

Abstract
Ewing sarcoma is the second most common bone cancer in pediatric patients. Although the primary cause of death in Ewing sarcoma is metastasis, the mechanism underlying tumor spread needs to be elucidated. To this end, the role of the CXCR4/SDF-1a chemokine axis as a mediator of Ewing sarcoma metastasis was investigated. CXCR4 expression status was measured in primary tumor specimens by immunohistochemical (IHC) staining and in multiple cell lines by quantitative RT-PCR and flow cytometry. Migration and invasion of CXCR4-positive Ewing sarcoma cells towards CXCL12/SDF-1a were also determined. Interestingly, while CXCR4 status was disparate among Ewing sarcoma cells, ranging from absent to high-level expression; its expression was found to be highly dynamic and responsive to changes in the microenvironment. In particular, up-regulation of CXCR4 occurred in cells that were subjected to growth factor deprivation, hypoxia, and space constraints. This up-regulation of CXCR4 was rapidly reversed upon removal of the offending cellular stress conditions. Functionally, CXCR4-positive cells migrated and invaded towards an SDF-1a gradient and these aggressive properties were impeded by both the CXCR4 small molecule inhibitor AMD3100, and by knockdown of CXCR4. In addition, CXCR4-dependent migration and invasion were inhibited by small molecule inhibitors of Cdc42 and Rac1, mechanistically implicating these Rho-GTPases as downstream mediators of the CXCR4-dependent phenotype.
Keywords: Ewing sarcoma, CXCR4, plasticity, metastasis
Introduction
Ewing sarcoma is an aggressive bone and soft tissue malignancy that primarily affects children and young adults [1]. Over the past several decades, overall survival has improved dramatically for patients who present with localized disease. Multi-agent systemic chemotherapy and aggressive local control measures have led to five-year event-free-survival rates of 70–80% in these patients [1, 2]. However, for the approximately 25% of patients who present with metastatic disease, the outcome is significantly worse. Event-free-survival for these patients remains less than 25%, and intensification of chemotherapeutic regimens has failed to improve outcome [1]. In addition, up to a third of patients who present with localized disease will relapse at distant sites following an initial clinical remission and outcomes for these patients are equally dismal. Innovative approaches to therapy and improved understanding of the metastatic process are needed to improve outcomes for patients with primary and relapsed metastatic Ewing sarcoma.
Despite its clinical importance, the biological mechanisms underlying Ewing sarcoma metastasis remain largely unknown. Chemokine receptors are seven transmembrane, G-protein-coupled cell surface proteins that are defined by their ability to induce chemotaxis through the binding of small chemoattractant cytokines, or chemokines [3]. Chemokine (C-X-C motif) receptor 4 (CXCR4) is the most commonly expressed chemokine receptor in human cancer, and increased expression of the CXCR4-encoding transcript was recently found to be associated with metastatic disease in Ewing sarcoma-derived cell lines and tumors [4]. Significantly, high CXCR4 expression has also been associated with metastatic disease and poor outcome in many other human cancers of both epithelial and non-epithelial origin [3, 5], including breast cancer [6], pancreatic cancer [7], leukemia [8], rhabdomyosarcoma [9–11] and osteosarcoma [12–14]. Interestingly, the ligand for CXCR4, CXCL12 (SDF-1α), is highly expressed in common sites of Ewing sarcoma metastasis including lung, bone, and bone marrow, further implicating the potential role of this axis in mediating the distant spread of primary tumor cells.
In this study, we evaluated the expression characteristics of CXCR4 in Ewing sarcoma primary tumors and cell lines, and specifically addressed whether the CXCR4/SDF-1α axis promotes tumor cell migration and invasion. Our findings demonstrate that expression of CXCR4 is both highly variable in Ewing sarcoma and highly dynamic, being reversibly induced in response to microenvironmental stresses, including growth factor deprivation, hypoxia and space constraints. Moreover, our studies confirm that Ewing sarcoma cells that express high levels of CXCR4 display increased chemotactic migration and invasion, which is mediated, at least in part by activation of the Rho-GTPases, Rac1 and Cdc42. Importantly, inhibition of the CXCR4/SDF-1α axis inhibits the aggressive cellular phenotype, thus revealing the potential contribution of CXCR4 signaling to Ewing sarcoma metastasis.
