First Hospital of Jilin University.
SmartChip real-time PCR analysis
Each formalin fixed paraffin embedded (FFPE) tissue using an RNA Extraction Kit (Qiagen, Valencia, CA) following the manufacturer’s instructions [13]. RNA concentration was determined using a NanoDrop (Thermo Scientific, Wilmington DE). The RNA samples were analyzed using the WaferGen’s SmartChip Real-Time PCR System using the SmartChip Human Oncology Panel V2 (WaferGen Biosystems, Fremont, CA) [9]. The reverse transcriptase reaction was performed prior to applying the sample to the chip using a High-Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA) following the recommendations of the manufacturer. A PCR cocktail containing SYBR Green I dye and the cDNA pool for each sample (an equivalent of 100 pg of starting RNA) was loaded onto a SmartChip Human Oncology Panel V2 in 100 nl/reaction volume. The SmartChip Human Oncology Panel V2 contains 1,243 unique real-time PCR reactions in quadruplicate for a total of 5,184 reactions/sample. Forty cycles of real-time PCR were performed on the SmartChip Cycler, followed by a melt curve analysis. Both raw Ct and Tm values for each assay and sample were collected for data analysis. A data quality screen based on amplification, Tm values from melting curves, and Ct and Tm variability was performed to remove any outlier data before delta-delta Ct calculations were used to determine fold change in mRNA levels.
The mean Ct and Tm values calculated from each of the 4 replicates on each SmartChip Panel were calculated by the SmartChip qPCR Software (WaferGen Biosystems, Fremont, CA). For the sake of ease of computation, those mRNA samples without Ct value generated (i.e., mRNAs absent or below the level of detection) were assigned Ct values of 30.0. Delta Ct values were computed using the mean of all replicate Ct values for each sample/chip, not including Cts of 30 [14]. The average delta Ct for the tumor samples (D1, D2, and D3) and the Normal samples (N1, N2, and N3) for each assay were then computed. From these, Delta-delta-Cts were computed (deltaCtTumor-deltaCtNormal).
Protein pathway array analysis
Briefly [15], 1 ml of 1× lysis buffer (Cell Signaling Technology, Danvers, MA) with 1× protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN) and 1× phosphatase inhibitor cocktail (Roche Applied Science, Indianapolis, IN) was added into each tissue sample and the lysate was sonicated twice for 15 seconds each time on ice, and then centrifuged at 14,000 rpm for 30 minutes at 4°C. The protein concentration was determined using BCA Protein Assay kit (PIERCE, Rockford, IL). Three hundred μg of protein lysate was loaded in one well across the entire width of 10% SDS polyacrylamide gel and separated by electrophoresis as described previously [15]. After electrophoresis, the proteins were transferred electrophoretically to a nitrocellulose membrane (Bio-Rad, Hercules, CA) which was then blocked for 1 hour with blocking buffer including 3% BSA in 1× TBST containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl and 0.1% Tween-20. Next, the membrane was clamped on a Western blotting manifold (Mini-PROTEAN II Multiscreen apparatus, Bio-Rad, Hercules, CA) which isolates 20 channels across the membrane. The multiplex immunoblot was performed using a total of 131 protein-specific or phosphorylation site-specific antibodies. Four sets of antibodies (a total of 30-36 protein-specific or phosphorylation site-specific antibodies) were individually used for each membrane and all of antibodies (from various companies) were validated independently before inclusion in Protein Pathway Array. For the first set of primary antibodies, a mixture of two antibodies in the blocking buffer were added into each channel and then incubated at 4°C overnight. The membrane was then washed with 1× TBS and 1× TBST, and was further incubated with secondary anti-rabbit (Bio-Rad, Hercules, CA), anti-mouse (Bio-Rad, Hercules, CA) or anti-goat (Santa Cruz Biotechnology, Santa Cruz, CA) antibody conjugated with horseradish peroxidase for 1 hour at room temperature. The membrane was developed with chemiluminescence substrate (Immun-StarTM HRP Peroxide Buffer/Immun-StarTM HRP Luminol Enhancer) (Bio-Rad, Hercules, CA), and chemiluminescent signals were captured using the ChemiDoc XRS System (Bio-Rad, Hercules, CA). The same membrane was then stripped off using stripping buffer (RestoreTM Western blot stripping buffer, Thermo Scientific, Rockford, IL) and then used to detect a second set of primary antibodies as described above.
