

Abstract
Recent lines of inquiry indicate sedatives can influence the immune system, leading to the concept of sedative-induced immunomodulation. It has been hypothesized that sedatives may alter immune responses by modulating the sympathetic nervous system, however, little information is known regarding the effects of sedatives on regulation of splenic sympathetic nerve discharge (SND), a significant omission based on the functional role that changes in splenic SND exert on splenic cytokine gene expression. The present investigation determined the effect of systemic Dexmedetomidine (Dex) administration on the level of directly-recorded splenic SND and tested the hypothesis that the intravenous administration of Dex would inhibit splenic SND in anesthetized rats. The present results demonstrate for the first time that intravenous Dex administration significantly reduces splenic sympathetic nerve outflow in baroreceptor-intact and sinoaortic-denervated rats, indicating that Dex administration alters the central regulation of splenic SND. The present results provide new information regarding the effect of a centrally-acting alpha2-adrenergic agonist on the level of sympathetic nerve outflow to a secondary lymphoid organ that plays a critical role in peripheral immune responses.
Keywords: Dexmedetomidine, splenic SND, renal SND, arterial blood pressure
INTRODUCTION
The management of critically ill patients often requires sedatives to establish and maintain appropriate levels of patient treatment, comfort, and cooperation. Sedatives influence a number of physiological systems and responses, including the immune system, leading to the concept of sedative-induced immunomodulation (Galley et al., 2000; Taniguchi et al., 2004; Maclaren, 2009; Qiao et al., 2009; Sanders et al., 2009; Kayhan et al., 2013). Because of the potential for infections and sepsis in intensive care settings, it has been suggested that immunomodulatory properties should be considered in the clinical decision-making process regarding the inclusion of sedatives in drug regimens (Sanders et al., 2009).
Dexmedetomidine (Dex), an alpha2-adrenergic receptor agonist, is used as a sedative agent in intensive care settings for critically-ill patients (Qiao et al., 2009; Kayhan et al., 2013), and has shown promising immunomodulatory contributions. Qiao and colleagues (Qiao et al., 2009) reported reduced plasma levels of the proinflammatory cytokines, tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), and reduced splenic caspase-3 expression, a marker of apoptosis, in Dex-treated compared with nontreated septic rats. Dex administration exerted positive effects in mice with inflammatory bowel disease, including increased levels of the anti-inflammatory cytokine IL-10 and reduced levels of the proinflammatory cytokine IL-23, a contributor to colitis-induced intestinal inflammation (Kayhan et al., 2013).
The sympathetic nervous system (SNS) plays a crucial role in physiological regulation. Sympathetic nerves innervating most target organs are tonically active, and the acute responsivity of the SNS is considered an important regulatory feature of this adaptive nervous system. Direct recordings of sympathetic nerve discharge (SND) provide an output measure of central sympathetic neural circuits. A fundamental SNS regulatory strategy involves selectively controlling the level of activity in nerves innervating different targets in response to specific physiological conditions and experimental interventions (Morrison, 2001). Alpha2-adrenergic receptor agonists influence SND regulation. Intravenous Dex administration reduces renal SND in anesthetized baroreceptor-intact (intact) and baroreceptor-denervated (SAD) rabbits (Oku et al., 1996; Xu et al., 1998), whereas intracerebroventricular Dex administration reduces plasma norepinephrine levels in conscious rats (Shirasaka et al., 2007). In addition, intravenous Dex abolishes cocaine-induced increases in skin SND in conscious humans (Menon et al., 2007; Kontak et al., 2013).
The nervous system and the immune system communicate via an extensive array of bidirectional pathways, and the SNS is an important component of the efferent arm mediating neural-immune interactions. For example, the sympathetic innervation to the spleen, via the splenic nerve, provides a neural communication pathway from central sympathetic neural circuits to immunocompetent cells in this lymphoid organ (Felten et al., 1985; Felten et al., 1987). Previous studies by Ganta et al. (2004, 2005) support the idea that alterations in the level of efferent splenic SND can modulate splenic cytokine gene expression. For example, hyperthermia produces a potent stimulus to visceral sympathetic nerve outflow, including pronounced splenic sympathoexcitation (Ganta et al., 2004). In rats with intact splenic nerves, splenic expression of IL-1β, IL-6, and growth-regulated oncogene 1 mRNA are higher in hyperthermic compared with normothermic subjects (Ganta et al., 2004). The splenic expression of IL-1β, IL-6, and growth-regulated oncogene 1 mRNA in response to heating is lower in splenic nerve-denervated compared with splenic nerve-intact rats, but is similar in heated splenic nerve-denervated and nonheated splenic nerve-intact rats (Ganta et al., 2004). Moreover, central administration of angiotensin II modulates splenic cytokine gene expression secondary to activation of splenic sympathetic nerve outflow (Ganta et al., 2005), supporting a functional relationship between central angiotensin II, splenic SND, and peripheral immune responses.
