

Abstract
The chemoreflexes exert significant control over respiration and sympathetic outflow. Abnormalities in chemoreflex function may contribute to various disease processes. Based on prior animal studies, we developed the hypothesis that acutely elevating circulating angiotensin II levels into the pathophysiological range increases chemoreflex responsiveness in healthy humans. Eighteen adults were studied before (Pre) and during (Post) low (protocol 1; 2ng/kg/min; n=9) or high (protocol 2; 5 ng/kg/min; n=9) dose angiotensin II infusion (study day 1). Chemoreflex responses were quantified by the pure nitrogen breathing method [slope of the minute ventilation vs. arterial oxygen saturation plot generated during a series (n=10) of 100% inspired nitrogen exposures (1–8 breaths)] and by measuring responses to hypercapnia (7% inspired carbon dioxide). Responses to a non-chemoreflex stimulus were also determined (cold pressor test). Measurements were repeated on a subsequent day (study day 2) before and during infusion of a control vasoconstrictor (phenylephrine) infused at a dose (0.6–1.2 μg/kg/min) sufficient to increase blood pressure to the same degree as that achieved during angiotensin II infusion. We found that despite increasing plasma angiotensin II levels to pathophysiological levels responses to pure nitrogen breathing, hypercapnia, and the cold pressor test were unchanged by low (2 ng/kg/min) and high dose (5 ng/kg/min) angiotensin II infusion (protocols 1 and 2). Similarly, responses measured during phenylephrine infusion (Post) were unchanged (from Pre). These findings indicate that acutely increasing plasma angiotensin II levels to levels observed in disease states, such as human heart failure, does not increase chemoreflex responsiveness in healthy humans.
Introduction
Chemoreflexes exert significant influences over ventilation and sympathetic outflow (Gonzalez et al., 1994; Marshall, 1994; Prabhakar et al., 2004). Chemoreflex function is of importance as it may influence the progression of numerous cardiovascular diseases (Prabhakar et al., 2004; Schultz et al., 2007b). Consistent with this suggestion chemoreflex responsiveness is increased in animal models of heart failure (Ding et al., 2009; Ding et al., 2010; Li et al., 2005; Li et al., 2006; Sun et al., 1999a; Sun et al., 1999b) and patients with the disease (Chua et al., 1996a; Chua et al., 1997; Chua et al., 1996b; Di Vanna et al., 2007; Narkiewicz et al., 1999; Ponikowski et al., 2001a; Schultz et al., 2007a). The consequences of enhanced chemoreflex responsiveness in heart failure may include exercise intolerance, as a result of exertional dyspnea (Chua et al., 1996a; Chua et al., 1997; Ciarka et al., 2006) and excess mortality (Giannoni et al., 2009; Ponikowski et al., 2001b). Thus, identification of mechanisms that modulate chemoreflex responsiveness in health, as well as contribute to abnormal function in disease, is of biomedical importance. In this context, angiotensin II (Ang II) may be important.
In healthy rabbits acute intravenous infusion of Ang II enhances chemoreflex control over ventilation (Li et al., 2006). This potentiation of chemoreflex responses by Ang II in healthy rabbits occurs rapidly (minutes) after plasma Ang II concentrations are increased to pathophysiological levels (Li et al., 2007; Li et al., 2006). Currently, whether or not Ang II modulates chemoreflex responsiveness in an analogous fashion in humans is unknown. Providing an answer to this basic physiological question would provide important information into the potential role of Ang II as an acute modulator of chemoreflex function in human health and possibly disease. Accordingly, we tested the hypothesis that acutely increasing circulating Ang II levels increases chemoreflex responsiveness in healthy humans. To test this hypothesis we employed infusion protocols specifically designed to increase plasma Ang II levels into the pathophysiological range (i.e., to levels observed in human heart failure). Furthermore, to enhance the significance of our findings we employed methods of altering chemoreceptor input that have previously been shown to have prognostic significance in humans (pure nitrogen breathing and hypercapnia) (Giannoni et al., 2009; Ponikowski et al., 2001b).
Materials and Methods
Subjects
A total of 18 male and female volunteers participated in the 2 research protocols associated with this study (Table 1). Inclusion criteria were: 1) age 21–40, 2) healthy (as assessed by history and physical examination), 3) non-smoker, 4) normotensive [blood pressure (BP) at rest <140/90 mmHg], and 5) non-obese (BMI<30 kg/m2) (Table 1). All volunteers were sedentary to recreationally active. The Institutional Review Board of the Penn State College of Medicine approved the experimental protocols. All experiments were conducted with the understanding and the consent of each subject.
Table 1.
Subject characteristics
| Protocol 1 (n=9) | Protocol 2 (n=9) | |
|---|---|---|
| Sex | 5 male/4 female | 5 male/4 female |
| Age, years | 27±1 | 26±1 |
| Height, cm | 181.8±3.2 | 174.3±3.2 |
| Body mass, kg | 78.4±4.7 | 76.4±4.0 |
| BMI, kg/m2 | 23.6±0.9 | 25.0±0.8 |
| Systolic BP, mmHg | 117±3 | 118±2 |
| Diastolic BP, mmHg | 65±2 | 64±2 |
| Mean BP, mmHg | 86±1 | 86±1 |
| Heart rate, bt/min | 57±3 | 56±3 |
Values are mean±SE
BMI = body mass index; BP = blood pressure
Measurements
BP and Heart Rate
BP was determined non-invasively with a semi-automated device (Dinamap) over the brachial artery and at the fingertip using photoplethysmography (Finometer). Heart rate was determined via ECG.
Respiration and Oxygen Saturation
Minute ventilation, partial end-tidal CO2, and arterial oxygen saturation (SaO2) were measured with a respiratory gas monitor (Cardiocap/5 GE Healthcare) via a 2-way non-rebreathing mask and pulse oximeter. The mask covered the subject’s nose and mouth and the pulse oximeter was positioned on an earlobe. Both devices were placed on the subject at least 10 minutes before data collection began.