For Protein Pathway Array data analysis, the signals of each protein were determined by densitometric scanning (Quantity One software package, Bio-Rad) and the background was locally subtracted from raw protein signal. The background-subtracted intensity was normalized by “global median subtraction” method to reduce the variations among different experiments, that is, the intensity of each protein from each sample divided by total intensities of all proteins from the same sample and then multiplied by average intensities of all proteins in all samples [15]. Significant Analysis of Microarray (SAM) tool (http://www-stat.stanford.edu/~tibs/SAM/) was used to select the differentially expressed proteins between tumors and normal tissues. A q value <0.05 was considered to be statistically significant.
Results
Differentially expressed mRNAs and proteins
SmartChip Human Oncology Panel contains a total of 1,243 gene-specific PCRs covering 16 functional groups, including signal transduction (383), apoptosis (256), angiogenesis (215), inflammation (181), kinase (154), transcription factors (127), cell cycle/proliferation (21), growth factors (5), and cancer related genes (262). A total of 854 mRNAs were detected, 395 mRNAs with >2 fold changes compared between tumors and normal tissues, 141 mRNAs were increased and 254 were down regulated in breast cancer as compared with normal tissues. Interestingly, 33 mRNAs only expressed in normal tissue and 66 mRNAs only expressed in tumor tissues (Supplementary Data 1).
A total of 131 proteins and phosphoproteins were tested using Protein Pathway Array. Of 131 proteins, 37 were phosphoproteins and 31 proteins had corresponding mRNAs in SmartChip panel. Among 131 proteins and phosphoproteins, 89 were detected and 57 showed significant changes between normal and tumor tissues as determined by SAM analysis. Among these proteins, 38 had increased expression in tumor tissues including MetRS, STAT1, CDK2, BAX, p-PDK1, PCNA, XIAP, p-PKCα, p-PTEN, PCNA, p-CREB, HSP90, β-catenin, CDC2 P34, PTEN, P38, CDK6, α-tubulin, RAP1, p-CDC2, E-cadherin, FOXM1, CDC42, P27, CDK4, cPKCα, p-STAT3, TWIST, NOTCH4, BCL2, TNF-α, p-P38, HIF-1α, ERβ, ERα, Mesothelin, NEU and SRC. In contrast, 19 had decreased expression in tumor tissues including Cyclin B1, Calretinin, NFkB50, BCL6, NFkB p65, FAS, TDP1, SK3, SLUG, p-FAK, CHK1, Cyclin E, CaMKKα, KLF6, Vimentin, p-CREB, TTF-1, AKT and CDC25C.
Concordance between protein level and mRNA expression
In comparing the changes between proteins and mRNAs, 12 proteins and/or mRNAs had significant differences (Figure 1), i.e. fold change, between tumors and control tissues (Table 1). Four proteins had concordant change with mRNAs, including ERα, BCL6, Vimentin and SLUG, suggesting that increased mRNAs lead to increase of protein expression. Four proteins including CDK4, BCL2, β-catenin and PCNA had no change between tumor and normal tissues but showed an increase of their corresponding mRNAs. The other 2 proteins, including p-STAT3 and BAX, showed dis-concordant changes, that is the protein expressions increased but their corresponding mRNAs decreased. These results suggest that the changes in the proteins may not be the result of increased mRNA levels. These results further suggested that there is a negative feedback between protein activation/expression and mRNA expression.
Figure 1.
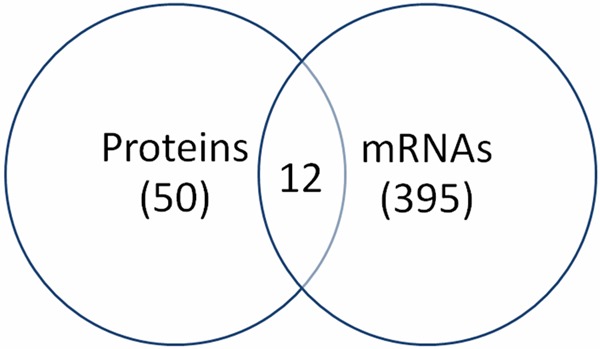
Differentially expressed proteins and mRNAs in breast cancer. 57 proteins and 395 mRNAs were altered in breast cancer. Only 12 genes had changes in both protein and mRNA level.