Materials and Methods
Cell Culture
Ewing sarcoma cell lines were kindly provided by Dr. Timothy Triche (CHLA, Los Angeles, CA, USA),) and the COG cell bank (www.cogcell.org) and identities confirmed by short tandem repeat profiling (courtesy of Dr. Patrick Reynolds, Texas Tech University, Lubbock, TX). Cells were maintained in RPMI-1640 media (Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Atlas Biologicals, Inc., Fort Collins, CO, USA) and 6mM L-glutamine (Life Technologies, Grand Island, NY, USA) in an incubator at 37°C in 5% CO2. For CHLA-25 studies, plates were coated with 0.2% gelatin prior to cell seeding. For serum-starved conditions cells were cultured in the same conditions without the addition of FBS. For hypoxia studies cells were incubated in 1% O2 in an xVivo system (Biospherix, Lacona, NY, USA) at 37°C and 5% CO2. For growth constraint conditions, cells were cultured under standard culture conditions and CXCR4 analyzed when cells reached 100% confluence.
Quantitative RT-PCR (qRT-PCR) and Western Blotting
RNA was isolated from cell lines (RNeasy® Mini, Qiagen, Valencia, CA, USA) and cDNA was generated (iScript, Bio-Rad, Hercules, CA, USA). qRT-PCR was performed using validated Taqman primers (CXCR4, 18S and B2M; Life Technologies, Grand Island, NY, USA). Analysis was performed in triplicate using the LightCycler® 480 System (Roche Applied Science, Indianapolis, IN, USA) and average Cp values were normalized relative to reference genes (18S and B2M) within each sample using ΔΔ Cp method. Levels of phospho-ERK (Cell Signaling, Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® Rabbit mAb #4370), phospho-Akt (Cell Signaling, Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb #4060), Akt (Cell Signaling, Akt (pan) (C67E7 Rabbit mAb #4691), Erk (Cell Signaling, p44/42 MAPK (Erk 1/2) #9102) and ACTIN (Abcam, Anti-beta Actin antibody (HRP) (ab20272)) were determined in whole cell lysates using standard western blot assays as previously described [15].
Cell sorting and assessment of Rac1 activation in sorted populations
Cells were dissociated with Accutase (EMD Millipore Corporation, Billerica, MA, USA) and re-suspended in staining media (L-15 media, 0.1% BSA, 10mM HEPES, Life Technologies, Grand Island, NY, USA) then blocked for 15 m at 4°C with agitation (in 0.5% FBS, Atlas Biologicals, Inc., Fort Collins, CO, USA). After blocking, human CXCR4 Alexa Fluor 488 monoclonal antibody (R&D Systems, clone 44717, Minneapolis, MN, USA) was added (5 uL per 1.0×106 cells) and incubated for 30 m at 4°C with agitation. After two washes, cells were re-suspended in staining media and passed through a 0.40 μm sterile nylon mesh strainer (Thermo Fisher Scientific, Waltham, MA, USA). Flow cytometry analysis was performed using a BD Accuri™ C6 Flow Cytometer (BD Biosciences, San Jose, CA, USA). FACS sorting of cells into CXCR4-positive and CXCR4-negative fractions (top 10% and bottom 10%) was done using a Beckman Coulter MoFlo Astrios (Flow Core, University of Michigan) with gating determined by analysis of unstained controls.
For evaluation of Rac1 activation, FACS-sorted TC-32 cells were serum starved overnight in the presence or absence of SDF-1α (200 ng/mL, R&D Systems). Levels of Rac1 activation, were determined using a G-LISA kit (Cytoskeleton, Denver, CO, USA) according to the manufacturer’s instructions.
Immunohistochemistry
For tumor immunohistochemistry, formalin-fixed, paraffin-embedded tumor microarray (TMA) slides were deparaffinized, hydrated, epitope retrieved and stained with an antibody against CXCR4 (dilution 1:500, Abcam, AB-2074, Cambridge, MA, USA) as previously validated and described [16]. Specificity of the antibody was confirmed in our hands by immunostaining of cell pellets collected from CXCR4-high, CXCR4-low as well as control and CXCR4 knockdown TC-32 cells. Adjacent TMA slides were incubated with CD99 (Mouse monoclonal antibody, clone 12E7, DAKO, Carpinteria, CA, Cat # M3601, 1:100) and hematoxylin and eosin in order to identify tumor cells. Sections were scored for the presence of CXCR4 using the Allred schema [17]. The proportion of tumor cells was assigned a score between 0 and 5, while the staining intensity was assigned a score between 0 and 3. These 2 values were added to produce a staining score. Given recent studies describing nuclear localization of CXCR4 in some cancers ([18], [19]) both cytoplasmic and nuclear staining was assessed and equally weighted. Nuclear staining of CXCR4 was evident in >10% of nuclei in 35% of cases.