Blood Samples
An intravenous catheter was placed in an antecubital vein of the left arm for administration of study drugs and another intravenous catheter was placed in the right arm for blood sampling. Blood samples were obtained at baseline (>30 minutes after assuming the supine position), 30 minutes after beginning infusions, and after completing the physiological stressors (~90 minutes after beginning the infusions). Plasma Ang II concentrations were quantified via radio-immunoassay (Alpco Diagnostics).
Experimental Design
General experimental protocol
Studies were conducted on supine subjects after a minimum 4-hour fast and 24 hour avoidance of caffeine and alcohol. After instrumenting the subjects, a 30-minute rest period occurred before any measurements were made. Subjects were exposed to 3 physiologic stressors separated by >15 minutes. BP and heart rate were measured in triplicate over the brachial artery before each stressor and throughout each trial on a beat-by-beat basis. Respiratory variables were obtained continuously.
Physiological stressors
Subjects were exposed twice to 3 physiological stressors on 2 separate study days (protocols 1 and 2; see below). The first exposure occurred before (Pre) and the second during (Post) intravenous infusion of Ang II (study day 1) or phenylephrine (study day 2).
Pure Nitrogen Breathing
After a 3-minute baseline, subjects were exposed to a series of 10 periods of 100% nitrogen inhalation (1–8 consecutive breaths). Transitions from room air to nitrogen breathing were accomplished using a 3-way valve positioned at the head of the bed (out of sight of the subject). Consecutive nitrogen exposures were separated by ~2 minutes to allow respiratory parameters to visually return to pre pure nitrogen exposure values. The number of breaths of nitrogen inhalation was varied and randomized to produce a wide range of arterial oxygen desaturations (~75–96%).
Hypercapnia
After a 3-minute baseline, subjects inspired hypercapnic gas (7% carbon dioxide, 30% oxygen, 63% nitrogen) for 3 minutes. Transition to hypercapnic gas was accomplished via a 3-way valve positioned out of the sight of the subject.
Cold Pressor Test
After a 3-minute baseline the subject’s right hand was submerged in ice water for 2 minutes. The cold pressor test like the other 2 stressors increases ventilation, but does so via a chemoreflex independent afferent pathway.
Protocol 1: Effect of low dose Ang II on chemoreflex responsiveness
After measuring responses to the physiological stressors (Pre), intravenous infusion of Ang II (2 ng/kg/min; Bachem AG, Switzerland; study day 1) or phenylephrine (0.6–1.2 μg/kg/min; Baxter Healthcare, Deerfield IL: study day 2) was begun and continued until the end of the study day. The dose of the control vasoconstrictor (phenylephrine) was titrated upward in 0.2 μg/kg/min increments at 3 min intervals until BP approximated levels achieved during the Ang II infusion on study day 1. Thus, infusion occurred in a single blind fashion. Thirty minutes after beginning the infusions the 3 physiological stressors were repeated (Post) in an identical manner to Pre (same order). Infusions were stopped after collection of the last blood sample (~90 min after they started), which occurred after completion of the last physiological stressor. At least 72 hours separated study days 1 and 2. The 2ng/kg/min dose of Ang II was chosen based on pilot studies in which we observed that this dose increases plasma Ang II levels to the lower end of the pathophysiological range in humans.
Protocol 2: Effect of high dose Ang II on chemoreflex responsiveness
Protocol 2 was identical to protocol 1 except the dose of Ang II used for intravenous infusion was 5 ng/kg/min. Measurements made and timing of events were identical between protocols 1 and 2. The 5ng/kg/min dose of Ang II was chosen based on pilot studies in which we observed that this dose increases plasma Ang II levels to the upper end of the pathophysiological range in humans.
Data Collection and Analysis
Physiological data were recorded (MacLab 8e, ADInstruments) at 400 Hz. Responses to hypercapnia and cold pressor test were quantified over 30 second intervals during the stressor. Responses to pure nitrogen breathing were assessed using standard methods as originally described (Edelman et al., 1973). During each pure nitrogen trial (10 trials Pre and 10 trials Post for each subject on each study day), the lowest measured SaO2 value (nadir) was plotted against the minute ventilation obtained from the 2 largest consecutive breaths after inhalation of 100% nitrogen. These single points (1/trial) were then used to develop a minute ventilation-SaO2 plot for each subject (Pre and Post) in each protocol (protocol 1 and 2).
Statistical Analysis
The slope of each individual’s minute ventilation-SaO2 plot was determined using linear regression analysis. The slope was used as an index of sensitivity only when the correlation coefficient was found to be significant (P<0.05) (Chua et al., 1995; Edelman et al., 1973). Effects of the different interventions were determined using a repeated measure ANOVA. Comparison of subject characteristics between protocols 1 and 2 was determined by unpaired t-tests. Statistical significance was established when P<0.05.
Results
Subject Characteristics
Subjects in Protocols 1 and 2 shared similar subject characteristics (Table 1).
Effect of low dose Ang II (Protocol 1)
Low dose Ang II infusion increased (P<0.05) plasma Ang II levels ~3-fold (Fig. 1) and increased BP at rest. As expected, increases in BP at rest during phenylephrine infusion were similar to those during Ang II infusion (Table 2). In contrast, heart rate at rest decreased during phenylephrine infusion, but not Ang II infusion (Table 2). Chemoreflex responsiveness determined during pure nitrogen breathing was unaltered by Ang II and phenylephrine infusion (Fig. 2). The correlation coefficients between minute ventilation and SaO2 were significant (P<0.05) in 8 of 9 subjects. In these 8 subjects the correlation coefficient (r-value) exceeded 0.80 at all conditions/time points (range 0.83–0.86). Similarly, responses to hypercapnia (Table 3 and Fig. 3) and the cold pressor test (Fig. 4) were unaltered by Ang II and phenylephrine infusion. Resting hemodynamic and respiratory parameters (baseline) before each physiological stressor were similar (Table 2).