Table 1.
Differentially expressed mRNA and proteins between tumor and normal tissues
| Protein | PPA | mRNA |
|---|---|---|
| CDK4 | +1.02 | +7.47 |
| ERα | +1.9 | +4.66 |
| BCL2 | +1 | +4.2 |
| β-catenin | +1 | +2.86 |
| PCNA | +1.02 | +2.72 |
| p-STAT3 | +1.83 | -2.1 |
| BAX | +2.33 | -3.6 |
| CDK2 | +1.01 | -4.9 |
| BCL6 | -1.9 | -5.6 |
| CDK6 | +1.03 | -6.7 |
| vimentin | -1.5 | -9.0 |
| SLUG | -1.5 | -15.5 |
Note: the numbers represent fold difference between tumor and normal tissues. +, represents increased expression. –, represents down-regulation.
Differential expression among different subtypes of breast cancer
We further examined the differentially expressed proteins in ER+, PR+ and HER2+ breast cancers. In our cohort of 33 breast cancer cases, 20 were PR+, 13 PR-, 25 ER+, 8 ER-, 11 HER2+, 22 HER2- and 3 triple negative. When compared between PR+ and PR- breast cancers, one phosphoprotein (p-CREB) showed increased expression and 2 proteins (Cyclin E and WT1) were down-regulated in PR+ tumors. In ER+ breast cancer, the levels of p-PDK1, E-cadherin and metRS were up-regulated but the levels of p-PKCα, TNFα, SLUG decreased. In HER2+ breast cancer, Cyclin E, WT1, NF-kBp65 and BCL6 were up-regulated but PCNA, KLF6, TDP1, CaMKKα, HIF-3α and P38 were down-regulated. In triple negative breast cancer, only p-CDC2 was down-regulated. These results demonstrated a distinct change in protein expression related to status of PR, ER and HER2.
Signaling pathways altered in breast cancer
In order to determine the pathways altered in breast cancer, the differentially expressed proteins and mRNAs were entered into Ingenuity Pathway Analysis (IPA, www.ingenuity.com), a computer based data system which contains all known interactions. The results showed that many important cancer-related signaling pathways were affected (Figure 2), including P53, ERK, ATM, PTEN, PIK3/AKT, PPAR and Wnt/β-catenin. In addition, the growth pathways (HGF, IGF, NGF, TGH, FGF), cytokines (IL6, IL9 and IL15), and inflammation pathway (NOS and NF-kB) were overwhelmingly affected. These results suggest that wide arrays of alterations occurred in breast cancer. It is interest to note that the pathways determined based on protein changes and mRNA levels were different. For example, the top signaling pathways as determined based on the protein expression were p53 (p=10), ILK (p=8), GNRH (p=8). However, the top signaling pathways based on mRNA expression were PTEN (p=23), p53 (p=19), PIK3/AKT (p=18). These differences may be due to the numbers of pathway components included in 2 different platforms as well as differences in regulation of these pathways between proteins and mRNAs.
Figure 2.
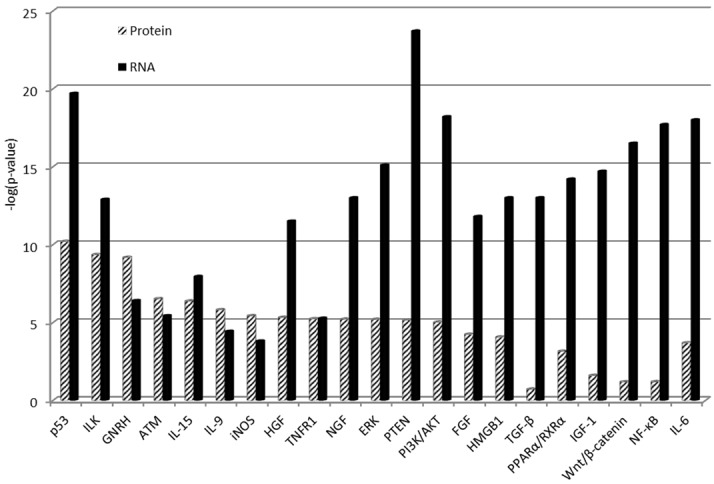
mRNA expression and protein abundance of signaling pathway genes altered in breast cancer. The higher the p values, the stronger association.