CXCR4 Knockdown
For CXCR4 knockdown studies cell lines were transduced with pLKO.1 puro vectors that contained one of two independent short hairpin RNAs targeted to CXCR4: shCXCR4-1: 5′-TGGAGGGGATCAGTATATACA-3′ and shCXCR4-2: 5′-GTTTTCACTCCAGCTAACACA-3′ (Addgene, plasmid 12271 and 12272, Cambridge, MA, USA) [20] or an inert non-silencing sequence: shNS: 5′-CAACAAGATGAAGAGCACCAA-3′. Cells were selected in puromycin (2 μg/mL, Sigma) for 72 h prior to subsequent experiments.
In vitro Migration and Invasion
Real-Time Cell Analysis (RTCA) of cell migration and invasion was monitored using a CIM-plate 16 and xCELLigence DP System (Acea Bioscience, Inc., San Diego, CA, USA). Cells were serum-starved overnight in RPMI-1640 with 0.2% Media Grade (K) Probumin (Millipore, Billerica, MA, USA). Prior to cell seeding, electrodes were coated with 0.2% gelatin and RPMI-1640 containing 0.2% Probumin was placed in the upper chamber and media containing SDF-1α (100 ng/mL, R&D Systems) was added to lower chambers. The CIM-plate was allowed to equilibrate for 1 hour in an incubator at 37°C in 5% CO2. For migration studies 1 × 105 cells/well were placed in the upper chamber of a CIM-16 plate and then the plate was equilibrated for 30 minutes at room temperature. For migration assays done with combination of stresses, cells were serum-starved and placed in either normoxic or hypoxic conditions overnight prior to evaluation of migration. For invasion studies, 1 × 105 cells/well were plated in the upper chamber of wells that had been previously coated with 5% (v/v) Growth Factor Reduced Matrigel™ Matrix (diluted 1:20 in basal RPMI media) (BD BioSciences, San Jose, CA, USA). Matrigel-coated plates were allowed to equilibrate for 4 hours in an incubator at 37°C in 5% CO2 prior to addition of cells. For compound assays, cells were pre-treated overnight with either 2.5 μg/mL AMD3100 (Sigma-Aldrich), 30 μM Rac1 inhibitor (NSC 23766 (hydrochloride), Cayman Chemical, Ann Arbor, MI, USA) or 7 μM Cdc42 inhibitor (ML 141, EMD Millipore) and then seeded in CIM-16 plates as above. Parallel migration assays were performed with 2 × 105 cells on 0.8 μm cell culture inserts (Thermo Fisher Scientific, Waltham, MA, USA) for 24 hours. After incubation, non-invading cells were removed from the upper surface and inserts were stained (Crystal Violet Stain; 0.5% crystal violet, 20% methanol) and migratory cells were imaged by light microscopy.
Statistical Analysis
Data are reported as mean ± SEM from three independent experiments and p-values were calculated using Student’s t-test.