It is intriguing to postulate that sedatives may influence immune responses by modulating the sympathetic nervous system, however, little to nothing is known regarding the effects of sedatives on regulation of splenic SND, a significant omission based on the functional role that changes in splenic SND can exert on splenic cytokine gene expression. The purpose of the present investigation was to determine the effect of systemic Dex administration on the level of directly-recorded splenic SND. We tested the hypothesis that the intravenous administration of Dex would inhibit splenic SND in intact and SAD rats. Experiments were completed in SAD rats to eliminate the influence of baroreceptor afferent feedback pathways that can influence changes in SND of central origin. Renal SND recordings provided a visceral nerve control.
METHODS
The procedures and protocols were performed in accordance with the American Physiological Society’s guiding principles for research involving animals and approved by the Institutional Animal Care and Use Committee at Kansas State University.
General Procedures
Experiments were completed using male Fischer 344 rats (300–400 grams). Anesthesia was induced by isoflurane (3–5%) and maintained during surgical procedures using isoflurane (1.25%–1.75%), α-chloralose (80 mg/kg ip), and urethane (800 mg/kg ip) (Hosking et al., 2009; Margiocco et al., 2010; Garcia et al., 2012). Two catheters (polyethylene-10 and polyethylene-50) were placed in the femoral vein. Isoflurane anesthesia was discontinued following completion of the surgical procedures. During experimental protocols, maintenance doses of α-chloralose (35–45 mg/kg/hr) were administered intravenously and maintenance doses of urethane (200 mg/kg every 4 hours) were administered intraperitoneally. The adequacy of anesthesia during the initial surgical procedures was indicated by the absence of a withdrawal reflex in response to mechanical stimulation of the tail or hindlimb. Femoral arterial pressure was monitored using a pressure transducer connected to a blood pressure analyzer. Core temperature was measured with a thermistor probe inserted approximately 3–4 cm into the colon and was kept at 37.5–37.8°C during surgical interventions by a temperature-controlled table.
To eliminate the influence of baroreceptor afferent feedback mechanisms that can alter SND responses of central origin, some experiments included in this study were completed in SAD rats. Bilateral denervation of the aortic arch was completed by sectioning the superior laryngeal nerve near its junction with the vagus nerve and removing the superior cervical ganglion. Bilateral carotid sinus denervation was completed by removing the adventitia from the area of the carotid sinus bifurcation. Sinoaortic-denervations were completed 3–4 hours before initiation of experimental protocols. Denervation was considered complete by the absence of a reflex change in SND during increases in mean arterial pressure (MAP) produced by phenylephrine hydrochloride (PE, 3–5 μg/kg iv) (see Results) and decreases in MAP produced by sodium nitroprusside (3–5 μg/kg iv), and loss of coherence between pulsatile arterial pressure and SND (Hirai et al., 1995). Coherence analysis provides a measure of the strength of linear correlation of two signals as a function of frequency (Kenney, 1994).
Sympathetic Nerve Recordings
Activity was recorded biphasically with a platinum bipolar electrode after capacity-coupled preamplification (bandpass 30–3000 Hz) from renal and splenic sympathetic nerves. Renal SND was recorded because the sympathetic neural innervation to the kidney influences physiological parameters involved in blood pressure regulation, including renal blood flow, renin release, and salt and water retention by the renal tubules (Koepke and DiBona, 1985; DiBona, 1994). Splenic SND was recorded because the sympathetic innervation to this peripheral lymphoid organ provides a link from central sympathetic neural circuits to splenic immune cells and influences immune function (Felten et al., 1985; Felten et al., 1987). Sympathetic nerves were isolated from a lateral incision and nerve-electrode preparations were covered with silicone gel to prevent exposure to room air. Filtered neurograms were routed to an oscilloscope and a nerve traffic analyzer where sympathetic nerve potentials were full wave rectified and integrated (time constant 10 ms). Total power in renal and splenic SND was quantified as microvolts x seconds (μV·s) (Hosking et al., 2009). SND recordings were corrected for background noise after ganglionic blockade (chlorisondamine, 5 mg/kg iv). The adequacy of anesthesia was demonstrated by an inability of mechanical stimulation of the hindlimb or tail to increase MAP or SND.