Integrated signaling network
In order to understand how these pathway components, as determined by protein and mRNA expression, interacted with each other, we overlapped the interactomes based on protein and mRNA inputs. As showed in Figure 3, there is significant overlap between proteins and mRNAs among different pathways. The only exception is that P38 was increased at protein level but decreased at mRNA levels, suggesting a different mechanism of regulation of protein and mRNA. Several important receptors including GNRH, EGFR, Integrin and TNFR and their associated pathways were involved in breast cancer, suggesting the important roles of hormones, growth factors and inflammation in breast cancer pathogenesis. Furthermore, there is significant interaction among different pathways, resulted in changes of many common down-stream effectors. Taking together, these data confirmed that breast cancer is a disease affected by multiple stimulants via different pathways.
Figure 3.
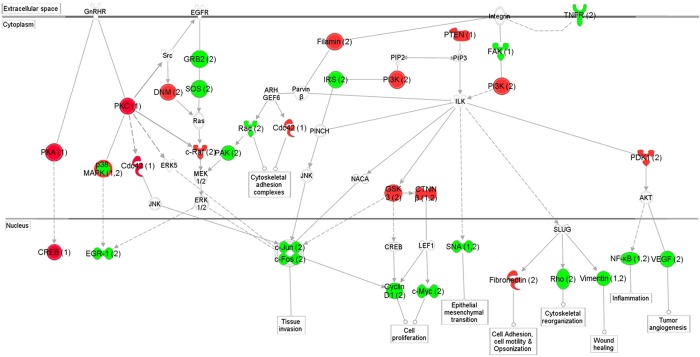
Schematic representation of the signaling networks affected in breast cancer as determined by protein and mRNA expressions. The network is displayed graphically as nodes (protein or mRNA) and edges (the biological relationship between the nodes). The nodes are represented using various shapes that represent the functional class of the protein products. The up- and down-regulation of each node is shaded in red and green, respectively. The nodes without color were not assessed or without difference in this study but identified by IPA as important nodes involved in the network. The number after each node indicates the changes determined by either protein (1) or mRNA (2). The P38 is up regulated in the protein level but down regulated in the mRNA level.
Discussion
Breast cancer is a complex disease that results from the dysregulation of signaling networks caused by the genetic and epigenetic alterations in cells. The understanding of pathways involved in breast cancer pathogenesis contributes to the identification of new targeted therapeutic agents as well and can also be utilized in diagnosis and prognosis. Although many studies based on global profiling of mRNA expression and protein levels of breast cancer have been published before [16-20], few studies have focused on the analysis of cancer-related genes and pathways. Furthermore, few studies have combined both genomic and proteomic approaches to study breast cancer. In this study, we were able to analyze 1,243 mRNA and 159 protein/phosphoprotein expression in the invasive breast cancer. Our results showed that 395 mRNAs and 57 proteins (Figure 1) were differentially expressed in cancer tissues. The discovery rate of differentially expressed mRNA and proteins in this study is about 31% for all genes analyzed and 43% for all proteins analyzed, which is significantly higher than global profiling which is about 2~6% [10].
Relatively little is known about the regulatory mechanisms that control the complex patterns of protein abundance and post-translational modification in tumors. It has been reported that there is a significant discordance between mRNA expression and protein levels. For example, Pascal et al reported that the discordance between CD antigen gene and protein expression in prostate cancer ranged from 32% to 54% and the correlation of expression levels was poor to moderate (Pearson correlations ranged from 0 to 0.63) [21]. Another study by Chen et al. found that only 17% of genes in lung adenocarcinoma had a significant correlation between mRNA and protein expression [22]. This study also found that the expression of individual isoforms of the same protein may or may not correlate with the mRNA, indicating that separate and likely post-translational mechanisms account for the regulation of isoform abundance [22]. In our study, 5 of the 12 proteins (41.7%) had changes discordant with their mRNAs. Therefore, assessment of mRNA levels will not adequately evaluate the protein changes. These results suggest that both post-transcriptional and/or post-translational modifications are important in regulation of protein expression or protein activation.