Results
CXCR4 expression is highly heterogeneous in Ewing sarcoma
Recent studies of gene expression showed that expression of the CXCR4 transcript varies among Ewing sarcoma cell lines and tumors, [4]. To determine if expression of the CXCR4 protein is equally heterogeneous, we assessed a panel of four well-established Ewing sarcoma cell lines. Quantitative RT-PCR analyses corroborated earlier studies and demonstrated a wide range of CXCR4 expression (Figure 1A). The variability in transcript expression was mirrored by flow cytometry studies of protein expression, with relatively low-levels of CXCR4 detected in TC-71 and A673 cells and high-level expression evident in CHLA-25 and TC-32 cells (Figure 1B). Analysis at the level of individual cells showed that the variation in CXCR4 signal intensity between the different cell lines was a result of different frequencies of CXCR4-positive cells within each culture (Figure 1B). Specifically, in the two low-expressing cell lines fewer than 5% of cells expressed CXCR4. Conversely, 20–40% of cells in CHLA-25 and TC-32 expressed detectable levels of the receptor at the cell surface. In addition, the level of expression in CXCR4-positive populations ranged from weak to robust, as demonstrated by the continuum of fluorescence intensities displayed by CXCR4-positive cells (Figure 1C). To evaluate whether this same heterogeneity in CXCR4 protein expression exists in primary tumors, we evaluated a tissue microarray (TMA) comprised of 64 Ewing sarcoma samples. Sufficient viable tumor was present to score 43 tumor samples from 32 unique patients. Consistent with cell line data, CXCR4 staining showed marked inter-tumor variability, ranging from absent (N=13) to strongly positive in the majority of tumor cells (N=13). The remainder of the samples (N=17) showed an intermediate staining pattern in which both CXCR4-positive and CXCR4-negative tumor cells were identified in the same core specimen (Figure 1D). No difference in staining pattern was identified between 28 samples that were obtained from primary tumor specimens and 15 that were isolated at the time of disease recurrence (Figure 1E). The average CXCR4 score was 5.0 in 4 diagnostic samples that were obtained from patients with metastatic disease and 3.8 in 17 localized tumor samples. Although this analysis showed a trend to increased expression in primary tumors of patients who present with metastatic disease, the sample size is inadequate to draw conclusions regarding associations between CXCR4 expression and clinical stage. Thus, like cell lines, CXCR4 protein expression is highly heterogeneous in Ewing sarcoma tumors, and individual cells within the same tumor also vary in CXCR4 expression.
Figure 1. Heterogeneous expression of CXCR4 in Ewing sarcoma.

A. Quantitative RT-PCR of CXCR4 expression in Ewing sarcoma cell lines. Expression was normalized to the housekeeping 18S rRNA in each sample. Individual replicates are shown and the horizontal line represents average expression of replicate experiments.
B. Flow cytometry of CXCR4 cell surface expression in Ewing sarcoma cell lines. Data are expressed as the percentage of positive cells. Gating was determined based on unstained control cells in the same experiment. Individual replicates are shown and the horizontal line represents average expression of replicate experiments.
C. Representative dot plots for each of the 4 cell lines are shown, demonstrating the heterogeneity of CXCR4 expression both between cell lines and also showing the range of positivity within the CXCR4-positive cell in each cell line.
D. Immunohistochemical (IHC) staining of 43 Ewing sarcoma tumor samples showed marked variability ranging from complete absence to robust staining in all tumor cells. Representative examples of robust (score 6–8), intermediate (score 2–5), and negative (score 0) tumors are shown along with the total number of tumors in each category. Both CXCR4-positive and -negative tumor cells were evident in intermediate tumors.
E. Summary of IHC scores for 28 primary tumor samples and 15 relapse samples shows no difference in CXCR4 expression between the two categories.
CXCR4 Expression is Dynamic and Induced in Response to Growth Factor Deprivation
Tumor cell heterogeneity is a key factor that contributes to drug resistance and tumor progression. We observed significant inter-experiment heterogeneity in CXCR4 expression in our in vitro studies of Ewing sarcoma cell lines (Figure 1A & B). In particular, we noted that the relative proportion of CXCR4-positive cells varied substantially between replicate experiments, particularly in the two high-expressing cell lines. This observation, together with the highly variable nature of expression in tumor samples, led us to hypothesize that expression of CXCR4 may be dynamic in Ewing sarcoma and subject to regulation in response to changes in the local microenvironment. To begin to address this possibility, we tested whether the variability in expression might be a consequence of the relative availability of growth factors. To achieve this, we measured CXCR4 expression in cells that had been deprived of serum. As shown, serum deprivation led to an increased frequency of CXCR4-positive cells in three of the four cell lines (Figure 2A). Only TC-71 cells remained unchanged with fewer than 2% of cells expressing CXCR4 in both serum-rich and serum-deprived conditions. To determine if the upregulation of CXCR4 protein expression was a consequence of increased CXCR4 transcription, we compared mRNA levels in the two conditions. Consistent with transcriptional upregulation, CXCR4 mRNA levels increased in all four cell lines following serum deprivation (Figure 2B). In addition, the degree of transcriptional induction corresponded to that of increased protein expression. TC-71 showed the least and TC-32 cells showed the most robust upregulation of transcript (Figure 2B). We next evaluated whether restoration of growth factor availability would reverse the induction of CXCR4. To achieve this, serum was added to the media of cells that had been starved for 24 hours. Following the addition of serum, rapid downregulation of CXCR4 expression was observed with levels being restored to baseline within 24 hours (Figure 2C).