Experimental Protocols
After completion of surgical procedures, anesthetized rats were allowed to stabilize for 60 min before initiation of experimental protocols. Following stabilization, MAP, renal SND, and splenic SND were measured and recorded for 15 min and control values were determined from the average of the final 5 min (baseline is represented as time 0 in Figure 2). Rats received Dex (0.5–1.0 μg/kg; 3 μg/kg; or 6–10 μg/kg) dissolved in 300 μl saline or vehicle (saline 300 μl). Dex and vehicle were administered as intravenous (iv) bolus doses via a femoral venous catheter. The range of Dex doses was selected based on previous studies in animals (Xu et al., 1998; 10 μg/kg iv: Oku et al., 1996; 3 μg/kg iv) and humans (Kontak et al., 2013; 0.1–1.0 μg/kg iv). Experiments were completed in baroreceptor-intact (intact) and SAD rats. MAP, renal SND, and splenic SND were recorded continuously for 60 min after completion of intravenous administration of Dex or vehicle. At the end of experiments rats were euthanized by an intravenous overdose of methohexital sodium (150 mg/kg iv).
Figure 2.

Mean arterial pressure (MAP, mmHg), renal sympathetic nerve discharge (SND, %Δ), and splenic SND (%Δ) responses to intravenous administration of saline (Intact, n=5; SAD, n=6; open triangles) or Dexmedetomidine (Dex) in baroreceptor-intact (Intact, left panels) (three Dex doses or dose ranges: 6–10 μg/kg, n=6, closed circles; 3 μg/kg, n=9, open circles; 0.5–1 μg/kg, n=6, closed triangles) and sinoaortic-denervated (SAD, right panels) (two Dex doses or dose ranges: 3 μg/kg, n=6, open circles; 0.5–1 μg/kg, n=3, closed triangles) F344 rats. Baseline levels are represented as Time 0. Recordings were maintained for 60 min after administration of Dex or saline. *Dex significantly different from control for the three Dex doses (6–10 μg/kg, 3 μg/kg, 0.5–1 μg/kg) administered to Intact rats and for the two Dex doses (3 μg/kg and 0.5–1 μg/kg) administered to SAD rats. **Dex 6–10 μg/kg and 3 μg/kg doses significantly different from the 0.5–1 μg/kg Dex dose in Intact rats. **Dex 3 μg/kg Dex dose significantly different from the 0.5–1 μg/kg Dex dose in SAD rats. ǂMAP significantly different from control values in response to each of the Dex doses administered to Intact and SAD rats.
Data Collection and Statistical Analysis
A computer-based ADInstruments Powerlab data acquisition system was used to collect all experimental data. Values are reported as means ± SE. Renal and splenic SND data are expressed as percentage change from control values. Statistical analyses included analysis of variance techniques with a repeated-measures design, followed by Bonferroni post hoc tests. The overall level of statistical significance was p<0.05.
RESULTS
Figure 1 shows representative traces of SND (renal and splenic) and pulsatile arterial blood pressure recorded before, during and after PE-induced increases in arterial blood pressure (depicted by arrows) in a rat with intact arterial baroreceptors (A) and in a SAD rat (B). Note the absence of a reflex inhibition of SND in response to PE-induced increases in arterial blood pressure in the SAD rat, demonstrating the functional efficacy of the arterial baroreceptor denervation procedure.
Figure 1.
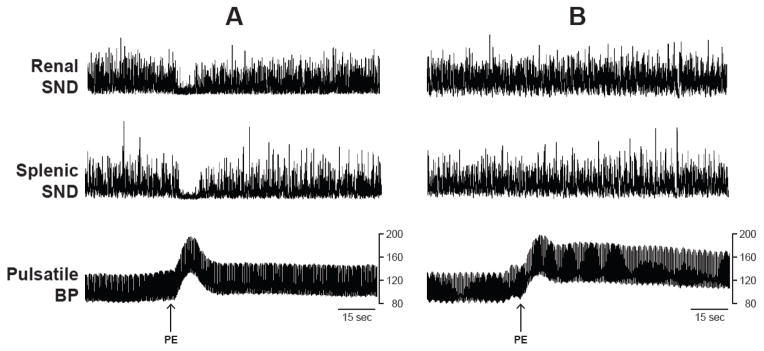
Renal sympathetic nerve discharge (SND), splenic SND, and pulsatile arterial blood pressure (BP) responses to the bolus administration of phenylephrine hydrochloride (PE, 5 μg/kg) in a baroreceptor-intact rat (A) and in a sinoaortic-denervated rat (B).