Figure 1.
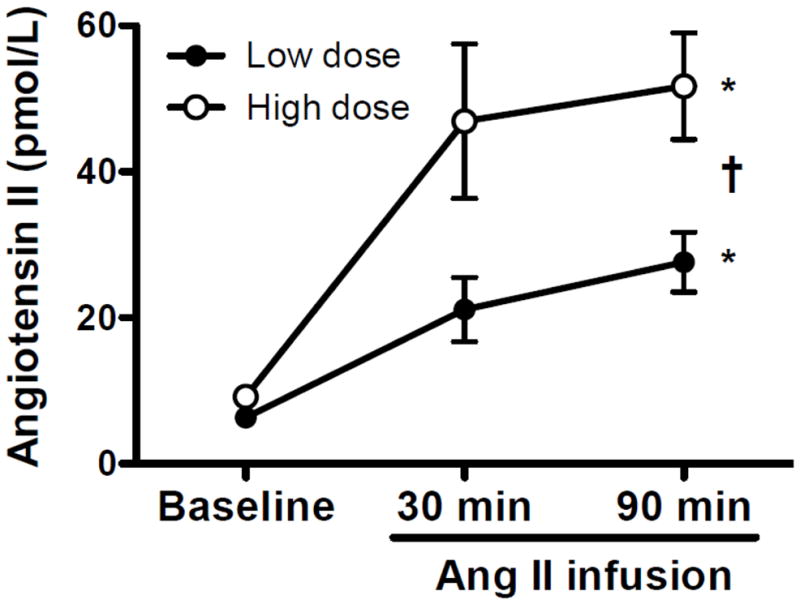
Plasma angiotensin II (Ang II) levels measured before (Baseline) and 30 and ~90 min after beginning intravenous infusion of low (2 ng/kg/min Ang II; closed symbols; protocol 1) or high dose Ang II (5 ng/kg/min; open symbol; protocol 2). Plasma Ang II levels were similar at baseline (pre-infusion) in both groups and increased in a time- (* P<0.05 time main effect) and dose-dependent fashion († P<0.05 dose x time interaction) with infusion. Values are mean±SE.
Table 2.
Hemodynamic and respiratory parameters at rest before each physiological stressor
| Protocol 1 (n=9)
|
Protocol 2 (n=9)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Angiotensin II
|
Phenylephrine
|
Angiotensin II
|
Phenylephrine
|
|||||
| Pre | Post | Pre | Post | Pre | Post | Pre | Post | |
| Pure nitrogen breathing | ||||||||
| Mean BP, mmHg | 87±1 | 92±1* | 86±1 | 91±1* | 86±1 | 98±2* | 86±1 | 95±2* |
| Heart rate, bt/min | 55±3 | 56±3 | 57±4 | 50±3*† | 56±3 | 57±4 | 55±3 | 47±3*† |
| VE, L/min | 5.8±0.5 | 6.2±0.5 | 6.3±0.5 | 6.3±0.5 | 5.5±0.4 | 5.9±0.4 | 5.9±0.5 | 6.4±0.4* |
| SaO2, % | 98±1 | 98±1 | 99±1 | 98±1 | 98±1 | 98±1 | 99±1 | 99±1 |
| Hypercapnia | ||||||||
| Mean BP, mmHg | 85±2 | 91±1* | 85±1 | 91±1* | 87±1 | 95±2* | 86±1 | 94±2* |
| Heart rate, bt/min | 61±3 | 60±3 | 66±4 | 53±3*† | 58±4 | 59±4 | 59±2 | 49±3*† |
| VE, L/min | 6.1±0.3 | 5.9±0.4 | 6.7±0.6 | 6.6±0.5 | 5.3±0.5 | 6.0±0.3 | 6.1±0.5 | 6.4±0.6 |
| SaO2, % | 99±1 | 99±1 | 98±1 | 98±1 | 98±1 | 98±1 | 99±1 | 98±1 |
| Cold pressor rest | ||||||||
| Mean BP, mmHg | 86±2 | 91±1* | 86±1 | 91±1* | 86±1 | 97±2* | 86±1 | 94±2* |
| Heart rate, bt/min | 62±3 | 60±3 | 62±4 | 52±3*† | 61±4 | 63±5 | 59±3 | 51±3*† |
| VE, L/min | 6.7±0.6 | 6.7±0.4 | 6.7±0.5 | 6.8±0.6 | 6.1±0.4 | 6.5±0.4 | 6.3±0.5 | 6.9±0.6* |
| SaO2, % | 99±1 | 98±1 | 98±1 | 98±1 | 99±1 | 98±1 | 99±1 | 99±1 |
Pre measures were made each study day before angiotensin II (2 or 5 ng/kg/min for protocols 1 and 2, respectively) or phenylephrine infusion (0.6–1.2 μg/kg/min). Post measurements were made at least 30 minutes after starting angiotensin II or phenylephrine infusion.
Values are mean±SE.
BP = blood pressure; VE = minute ventilation; SaO2 = arterial oxygen saturation
P<0.05 vs. Pre;
P<0.05 drug (angiotensin II vs. phenylephrine) x condition (pre vs. post) interaction
Figure 2.

Responses to pure nitrogen breathing measured before (Pre) and during (Post) low (upper panels) or high dose (lower panels) angiotensin II (left panels) and phenylephrine infusion (right panels). Subjects were exposed to a series of 10 trials of 100% inspired nitrogen (1–8 breaths). Consecutive pure nitrogen trials were separated by ~ 2 min. For analysis, after each nitrogen exposure the nadir in arterial oxygen saturation (SaO2) was obtained and plotted as a function of the minute ventilation of the 2 largest consecutive breaths to develop a minute ventilation-SaO2 plot. The slope of this plot was used as an index of sensitivity. Neither low nor high dose angiotensin II or phenylephrine infusion had an effect on sensitivity. Values are mean±SE.