Many signaling pathways are implicated in breast cancer pathogenesis. Some important examples of this are the HER-2 tyrosine kinase pathway [5], the hedgehog signaling pathway [23], p53 pathway, and PTEN pathway. Our results showed the p53 pathway is most significantly altered in breast cancer at both protein and mRNA levels (Figure 2), suggesting its important role in carcinogenesis. The p53 pathway is very important in the repair of DNA damage and it has been reported that the p53 mutation is relatively common in breast cancer (approximately 20% of tumors harbor this mutation) [6,24]. PTEN and PI3K/AKT pathways were significantly altered but mostly at mRNA level, suggesting a transcriptional level change [25]. Alteration of both PTEN and PIK3CA is commonly found in breast cancer and alteration of PTEN has been reported to be involved in breast cancer resistance to treatment [25,26]. Several cell growth receptor pathways were altered in breast cancer, including HGF, NGF, FGF and IGF, suggesting that many growth factors are interacting with breast cancer cells, resulting in uncontrolled proliferation and survival advantage of cancer cells. Our study also showed that many cytokine and inflammatory pathways are also altered in breast cancer including LI6, NF-kB, TGF-β, TNF, iNOS, IL9 and IL15, suggesting the critical role of inflammation in breast cancer carcinogenesis. The study has shown that the transforming growth factor (TGF)-β [27] pathway is activated either chemically or genetically as determined by activated signature of the pathway.
Finally, we also evaluated the expression patterns among different subtypes of breast cancer. We found a distinct change in protein expression related to different status of PR, ER and HER2. In the triple negative breast cancer phosphorylated-CDC2 was down-regulated. This is a particularly interesting finding because CDC2 functions in conjunction with Cyclin B1 in the regulation of mitosis and stimulate cell cycle progression, a process that is dysregulated in breast cancer [28]. In HER2+ breast cancer, Cyclin E, WT1, NF-kBp65 and BCL6 were up-regulated, suggesting that these proteins are regulated by HER2 pathway. It has been reported that HER2/neu engages Akt to increase WT1 expression, and that WT1 protein plays a vital role in regulating cell cycle progression and apoptosis in HER2/neu-overexpressing breast cancer cells [29]. In addition, the NF-kappaB pathway has been shown to be activated downstream of Her2 overexpression [30]. Cyclin E overexpression has been described to be a mechanism of trastuzumab resistance in HER2+ breast cancer patients [15]. In ER+ breast cancer, the levels of p-PDK1, E-cadherin and metRS were up-regulated, suggesting the involvement of these proteins in ER pathway.
In conclusion, this study evaluated a multitude of genes and proteins to evaluate the differential expression between breast carcinoma and benign tissue. This elucidated several important genes and proteins involved in the signaling pathways of breast cancer pathogenesis. The study also found that mRNA expression did not correlate with protein level therefore suggesting different regulation mechanisms between proteins and mRNAs. Future studies will focus on understanding the role of these proteins and pathways in breast cancer pathogenesis.
Disclosure of conflict of interest
The authors declare that they have no competing interests.