Figure 2 shows summarized MAP, renal SND, and splenic SND data recorded before (time 0) and for 60 min after intravenous administration of saline (intact, n=5; SAD, n=6) or Dex in baroreceptor-intact (left panels) (three Dex doses or dose ranges: 6–10 μg/kg, n=6; 3 μg/kg, n=9; 0.5–1 μg/kg, n=6) and SAD (right panels) (two Dex doses or dose ranges: 3 μg/kg, n=6; 0.5–1 μg/kg, n=3) F344 rats. Data are presented at one-min intervals for the first 15 min following intravenous Dex or saline administration and at 5 min intervals for the duration of the 60 min recovery period.
In baroreceptor-intact rats (Figure 2, left panels) Dex administration produced immediate, transient (1–2 min) and significant increases in MAP, followed by abrupt (2–5 min after Dex administration) and significant reductions in MAP from control levels in response to each Dex dose (6–10 μg/kg, 3 μg/kg, 0.5–1 μg/kg) (designated collectively by the single asterisk). Dex-induced reductions in MAP were significantly greater after the 6–10 μg/kg and 3 μg/kg Dex doses compared to the 0.5–1 μg/kg Dex dose (designated by the double asterisk). MAP remained at nadir levels for approximately 10–15 min, and then demonstrated a progressive recovery towards control values, although MAP remained significantly reduced from control for the duration of the recovery period after the 6–10 μg/kg and 3 μg/kg Dex doses. Renal SND and splenic SND were significantly reduced from control levels immediately following Dex administration at all three doses (designated collectively by the single asterisks), with the magnitude of the renal and splenic sympathoinhibitory responses significantly greater following Dex administration at the 6–10 μg/kg and 3 μg/kg doses compared to the 0.5–1 μg/kg dose (designated by the double asterisks). Renal and splenic SND remained at nadir levels for up to 15 min, followed by a progressive recovery towards control values. The level of activity in both nerves remained significantly reduced from control for the duration of the recovery period after the 6–10 μg/kg and 3 μg/kg Dex doses, and for 35–40 min after Dex administration at the 0.5–1 μg/kg dose. Each of the measured variables remained unchanged from control values for 60 min after intravenous saline administration in baroreceptor-intact rats.
The response profiles for each of the measured variables to Dex administration in SAD rats (Figure 2, right panels) were similar in direction, magnitude and timing to those observed in baroreceptor-intact rats (Figure 2, left panels). The level of splenic sympathoinhibition in SAD rats was significantly greater following Dex at the 3 μg/kg dose compared to the 0.5–1 μg/kg dose. Each of the measured variables remained unchanged from control values for 60 min after intravenous saline administration in SAD rats (Figure 2).
DISCUSSION
The present results demonstrate that intravenous Dex administration produces marked inhibition of splenic sympathetic nerve outflow in baroreceptor-intact rats, a response paralleled by pronounced Dex-induced inhibition of renal SND. As expected, Dex administration produced an immediate, yet transient, increase in MAP, an effect that may have contributed to the reduction in visceral SND secondary to activation of the arterial baroreflex. However, Dex administration produced similar splenic and renal sympathoinhibitory responses in SAD and baroreceptor-intact rats, supporting the notion that Dex administration alters the central regulation of visceral SND. Moreover, the lack of visceral sympathoexcitation during Dex-induced hypotension in baroreceptor-intact rats indicates that SND responses to Dex were not markedly influenced by unloading of the arterial baroreceptors, providing additional support for altered regulation of central sympathetic nerve outflow in response to Dex administration. As expected, each of the measured variables remained unchanged from control values in response to intravenous vehicle infusion.