Table 3.
Hemodynamic and respiratory responses to hypercapnia (protocol 1; n=9)
| Baseline | Hypercapnia
|
||||||
|---|---|---|---|---|---|---|---|
| 30 | 60 | 90 | 120 | 150 | 180 | ||
| Angiotensin II | |||||||
| Pre | |||||||
| Heart rate, bt/min | 61±3 | 63±3 | 63±3 | 66±3 | 68±4 | 67±4 | 68±4† |
| Mean BP, mmHg | 85±2 | 86±2 | 87±2 | 90±2 | 94±2 | 96±2 | 96±2† |
| VE, L/min | 6.1±0.3 | 6.7±0.7 | 8.7±0.6 | 12.1±1.2 | 14.8±1.5 | 17.2±1.8 | 17.1±1.8† |
| PetCO2, mmHg | 45±1 | 54±1 | 56±1 | 57±1 | 58±1 | 59±1 | 59±1† |
| Post | |||||||
| Heart rate, bt/min | 60±3 | 64±3 | 64±3 | 65±3 | 61±3 | 62±3 | 62±3 |
| Mean BP, mmHg | 91±1* | 91±1 | 91±1 | 95±1 | 98±2 | 98±2 | 99±2† |
| VE, L/min | 5.9±0.4 | 6.8±0.5 | 10.5±1.4 | 13.6±1.7 | 15.4±1.8 | 16.6±1.9 | 16.9±2.1† |
| PetCO2, mmHg | 44±1 | 54±1 | 59±2 | 59±2 | 59±2 | 58±1 | 59±1† |
| Phenylephrine | |||||||
| Pre | |||||||
| Heart rate, bt/min | 65±4 | 67±4 | 67±4 | 70±4 | 71±3 | 71±3 | 71±4† |
| Mean BP, mmHg | 85±1 | 85±2 | 88±1 | 91±2 | 92±2 | 92±2 | 93±2† |
| VE, L/min | 6.7±0.6 | 6.5±0.5 | 9.7±0.6 | 13.6±1.0 | 17.0±1.5 | 19.0±2.0 | 19.3±2.1† |
| PetCO2, mmHg | 44±1 | 53±1 | 56±2 | 57±1 | 57±1 | 58±1 | 58±1† |
| Post | |||||||
| Heart rate, bt/min | 53±3*‡ | 53±3 | 53±4 | 56±4 | 57±3 | 59±3 | 60±3† |
| Mean BP, mmHg | 91±1* | 93±1 | 96±2 | 87±2 | 98±2 | 100±2 | 100±2† |
| VE, L/min | 6.6±0.5 | 7.5±0.5 | 11.0±0.9 | 14.5±1.3 | 17.9±2.1 | 20.2±1.9 | 20.0±22† |
| PetCO2, mmHg | 45±1 | 55±1 | 57±2 | 58±1 | 58±1 | 59±1 | 59±1† |
Responses to hypercapnia (7% inspired carbon dioxide) measured before (Pre) and during (Post) low dose angiotensin II (2 ng/kg/min) or phenylephrine infusion (0.6–1.2 μg/kg/min). Post measurements were made at least 30 minutes after starting angiotensin II or phenylephrine infusion. Values were determined in the 3 min period before hypercapnia (Baseline) as well as at 30 s intervals during hypercapnia. Subjects were exposed to both infusions on separate study days. Responses measured Pre and Post were similar on both study days.
Values are mean±SE.
BP = blood pressure; VE = minute ventilation; PetCO2 = end tidal carbon dioxide
P<0.05 vs. Baseline (Pre);
P<0.05 time main effect;
P<0.05 drug (angiotensin II vs. phenylephrine) x condition (pre vs. post) interaction (Baseline)
Figure 3.

Mean arterial blood pressure (MAP) and minute ventilation (VE) responses to hypercapnia (7% inspired carbon dioxide). Responses are presented as change from baseline levels (room air breathing) in protocol 1 (upper panels) and 2 (lower panels) before (Pre; closed symbols) and during (Post; open symbols) angiotensin II (left panels) or phenylephrine (right panels) infusion. Data are presented as 30 s mean values in response to the 3 min exposure. Both MAP and VE increased during hypercapnia. However, responses were not altered by infusion of low or high dose angiotensin II or phenylephrine. Values are mean±SE.
* P<0.05 time main effect
Figure 4.

Mean arterial blood pressure (MAP) and minute ventilation (VE) responses to the cold pressor test (placing hand in ice water). Responses are presented as change from baseline levels in protocol 1 (upper panels) and 2 (lower panels) before (Pre; closed symbols) and during (Post; open symbols) angiotensin II (left panels) or phenylephrine (right panels) infusion. Data are presented as 30 s mean values in response to the 2 min exposure. Both MAP and VE increased during the cold pressor test. However, responses were not altered by infusion of low or high dose angiotensin II or phenylephrine. Values are mean±SE.