Supporting Information
References
- 1.Gage M, Wattendorf D, Henry LR. Translational advances regarding hereditary breast cancer syndromes. J Surg Oncol. 2012;105:444–451. doi: 10.1002/jso.21856. [DOI] [PubMed] [Google Scholar]
- 2.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM, Pisansky TM, Fehrenbacher L, Kutteh LA, Vogel VG, Visscher DW, Yothers G, Jenkins RB, Brown AM, Dakhil SR, Mamounas EP, Lingle WL, Klein PM, Ingle JN, Wolmark N. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 3.Eswaran J, Cyanam D, Mudvari P, Reddy SD, Pakala SB, Nair SS, Florea L, Fuqua SA, Godbole S, Kumar R. Transcriptomic landscape of breast cancers through mRNA sequencing. Sci Rep. 2012;2:264. doi: 10.1038/srep00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 5.Pietras RJ, Arboleda J, Reese DM, Wongvipat N, Pegram MD, Ramos L, Gorman CM, Parker MG, Sliwkowski MX, Slamon DJ. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone independent growth in human breast cancer cells. Oncogene. 1995;10:2435–2446. [PubMed] [Google Scholar]
- 6.Pharoah PD, Day NE, Caldas C. Somatic mutations in the p53 gene and prognosis in breast cancer: a meta-analysis. Br J Cancer. 1999;80:1968–1973. doi: 10.1038/sj.bjc.6690628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tilghman SL, Townley I, Zhong Q, Carriere PP, Zou J, Llopis SD, Preyan LC, Williams CC, Skripnikova E, Bratton MR, Zhang Q, Wang G. Proteomic signatures of acquired letrozole resistance in breast cancer: suppressed estrogen signaling and increased cell motility and invasiveness. Mol Cell Proteomics. 2013;12:2440–2455. doi: 10.1074/mcp.M112.023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schramm G, Kannabiran N, Konig R. Regulation patterns in signaling networks of cancer. BMC Syst Biol. 2010;4:162. doi: 10.1186/1752-0509-4-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen HJ, Edwards R, Tucci S, Bu P, Milsom J, Lee S, Edelmann W, Gumus ZH, Shen X, Lipkin S. Chemokine 25-induced signaling suppresses colon cancer invasion and metastasis. J Clin Invest. 2012;122:3184–3196. doi: 10.1172/JCI62110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang DY, Ye F, Gao L, Liu X, Zhao X, Che Y, Wang H, Wang L, Wu J, Song D, Liu W, Xu H, Jiang B, Zhang W, Wang J, Lee P. Proteomics, pathway array and signaling network-based medicine in cancer. Cell Div. 2009;4:20. doi: 10.1186/1747-1028-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W, Ye F, Wang D, Sun X, Tong W, Lian G, Jiang J, Suo J, Zhang DY. Protein predictive signatures for lymph node metastasis of gastric cancer. Int J Cancer. 2013;132:1851–1859. doi: 10.1002/ijc.27864. [DOI] [PubMed] [Google Scholar]
- 12.Wang D, Ye F, Sun Y, Li W, Liu H, Jiang J, Zhang Y, Liu C, Tong W, Gao L, Sun Y, Zhang W, Seetoe T, Lee P, Suo J, Zhang DY. Protein signatures for classification and prognosis of gastric cancer a signaling pathway-based approach. Am J Pathol. 2011;179:1657–1666. doi: 10.1016/j.ajpath.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deben C, Zwaenepoel K, Boeckx C, Wouters A, Pauwels P, Peeters M, Lardon F, Baay M, Deschoolmeester V. Expression analysis on archival material revisited: isolation and quantification of RNA extracted from FFPE samples. Diagn Mol Pathol. 2013;22:59–64. doi: 10.1097/PDM.0b013e318269de3b. [DOI] [PubMed] [Google Scholar]
- 14.Mestdagh P, Fredlund E, Pattyn F, Schulte JH, Muth D, Vermeulen J, Kumps C, Schlierf S, De Preter K, Van Roy N, Noguera R, Laureys G, Schramm A, Eggert A, Westermann F, Speleman F, Vandesompele J. MYCN/c-MYC-induced microRNAs repress coding gene networks associated with poor outcome in MYCN/c-MYC-activated tumors. Oncogene. 2010;29:1394–1404. doi: 10.1038/onc.2009.429. [DOI] [PubMed] [Google Scholar]