The SNS plays a critical role in regulating processes required for maintaining physiological homeostasis and responding to acute stressors, and has often been considered to function rather independently of other adaptive systems. However, there is an evolving concept of cooperation regarding the SNS and integrative physiology. Recent lines of inquiry have expanded the functional repertoire of the SNS by establishing an essential role for this system in regulating and integrating processes between diverse physiological systems (Elenkov et al., 2000; Ganta et al., 2004; Cirmanova et al., 2008; Hamrick and Ferrari, 2008; Confravreux et al., 2009) including a critical role for the SNS in mediating interactions between the nervous and immune systems. A key principle for defining integrative sympathoimmune physiological processing is determining how changes in sympathetic nerve outflow influence target immune organ and cell function. An emerging line of inquiry indicates that Dex administration can affect immune function, including reducing levels of pro-inflammatory cytokines, interleukins, and apoptosis factors (Qiao et al., 2009), as well as altering immune regulation in septic and colitis-induced rats (Qiao et al., 2009; Kayhan et al., 2013). Mechanisms mediating the effects of Dex and other sedatives on peripheral immune responses are not well-established, although it has been hypothesized that these drugs may influence immune function by modulating the SNS. The SNS innervation to the spleen via the splenic nerve serves as an important communication pathway between the central nervous system and the immunocompetent cells of the spleen (Felten et al., 1985; Felten et al., 1987), and alterations in the level of efferent splenic SND can change splenic cytokine gene expression (Ganta et al., 2004; Ganta et al., 2005). For example, heating- and Ang II-induced activation of central sympathetic nerve outflow can modulate splenic cytokine gene expression secondary to increased splenic SND (Ganta et al., 2004; Ganta et al., 2005). The present study is the first to determine the effects of Dex on the efferent sympathetic nerve outflow targeting the spleen, and it is tempting to speculate the Dex-induced inhibition of splenic SND may reduce splenic pro-inflammatory cytokine gene expression, thereby promoting improved immune function. Although additional studies are required to determine the extent of Dex-induced changes on splenic cytokine gene expression and cytokine levels, the present results provide new information regarding the effect of a centrally-acting alpha2-adrenergic agonist on the level of sympathetic nerve outflow to a secondary lymphoid organ that plays a critical role in peripheral immune responses.
A fundamental regulatory strategy of the SNS involves selectively controlling the level of activity in nerves innervating different targets (Hirai et al., 1995; Morrison, 2001; Miki and Yoshimoto, 2010; Mueller et al., 2011). The results of previous studies (Oku et al., 1996; Xu et al., 1998) demonstrate that Dex administration produces renal sympathoinhibition. Whether this represents the selective inhibition of sympathetic outflow to a specific target, or is indicative of a generalized inhibition of sympathetic nerve activity to other targets, in particular other visceral targets, especially the spleen, has remained unknown. The present results extend the current literature by demonstrating that Dex administration markedly reduces splenic SND in both baroreceptor-intact and baroreceptor-denervated rats. Together, these findings demonstrate that Dex administration modulates sympathetic nerve outflow to visceral organs, and suggest that alpha2-adrenergic agonists may produce a generalized attenuation in peripheral sympathetic nerve outflow. Importantly, changes in the level of efferent sympathetic nerve outflow provide a mechanism whereby the sympathetic nervous system regulates target organ responses.
Although the anesthetic regimen employed is used often in studies designed to investigate the central regulation of sympathetic nerve outflow, anesthesia can modulate SND regulation (Shimokawa et al., 1998; Ramchandra et al., 2006). However, determining if Dex produces nonuniform visceral SND regulation requires the simultaneous recording of discharges in two or more sympathetic nerves, an experimental methodology that, at least in the hands of the present investigators, is best completed in anesthetized preparations. In addition, Dex is often used in patients that are critically-ill and likely are receiving other analgesic, anesthetic, or sedative agents, therefore, completion of the current study in anesthetized rats may have some translational significance. It must be considered that the marked Dex-induced reductions in MAP observed in the present study may be an unwanted side effect, especially in critically-ill patients, although, Kontak et al. (2013) reported significant dose-dependent reductions in MAP in human subjects receiving Dex (0.1–1.0 μg/kg, iv) in a dose range used for clinical applications.
Further understanding of regulatory mechanisms linking Dex administration, the level of splenic sympathetic nerve outflow, and ultimately immune system function, may provide mechanistic information regarding the use of Dex or other central acting alpha2-adrenergic agonists as adjunct sedatives in critically-ill patients.
Acknowledgments
Supported by NIH grant AG-041948.