* P<0.05 time main effect
Effect of high dose Ang II (Protocol 2)
High dose Ang II infusion increased (P<0.05) plasma Ang II levels ~5-fold (Fig. 1) and increased BP at rest (Table 2). As expected, increases in plasma Ang II and BP at rest were greater after high versus low dose Ang II infusion (Fig. 1 and Table 2). BP at rest increased similarly during phenylephrine and Ang II infusion. As in protocol 1, heart rate at rest decreased during phenylephrine infusion, but not Ang II infusion (Table 2). Chemoreflex responsiveness determined during pure nitrogen breathing was unaltered by Ang II and phenylephrine infusion (Fig. 2). The correlation coefficients (r-value) between minute ventilation and SaO2 were significant (P<0.05) in 8 of 9 subjects. In these 8 subjects the correlation coefficient exceeded 0.70 at all conditions/time points (range 0.73–0.78). Similarly, responses to hypercapnia (Table 4 and Fig. 3) and the cold pressor test (Fig. 4) were generally unaltered by Ang II or phenylephrine infusion. However, there was a significant drug (Ang II vs. phenylephrine) x condition (Pre vs. Post) interaction observed for absolute heart rate during the hypercapnia and cold pressor test trials (absolute value data not shown), which reflects the downward shift in heart rate observed during phenylephrine infusion that persisted throughout the stressor. As in protocol 1, resting hemodynamic and respiratory parameters before each physiological stressor were similar (Table 2).
Table 4.
Hemodynamic and respiratory responses to hypercapnia (protocol 2; n=9)
| Baseline | Hypercapnia
|
||||||
|---|---|---|---|---|---|---|---|
| 30 | 60 | 90 | 120 | 150 | 180 | ||
| Angiotensin II | |||||||
| Pre | |||||||
| Heart rate, bt/min | 58±4 | 63±5 | 63±5 | 63±4 | 64±4 | 63±4 | 63±4† |
| Mean BP, mmHg | 87±1 | 87±2 | 90±2 | 91±2 | 92±2 | 94±2 | 94±2† |
| VE, L/min | 5.3±0.5 | 6.9±0.7 | 10.3±1.1 | 12.3±1.5 | 14.2±2.0 | 15.4±2.1 | 15.2±2.1† |
| PetCO2, mmHg | 46±2 | 56±1 | 58±1 | 58±1 | 62±2 | 60±1 | 60±2† |
| Post | |||||||
| Heart rate, bt/min | 59±4 | 63±4 | 63±4 | 64±4 | 64±4 | 63±4 | 63±3†§ |
| Mean BP, mmHg | 95±2* | 95±2 | 98±2 | 100±2 | 103±2 | 102±2 | 104±2† |
| VE, L/min | 6.0±0.3 | 6.8±0.5 | 10.5±1.4 | 13.6±1.7 | 15.4±1.8 | 16.6±1.9 | 16.9±2.1† |
| PetCO2, mmHg | 46±3 | 55±1 | 59±3 | 57±1 | 58±2 | 58±1 | 58±1† |
| Phenylephrine | |||||||
| Pre | |||||||
| Heart rate, bt/min | 59±2 | 62±3 | 62±3 | 62±3 | 63±4 | 64±3 | 65±3† |
| Mean BP, mmHg | 86±1 | 86±1 | 89±2 | 90±2 | 91±2 | 92±2 | 92±2† |
| VE, L/min | 6.1±0.5 | 7.6±0.8 | 10.3±1.1 | 12.9±1.6 | 14.2±2.1 | 14.7±2.5 | 15.3±1.9† |
| PetCO2, mmHg | 45±1 | 55±2 | 56±2 | 57±2 | 58±1 | 59±1 | 59±1† |
| Post | |||||||
| Heart rate, bt/min | 49±3*‡ | 52±3 | 52±3 | 53±3 | 56±3 | 57±4 | 58±4†§ |
| Mean BP, mmHg | 94±2* | 99±3 | 99±3 | 101±4 | 100±3 | 102±2 | 102±2† |
| VE, L/min | 6.4±0.6 | 8.4±1.0 | 12.1±1.1 | 14.8±1.5 | 17.0±2.0 | 17.5±1.8 | 18.3±1.9† |
| PetCO2, mmHg | 44±1 | 56±1 | 57±1 | 58±1 | 58±1 | 59±1 | 61±2† |
Responses to hypercapnia (7% inspired carbon dioxide) measured before (Pre) and during (Post) high dose angiotensin II (5 ng/kg/min) or phenylephrine infusion (0.6–1.2 μg/kg/min). Post measurements were made at least 30 minutes after starting angiotensin II or phenylephrine infusion. Values were determined in the 3 min period before hypercapnia (Baseline) as well as at 30 s intervals during hypercapnia. Subjects were exposed to both infusions on separate study days. Responses measured Pre and Post were similar on both study days.
Values are mean±SE.
BP = blood pressure; VE = minute ventilation; PetCO2 = end tidal carbon dioxide
P<0.05 vs. Baseline (Pre);
P<0.05 time main effect;
P<0.05 drug (angiotensin II vs. phenylephrine) x condition (pre vs. post) interaction (Baseline);
P<0.05 drug (angiotensin II vs. phenylephrine) x condition (pre vs. post) interaction
Discussion
The present findings indicate that acutely increasing circulating Ang II to levels observed in pathophysiological states, such as human heart failure, does not increase chemoreflex responsiveness to pure nitrogen breathing or hypercapnia in healthy adults. These results contrast previous findings in healthy animals (rabbits) that demonstrated pronounced rapid increases in chemoreflex responsiveness under very similar experimental conditions (Li et al., 2007; Li et al., 2006). Below we will discuss the significance of the present findings and address discrepancies with prior animal studies.
Previous studies indicate that Ang II increases chemoreflex sensitivity in healthy animals. Specifically, acute intravenous infusion of Ang II in healthy rabbits, at a dose sufficient to produce plasma Ang II levels comparable to those observed in heart failure rabbits, produced rapid and pronounced increases in ventilatory and sympathetic responses to alterations in chemoreflex input (Li et al., 2007; Li et al., 2006). Moreover, in heart failure rabbits, administration of an angiotensin type 1 receptor antagonist decreased chemoreflex responsiveness (Li et al., 2006). The specific mechanism(s) by which Ang II mediates its effects likely involve signaling processes via the angiotensin type 1 receptor subtype and induction of oxidative stress (Allen, 1998; Li et al., 2007). Collectively, these results strongly suggest that Ang II rapidly and powerfully affect chemoreflex function in both health and disease in animals (i.e., rabbits). However, the present study was unable to translate these prior findings in rabbits to healthy humans despite numerous similarities between the studies, such as duration of Ang II infusion, plasma Ang II levels obtained, and achievement of plasma Ang II levels similar to those observed in heart failure in the respective species.