- 15.Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J, Chandarlapaty S, Serra V, Prat A, Ibrahim YH, Guzman M, Gili M, Rodriguez O, Rodriguez S, Perez J, Green SR, Mai S, Rosen N, Hudis C, Baselga J. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci U S A. 2011;108:3761–3766. doi: 10.1073/pnas.1014835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kittaneh M, Montero AJ, Gluck S. Molecular Profiling for Breast Cancer: A Comprehensive Review. Biomark Cancer. 2013;5:61–70. doi: 10.4137/BIC.S9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dey N, Barwick BG, Moreno CS, Ordanic-Kodani M, Chen Z, Oprea-Ilies G, Tang W, Catzavelos C, Kerstann KF, Sledge GW Jr, Abramovitz M, Bouzyk M, De P, Leyland-Jones BR. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer. 2013;13:537. doi: 10.1186/1471-2407-13-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaheed SU, Rustogi N, Scally A, Wilson J, Thygesen H, Loizidou MA, Hadjisavvas A, Hanby A, Speirs V, Loadman P, Linforth R, Kyriacou K, Sutton CW. Identification of stage-specific breast markers using quantitative proteomics. J Proteome Res. 2013;12:5696–5708. doi: 10.1021/pr400662k. [DOI] [PubMed] [Google Scholar]
- 19.Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378:1812–1823. doi: 10.1016/S0140-6736(11)61539-0. [DOI] [PubMed] [Google Scholar]
- 20.Goncalves A, Bertucci F. Clinical application of proteomics in breast cancer: state of the art and perspectives. Med Princ Pract. 2011;20:4–18. doi: 10.1159/000319544. [DOI] [PubMed] [Google Scholar]
- 21.Pascal LE, True LD, Campbell DS, Deutsch EW, Risk M, Coleman IM, Eichner LJ, Nelson PS, Liu AY. Correlation of mRNA and protein levels: cell type-specific gene expression of cluster designation antigens in the prostate. BMC Genomics. 2008;9:246. doi: 10.1186/1471-2164-9-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, Gharib TG, Huang CC, Taylor JM, Misek DE, Kardia SL, Giordano TJ, Iannettoni MD, Orringer MB, Hanash SM, Beer DG. Discordant protein and mRNA expression in lung adenocarcinomas. Mol Cell Proteomics. 2002;1:304–313. doi: 10.1074/mcp.m200008-mcp200. [DOI] [PubMed] [Google Scholar]
- 23.Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, Nomura M, Kuroki S, Katano M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004;64:6071–6074. doi: 10.1158/0008-5472.CAN-04-0416. [DOI] [PubMed] [Google Scholar]
- 24.Boyault S, Drouet Y, Navarro C, Bachelot T, Lasset C, Treilleux I, Tabone E, Puisieux A, Wang Q. Mutational characterization of individual breast tumors: TP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res Treat. 2012;132:29–39. doi: 10.1007/s10549-011-1518-y. [DOI] [PubMed] [Google Scholar]
- 25.Craig DW, O’Shaughnessy JA, Kiefer JA, Aldrich J, Sinari S, Moses TM, Wong S, Dinh J, Christoforides A, Blum JL, Aitelli CL, Osborne CR, Izatt T, Kurdoglu A, Baker A, Koeman J, Barbacioru C, Sakarya O, De La Vega FM, Siddiqui A, Hoang L, Billings PR, Salhia B, Tolcher AW, Trent JM, Mousses S, Von Hoff D, Carpten JD. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol Cancer Ther. 2013;12:104–116. doi: 10.1158/1535-7163.MCT-12-0781. [DOI] [PubMed] [Google Scholar]
- 26.Hernandez-Aya LF, Gonzalez-Angulo AM. Targeting the phosphatidylinositol 3-kinase signaling pathway in breast cancer. Oncologist. 2011;16:404–414. doi: 10.1634/theoncologist.2010-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang Y, Chen CR, Massague J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11:915–926. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- 28.Chae SW, Sohn JH, Kim DH, Choi YJ, Park YL, Kim K, Cho YH, Pyo JS, Kim JH. Overexpressions of Cyclin B1, cdc2, p16 and p53 in human breast cancer: the clinicopathologic correlations and prognostic implications. Yonsei Med J. 2011;52:445–453. doi: 10.3349/ymj.2011.52.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuna M, Chavez-Reyes A, Tari AM. HER2/neu increases the expression of Wilms’ Tumor 1 (WT1) protein to stimulate S-phase proliferation and inhibit apoptosis in breast cancer cells. Oncogene. 2005;24:1648–1652. doi: 10.1038/sj.onc.1208345. [DOI] [PubMed] [Google Scholar]
- 30.Merkhofer EC, Cogswell P, Baldwin AS. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene. 2010;29:1238–1248. doi: 10.1038/onc.2009.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.