Footnotes
DISCLOSURES
No conflicts of interest are declared by the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cirmanova V, Bayer M, Starka L, Zajickova K. The effect of leptin on bone-an evolving concept of action. Physiol Res. 2008;57:S143–S151. doi: 10.33549/physiolres.931499. [DOI] [PubMed] [Google Scholar]
- 2.Confravreux CB, Levine RL, Karsenty G. A paradigm of integrative physiology, the crosstalk between bone and energy metabolisms. Mol Cell Endocrin. 2009;310:21–29. doi: 10.1016/j.mce.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiBona GF. Neural control of renal function in health disease. Clin Auton Res. 1994;4:69–74. doi: 10.1007/BF01828841. [DOI] [PubMed] [Google Scholar]
- 4.Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve-an integrative interface between two supersystems: the brain and the immune system. Pharmcol Rev. 2000;52:595–638. [PubMed] [Google Scholar]
- 5.Felten DL, Felten SY, Carlson SL, Olschowka JA, Livnat S. Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol. 1985;135:755S–765S. [PubMed] [Google Scholar]
- 6.Felten SY, Olschowka J. Noradrenergic sympathetic innervation of the spleen: II. Tyrosine hydroxylase (TH)-positive nerve terminals form synapticlike contacts on lymphocytes in the splenic white pulp. J Neurosci Res. 1987;18:37–48. doi: 10.1002/jnr.490180108. [DOI] [PubMed] [Google Scholar]
- 7.Galley HF, DiMatteo MA, Webster NR. Immunomodulation by anaesthetic, sedative and analgesic agents: Does it matter? Intensive Care Med. 2000;26:267–274. doi: 10.1007/s001340051149. [DOI] [PubMed] [Google Scholar]
- 8.Ganta CK, Blecha F, Ganta RR, Helwig BG, Parimi S, Lu N, Fels RJ, Musch TI, Kenney MJ. Hyperthermia-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Physiol Genomics. 2004;19:175–183. doi: 10.1152/physiolgenomics.00109.2004. [DOI] [PubMed] [Google Scholar]
- 9.Ganta CK, Lu N, Blecha F, Ganta RR, Zheng L, Fels RJ, Kenney MJ. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol. 2005;289:H1683–H1691. doi: 10.1152/ajpheart.00125.2005. [DOI] [PubMed] [Google Scholar]
- 10.Garcia AA, Fels RJ, Mosher LJ, Kenney MJ. Bacillus anthracis lethal toxin alters regulation of visceral sympathetic nerve discharge. J Appl Physiol. 2012;112:1033–1040. doi: 10.1152/japplphysiol.01105.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamrick MW, Ferrari SL. Leptin and the sympathetic connection of fat to bone. Osteoporos Int. 2008;19:905–912. doi: 10.1007/s00198-007-0487-9. [DOI] [PubMed] [Google Scholar]
- 12.Hirai T, Musch TI, Morgan DA, Kregel KC, Claassen DE, Pickar JG, Lewis SJ, Kenney MJ. Differential sympathetic nerve responses to nitric oxide synthase inhibition in anesthetized rats. Am J Physiol. 1995;269:R807–813. doi: 10.1152/ajpregu.1995.269.4.R807. [DOI] [PubMed] [Google Scholar]
- 13.Hosking KG, Fels RJ, Kenney MJ. Inhibition of RVLM synaptic activation at peak hyperthermia reduces visceral sympathetic nerve discharge. Auton Neurosci. 2009;150:104–110. doi: 10.1016/j.autneu.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kayhan GE, Gul M, Kayhan B, Gedik E, Ozgul U, Kurtoglu EL, Durmus M, Ersoy MO. Dexmedetomidine ameliorates TNBS-induced colitis by inducing immunomodulator effects. J Surg Res. 2013;183:733–741. doi: 10.1016/j.jss.2013.03.028. [DOI] [PubMed] [Google Scholar]
- 15.Kenney MJ. Frequency characteristics of sympathetic nerve discharge in anesthetized rats. Am J Physiol. 1994;267:R830–840. doi: 10.1152/ajpregu.1994.267.3.R830. [DOI] [PubMed] [Google Scholar]
- 16.Koepke JP, DiBona GR. Functions of the renal nerves. Physiologist. 1985;28:47–52. [PubMed] [Google Scholar]
- 17.Kontak AC, Victor RG, Vonpatanasin W. Dexmedetomidine as a novel countermeasure for cocaine-induced central sympathoexcitation in cocaine-addicted humans. Hypertension. 2013;61:388–394. doi: 10.1161/HYPERTENSIONAHA.112.203554. [DOI] [PubMed] [Google Scholar]