The stated aim of this study was to determine if acutely increasing plasma Ang II levels to those observed in human disease states, such as heart failure, increases measures of chemoreflex responsiveness with prognostic significance in healthy humans. To address this aim we performed 2 separate experimental protocols in which low (protocol 1; 2 ng/kg/min Ang II) and high (protocol 2; 5 ng/kg/min Ang II) doses of Ang II were infused in healthy adults to allow examination of responses across the pathophysiological range of Ang II levels (Dzau et al., 1981; MacFadyen et al., 1999; Roig et al., 2000; Swedberg et al., 1990; van de Wal et al., 2006). Effectiveness of our infusion protocols was carefully documented through definitive biochemical measurements of plasma Ang II. Both the present study, and the prior study in healthy rabbits (Li et al., 2006), achieved similar plasma Ang II levels during infusion (~50 pmol/L) that approximate levels observed in disease (i.e., heart failure) in the respective species (Dzau et al., 1981; Li et al., 2006; Liu et al., 2000; MacFadyen et al., 1999; Roig et al., 2000; Swedberg et al., 1990; van de Wal et al., 2006). We acknowledge that the dose of Ang II infused in the present human study (2–5 ng/kg/min) was considerably smaller than that in the previous animal study (20 ng/kg/min) (Li et al., 2006), but the plasma level of Ang II achieved rather than the dose infused is the critical variable to consider here. However, the fact that a much lower dose of Ang II was able to achieve similar plasma Ang II level in humans versus rabbits does suggest the presence of a species differences in Ang II kinetics.
Developing a better understanding of the mechanisms underlying chemoreflex responsiveness in both health and disease is important. For example, chemoreflex responsiveness is increased in heart failure patients (Chua et al., 1996a; Chua et al., 1997; Chua et al., 1996b; Di Vanna et al., 2007; Narkiewicz et al., 1999; Ponikowski et al., 2001a; Prabhakar et al., 2004; Schultz et al., 2007a; Schultz et al., 2007b) and these increases may contribute to excess morbidity/mortality. A consequence of increased chemoreflex responsiveness in heart failure patients could be greater ventilatory responses to exertion (i.e., exercise) (Chua et al., 1996a; Chua et al., 1997; Ciarka et al., 2006) resulting in excessive exertional dyspnea/exercise intolerance, which are cardinal symptoms in heart failure patients (Chua et al., 1997; Chugh et al., 1996). Additionally, increased chemoreflex responsiveness may contribute to central sleep apnea (Hanly et al., 1993; Wilcox et al., 1993) and arrhythmogenesis (Chua et al., 1997). Collectively, these findings may help explain how increased chemoreflex responsiveness, as measured by a potentiated ventilatory response to chemoreceptor stimulation, helps to identify heart failure patients at elevated risk (Giannoni et al., 2009; Ponikowski et al., 2001b). For these reasons, identification of the mechanism(s) underlying increased ventilatory chemoreflex sensitivity may prove useful in identifying effective therapies/modalities to reduce significant morbidity/mortality associated with diseases such as heart failure (Roger et al., 2012). Moreover, better understanding of the mechanism underlying normal physiological function in health may allow alterations/abnormalities present in disease (i.e., pathophysiology) to be more readily identified and targeted.
Important interactions between the baroreflexes and chemoreflexes may occur. Studies in humans suggest that when chemoreflex input is altered the baroreflexes reset to operate around a higher level BP, without an apparent effect on their gain (Halliwill et al., 2002; Halliwill et al., 2003; Simmons et al., 2007). Additionally, studies in humans (Somers et al., 1991) suggest that large increases in baroreceptor input impair chemoreflex responsiveness. These findings are consistent with finding in animal studies (Heistad et al., 1975) that indicate that the site of interaction is of central origin (Heistad et al., 1974). These data (Heistad et al., 1975; Heistad et al., 1974; Somers et al., 1991) suggest that our ability to detect increases in chemoreflex responsiveness during Ang II infusion may have been masked by an increase in BP (i.e., baroreceptor activation). However, comparison of responses to Ang II infusion with those obtained on another study day, in which a control vasoconstrictor (phenylephrine) was infused at levels sufficient to approximate the increase in BP observed during Ang II infusion, strongly suggest that a sensitizing effect of Ang II on chemoreflex responsiveness was not masked by a modest increase in BP (i.e., baroreceptor activation). An alternative experimental approach to possibly minimize concerns related to reflex interactions would have been to co-infuse a vasodilator drug with Ang II, in an attempt to avoid changes in baroreceptor input. However, as with all approaches in humans the assumption that changes in baroreceptor input would not occur simply because BP levels were unchanged may not be valid (Taylor et al., 1995).
This study has several limitations. First, chemoreflex control of ventilation and not sympathetic outflow was studied. This was done as the former (Chua et al., 1996a; Li et al., 2005; Ponikowski et al., 2001b), but not the latter (Narkiewicz et al., 1999) is potentiated in human heart failure and carries prognostic significance (Giannoni et al., 2009; Ponikowski et al., 2001b). Second, prior animal data demonstrating potentiation of chemoreflex responses by Ang II were collected under isocapnic hypoxic conditions (Li et al., 2007; Li et al., 2006). The fact that responses to pure nitrogen breathing were not collected under identical conditions may contribute to the divergent findings (Duffin, 2007) in humans and animals. Lastly, the duration of our Ang II infusion was short (~90 min). However, the infusion duration was at least as long as that previously used to demonstrate potentiated chemoreflex responses in healthy rabbits (Li et al., 2006). Future studies could address concerns related to the short exposure to increased circulating levels of Ang II by studying groups that chronically differ in Ang II production and or by measuring responses before and after long-term administration of angiotensin converting enzyme inhibitors or AT1 receptor antagonists.