Figure 2. CXCR4 expression is reversibly induced in response to growth factor deprivation.

A. Surface expression of CXCR4 was determined by flow cytometry as in Figure 1 for Ewing sarcoma cells plated under standard (10% FBS) and serum deprived (Serum free media, SFM) conditions. Exposure of cells to SFM for 24 hours resulted in upregulation of CXCR4. Each line and pair of data points represents the data for an independent experiment.
B. Quantitative RT-PCR analysis of CXCR4 expression in Ewing sarcoma cells grown in SFM conditions for 24 hours. Expression in each sample was normalized to the housekeeping beta 2 microglobulin (B2M) and expressed as fold-change relative to expression in standard 10% FBS conditions. Results are shown as mean ± SEM from three independent experiments.
C. Flow cytometry of CXCR4 expression in serum-starved Ewing sarcoma cells (SFM) after being returned to standard culture conditions (10%) shows reversion of expression to baseline state. Each line and pair of data points represents the data for an independent experiment.
D. TC32 cells were FACS-sorted into CXCR4 high (top 10%) and CXCR4 low (bottom 10%) populations and then both populations were maintained in standard culture conditions for 3 weeks. CXCR4 expression was monitored by flow cytometry on days 5, 12, 16 and 21 post-sorting revealing reversion over time to baseline heterogeneity. Results are shown as mean ± SEM from three independent experiments.
Next we addressed whether the reversible changes in CXCR4 expression seen in heterogeneous cell populations reflected dynamic regulation at the level of individual cells. TC-32 cells were FACS-sorted into pure populations of CXCR4-positive and CXCR4-negative cells, and then monitored over three weeks in ambient culture conditions to determine if positive cells would become negative and vice versa. Consistent with dynamic and bi-directional regulation of CXCR4, both populations of FACS-sorted TC-32 cells gradually reverted to their basal pattern of CXCR4 expression (Figure 2D). Specifically, the initial CXCR4-positive population generated CXCR4-negative cells and the initial CXCR4-negative population generated CXCR4-positive cells with both cultures re-establishing the baseline equilibrium state of ~30–40% CXCR4-positive cells within three weeks.
Thus, CXCR4 expression in Ewing sarcoma cells is dynamic and is rapidly and reversibly induced in response to growth factor deprivation. Moreover, Ewing sarcoma cells in standard tissue culture transition back and forth between CXCR4-negative and CXCR4-positive cell states in response to changes in the microenvironment, ultimately maintaining a basal equilibrium state that is specific for each cell line and condition.
CXCR4 is induced in Ewing sarcoma cells that are exposed to hypoxia and growth constraints
Having established that growth factor deprivation leads to induction of CXCR4, we next questioned whether other stresses that might be encountered by a growing Ewing sarcoma, such as hypoxia and space constraints, would also impact CXCR4 expression. CXCR4 is induced by hypoxia inducible factor alpha (HIF1-α) in mesenchymal stem cells and cancer cells that are exposed to hypoxic environments [21, 22]. Consistent with these observations, we discovered that exposure of Ewing sarcoma cells to hypoxia resulted in an increase in CXCR4 transcript (Figure 3A) and an increased frequency of CXCR4+ cells (Figure 3B). Removal of the hypoxic insult resulted in a return to basal levels within 48 hours (Figure 3B). Interestingly, in direct contrast to growth factor deprivation, TC-71 cells were more susceptible to hypoxia-induced changes than were TC-32 cells, indicating that the inherent plasticity of CXCR4 expression in response to different stimuli varies among the different cell lines. Finally, subjecting cells to space constraints, by growing them to confluence, also resulted in reproducible upregulation of CXCR4-transcript (Figure 3C) and protein expression (Figure 3D) that was reversed when cells were returned to sub-confluent, log-phase growth conditions (Figure 3D).