- 18.Maclaren R. Immunosedation: a consideration for sepsis. Crit Care. 2009;13:191. doi: 10.1186/cc8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Margiocco ML, Borgarelli M, Musch TI, Hirai DM, Hageman KS, Fels RJ, Garcia AA, Kenney MJ. Effects of combined aging and heart failure on visceral sympathetic nerve and cardiovascular responses to progressive hyperthermia in F344 rats. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1555–63. doi: 10.1152/ajpregu.00434.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menon DV, Wang Z, Fadel PJ, Arbique D, Leonard D, Li JL, Victor RG, Vongpatanasin W. Central sympatholysis as a novel countermeasure for cocaine-induced sympathetic activation and vasoconstriction in humans. J Am Coll Cardiol. 2007;50:626–633. doi: 10.1016/j.jacc.2007.03.060. [DOI] [PubMed] [Google Scholar]
- 21.Miki K, Yoshimoto M. Role of differential changes in sympathetic nerve activity in the preparatory adjustments of cardiovascular functions during freezing behavior in rats. Exp Physiol. 2010;95:56–60. doi: 10.1113/expphysiol.2009.050187. [DOI] [PubMed] [Google Scholar]
- 22.Morrison SF. Differential control of sympathetic outflow. Am J Physiol Regul Integr Comp Physiol. 2001;281:R683–R698. doi: 10.1152/ajpregu.2001.281.3.R683. [DOI] [PubMed] [Google Scholar]
- 23.Mueller PJ, Mischel NA, Scislo TJ. Differential activation of adrenal, renal, and lumbar sympathetic nerves following stimulation of the rostral ventrolateral medulla of the rat. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1230–40. doi: 10.1152/ajpregu.00713.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oku S, Benson KT, Hirakawa M, Goto H. Renal sympathetic nerve activity after dexmedetomidine in nerve-intact and baroreceptor-denervated rabbits. Anesth Analg. 1996;83:477–481. doi: 10.1097/00000539-199609000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Qiao H, Sanders RD, Ma D, Wu X, Maze M. Sedation Improves early outcome in severely septic Sprague Dawley rats. Crit Care. 2009;13:R136–143. doi: 10.1186/cc8012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramchandra R, Barrett CJ, Guild SJ, Malpas SC. Evidence of differential control of renal and lumbar sympathetic nerve activity in conscious rabbits. Am J Physiol Regul Integr Comp Physiol. 2006;290:R701–R708. doi: 10.1152/ajpregu.00504.2005. [DOI] [PubMed] [Google Scholar]
- 27.Sanders RD, Hussell T, Maze M. Sedation & Immunomodulation. Crit Care Clin. 2009;25:551–570. doi: 10.1016/j.ccc.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Shimokawa A, Kunitake T, Takasaki M, Kannan H. Differential effects of anesthetics on sympathetic nerve activity and arterial baroreceptor reflex in chronically instrumented rats. J Auton Nerv Syst. 1998;72:46–54. doi: 10.1016/s0165-1838(98)00084-8. [DOI] [PubMed] [Google Scholar]
- 29.Shirasaka T, Qui DL, Kannan H, Takasaki M. The effects of centrally administered dexmedetomidine on cardiovascular and sympathetic function in conscious rats. Anesth Analg. 2007;105:1722–1728. doi: 10.1213/01.ane.0000286230.02948.77. [DOI] [PubMed] [Google Scholar]
- 30.Taniguchi T, Kidani Y, Kanakura H, Takemoto Y, Yamamoto K. Effects of dexmedetomidine on mortality rate and inflammatory responses to endotoxin-induced shock in rats. Crit Care Med. 2004;32:1322–1326. doi: 10.1097/01.ccm.0000128579.84228.2a. [DOI] [PubMed] [Google Scholar]
- 31.Xu H, Aibiki M, Seki K, Ogli K. Effects of dexmedetomidine, an α2-adrenoceptor agonist, on renal sympathetic nerve activity, blood pressure, heart rate and central venous pressure in urethane-anesthetized rabbits. J Auton Nerv Sci. 1998;71:48–54. doi: 10.1016/s0165-1838(98)00061-7. [DOI] [PubMed] [Google Scholar]