In conclusion, despite achieving acute increases in plasma Ang II concentrations across the pathophysiological range (~3–5-fold increase from basal levels) we were unable to detect any effect on ventilatory responsiveness to several distinct chemoreflex stimuli with prognostic significance (Giannoni et al., 2009; Ponikowski et al., 2001b) in healthy humans. These data contrast prior animal studies, which demonstrate rapid and pronounced increases in chemoreflex sensitivity when plasma Ang II levels are raised to nearly identical levels.
Acknowledgments
Funding Sources: Grants from the National Institutes of Health (HL092309, AG024420, HL096570 and UL1 TR000127) supported this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen AM. Angiotensin AT1 receptor-mediated excitation of rat carotid body chemoreceptor afferent activity. J Physiol. 1998;510 (Pt 3):773–781. doi: 10.1111/j.1469-7793.1998.773bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua TP, Clark AL, Amadi AA, Coats AJ. Relation between chemosensitivity and the ventilatory response to exercise in chronic heart failure. J Am Coll Cardiol. 1996a;27:650–657. doi: 10.1016/0735-1097(95)00523-4. [DOI] [PubMed] [Google Scholar]
- Chua TP, Coats AJ. The reproducibility and comparability of tests of the peripheral chemoreflex: comparing the transient hypoxic ventilatory drive test and the single-breath carbon dioxide response test in healthy subjects. Eur J Clin Invest. 1995;25:887–892. doi: 10.1111/j.1365-2362.1995.tb01962.x. [DOI] [PubMed] [Google Scholar]
- Chua TP, Ponikowski P, Webb-Peploe K, Harrington D, Anker SD, Piepoli M, Coats AJ. Clinical characteristics of chronic heart failure patients with an augmented peripheral chemoreflex. Eur Heart J. 1997;18:480–486. doi: 10.1093/oxfordjournals.eurheartj.a015269. [DOI] [PubMed] [Google Scholar]
- Chua TP, Ponikowski PP, Harrington D, Chambers J, Coats AJ. Contribution of peripheral chemoreceptors to ventilation and the effects of their suppression on exercise tolerance in chronic heart failure. Heart. 1996b;76:483–489. doi: 10.1136/hrt.76.6.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh SS, Chua TP, Coats AJ. Peripheral chemoreflex in chronic heart failure: friend and foe. Am Heart J. 1996;132:900–904. doi: 10.1016/s0002-8703(96)90333-6. [DOI] [PubMed] [Google Scholar]
- Ciarka A, Cuylits N, Vachiery JL, Lamotte M, Degaute JP, Naeije R, van de Borne P. Increased peripheral chemoreceptors sensitivity and exercise ventilation in heart transplant recipients. Circulation. 2006;113:252–257. doi: 10.1161/CIRCULATIONAHA.105.560649. [DOI] [PubMed] [Google Scholar]
- Di Vanna A, Braga AM, Laterza MC, Ueno LM, Rondon MU, Barretto AC, Middlekauff HR, Negrao CE. Blunted muscle vasodilatation during chemoreceptor stimulation in patients with heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H846–852. doi: 10.1152/ajpheart.00156.2007. [DOI] [PubMed] [Google Scholar]
- Ding Y, Li YL, Zimmerman MC, Davisson RL, Schultz HD. Role of CuZn superoxide dismutase on carotid body function in heart failure rabbits. Cardiovascular research. 2009;81:678–685. doi: 10.1093/cvr/cvn350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Li YL, Zimmerman MC, Schultz HD. Elevated mitochondrial superoxide contributes to enhanced chemoreflex in heart failure rabbits. Am J Physiol Regul Integr Comp Physiol. 2010;298:R303–311. doi: 10.1152/ajpregu.00629.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffin J. Measuring the ventilatory response to hypoxia. J Physiol. 2007;584:285–293. doi: 10.1113/jphysiol.2007.138883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzau VJ, Colucci WS, Hollenberg NK, Williams GH. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation. 1981;63:645–651. doi: 10.1161/01.cir.63.3.645. [DOI] [PubMed] [Google Scholar]
- Edelman NH, Epstein PE, Lahiri S, Cherniack NS. Ventilatory responses to transient hypoxia and hypercapnia in man. Respir Physiol. 1973;17:302–314. doi: 10.1016/0034-5687(73)90005-4. [DOI] [PubMed] [Google Scholar]
- Giannoni A, Emdin M, Bramanti F, Iudice G, Francis DP, Barsotti A, Piepoli M, Passino C. Combined increased chemosensitivity to hypoxia and hypercapnia as a prognosticator in heart failure. J Am Coll Cardiol. 2009;53:1975–1980. doi: 10.1016/j.jacc.2009.02.030. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Halliwill JR, Minson CT. Effect of hypoxia on arterial baroreflex control of heart rate and muscle sympathetic nerve activity in humans. J Appl Physiol. 2002;93:857–864. doi: 10.1152/japplphysiol.01103.2001. [DOI] [PubMed] [Google Scholar]
- Halliwill JR, Morgan BJ, Charkoudian N. Peripheral chemoreflex and baroreflex interactions in cardiovascular regulation in humans. J Physiol. 2003;552:295–302. doi: 10.1113/jphysiol.2003.050708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanly P, Zuberi N, Gray R. Pathogenesis of Cheyne-Stokes respiration in patients with congestive heart failure. Relationship to arterial PCO2. Chest. 1993;104:1079–1084. doi: 10.1378/chest.104.4.1079. [DOI] [PubMed] [Google Scholar]
- Heistad D, Abboud FM, Mark AL, Schmid PG. Effect of baroreceptor activity on ventilatory response to chemoreceptor stimulation. J Appl Physiol. 1975;39:411–416. doi: 10.1152/jappl.1975.39.3.411. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Abboud FM, Mark AL, Schmid PG. Interaction of baroreceptor and chemoreceptor reflexes. Modulation of the chemoreceptor reflex by changes in baroreceptor activity. J Clin Invest. 1974;53:1226–1236. doi: 10.1172/JCI107669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Gao L, Zucker IH, Schultz HD. NADPH oxidase-derived superoxide anion mediates angiotensin II-enhanced carotid body chemoreceptor sensitivity in heart failure rabbits. Cardiovascular research. 2007;75:546–554. doi: 10.1016/j.cardiores.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Li YF, Liu D, Cornish KG, Patel KP, Zucker IH, Channon KM, Schultz HD. Gene transfer of neuronal nitric oxide synthase to carotid body reverses enhanced chemoreceptor function in heart failure rabbits. Circ Res. 2005;97:260–267. doi: 10.1161/01.RES.0000175722.21555.55. [DOI] [PubMed] [Google Scholar]
- Li YL, Xia XH, Zheng H, Gao L, Li YF, Liu D, Patel KP, Wang W, Schultz HD. Angiotensin II enhances carotid body chemoreflex control of sympathetic outflow in chronic heart failure rabbits. Cardiovascular research. 2006;71:129–138. doi: 10.1016/j.cardiores.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Liu JL, Irvine S, Reid IA, Patel KP, Zucker IH. Chronic exercise reduces sympathetic nerve activity in rabbits with pacing-induced heart failure: A role for angiotensin II. Circulation. 2000;102:1854–1862. doi: 10.1161/01.cir.102.15.1854. [DOI] [PubMed] [Google Scholar]
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart. 1999;82:57–61. doi: 10.1136/hrt.82.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM. Peripheral chemoreceptors and cardiovascular regulation. Physiol Rev. 1994;74:543–594. doi: 10.1152/physrev.1994.74.3.543. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, Pesek CA, van de Borne PJ, Kato M, Somers VK. Enhanced sympathetic and ventilatory responses to central chemoreflex activation in heart failure. Circulation. 1999;100:262–267. doi: 10.1161/01.cir.100.3.262. [DOI] [PubMed] [Google Scholar]
- Ponikowski P, Banasiak W. Chemosensitivity in chronic heart failure. Heart failure monitor. 2001a;1:126–131. [PubMed] [Google Scholar]
- Ponikowski P, Chua TP, Anker SD, Francis DP, Doehner W, Banasiak W, Poole-Wilson PA, Piepoli MF, Coats AJ. Peripheral chemoreceptor hypersensitivity: an ominous sign in patients with chronic heart failure. Circulation. 2001b;104:544–549. doi: 10.1161/hc3101.093699. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ. Peripheral chemoreceptors in health and disease. J Appl Physiol. 2004;96:359–366. doi: 10.1152/japplphysiol.00809.2003. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig E, Perez-Villa F, Morales M, Jimenez W, Orus J, Heras M, Sanz G. Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur Heart J. 2000;21:53–57. doi: 10.1053/euhj.1999.1740. [DOI] [PubMed] [Google Scholar]
- Schultz HD, Li YL. Carotid body function in heart failure. Respir Physiol Neurobiol. 2007a;157:171–185. doi: 10.1016/j.resp.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz HD, Li YL, Ding Y. Arterial chemoreceptors and sympathetic nerve activity: implications for hypertension and heart failure. Hypertension. 2007b;50:6–13. doi: 10.1161/HYPERTENSIONAHA.106.076083. [DOI] [PubMed] [Google Scholar]
- Simmons GH, Manson JM, Halliwill JR. Mild central chemoreflex activation does not alter arterial baroreflex function in healthy humans. J Physiol. 2007;583:1155–1163. doi: 10.1113/jphysiol.2007.137216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Abboud FM. Interaction of baroreceptor and chemoreceptor reflex control of sympathetic nerve activity in normal humans. J Clin Invest. 1991;87:1953–1957. doi: 10.1172/JCI115221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced activity of carotid body chemoreceptors in rabbits with heart failure: role of nitric oxide. J Appl Physiol. 1999a;86:1273–1282. doi: 10.1152/jappl.1999.86.4.1273. [DOI] [PubMed] [Google Scholar]
- Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced peripheral chemoreflex function in conscious rabbits with pacing-induced heart failure. J Appl Physiol. 1999b;86:1264–1272. doi: 10.1152/jappl.1999.86.4.1264. [DOI] [PubMed] [Google Scholar]
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation. 1990;82:1730–1736. doi: 10.1161/01.cir.82.5.1730. [DOI] [PubMed] [Google Scholar]
- Taylor JA, Halliwill JR, Brown TE, Hayano J, Eckberg DL. ‘Non-hypotensive’ hypovolaemia reduces ascending aortic dimensions in humans. J Physiol. 1995;483 (Pt 1):289–298. doi: 10.1113/jphysiol.1995.sp020585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wal RM, Plokker HW, Lok DJ, Boomsma F, van der Horst FA, van Veldhuisen DJ, van Gilst WH, Voors AA. Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. Int J Cardiol. 2006;106:367–372. doi: 10.1016/j.ijcard.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Wilcox I, Grunstein RR, Collins FL, Berthon-Jones M, Kelly DT, Sullivan CE. The role of central chemosensitivity in central apnea of heart failure. Sleep. 1993;16:S37–38. doi: 10.1093/sleep/16.suppl_8.s37. [DOI] [PubMed] [Google Scholar]