Figure 3. CXCR4 expression is reversibly induced in response to hypoxia and cell confluence.
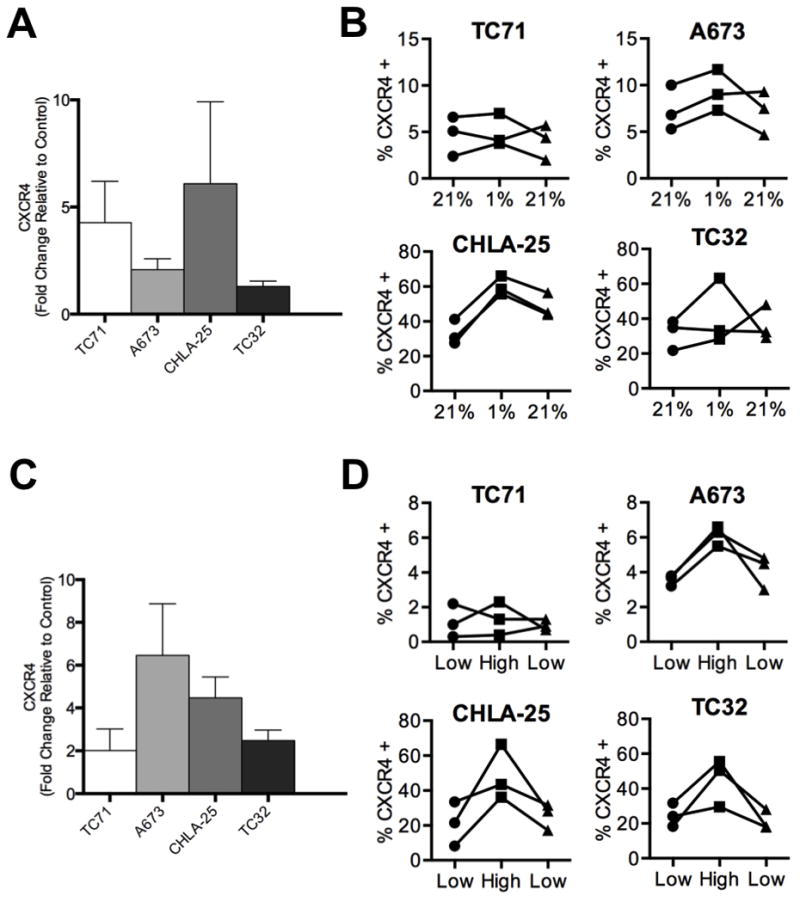
A. Quantitative RT-PCR analysis of CXCR4 expression in Ewing sarcoma cells grown in hypoxic conditions for 24 hours shows upregulation of the transcript. Gene expression calculated in each sample was normalized to the housekeeping beta 2 microglobulin (B2M) and expressed as fold-change in hypoxia relative to expression in normoxia (control). Histograms represent mean fold-change ± SEM for three independent experiments.
B. Flow cytometry of CXCR4 expression in Ewing sarcoma cells before (21%) and after (1%) exposure to hypoxia for 24 hours shows upregulation of CXCR4 expression in hypoxic conditions. The CXCR4-positive cell frequency reverted to baseline 48 hrs. after cells were returned to ambient (21%) conditions.
C. Quantitative RT-PCR analysis of CXCR4 expression in Ewing sarcoma cells grown in log phase, low density (low) compared to confluent, high density (high) conditions for 48 hours. Gene expression calculated as in Figure 3A and expressed as mean fold change ± SEM in high density cells relative to low density (control) cells.
D. Flow cytometry of CXCR4 expression in log phase (low) and confluent (high) conditions shows upregulation of CXCR4 expression that is then reversed when cells are returned to low density growth conditions after 48 hours. For panels A and C, results are shown as mean ± SEM from three independent experiments. For panels B and D, each line and pair of data points represents the data for an independent experiment.
Thus, like growth factor deprivation, exposing Ewing sarcoma cells to hypoxia and space constraints also results in upregulation of CXCR4 transcription and an increased frequency of CXCR4-positive cells. These changes are reversed when these microenvironment stresses are removed, demonstrating the highly plastic and dynamic nature of CXCR4 regulation in Ewing sarcoma cells.
CXCR4 promotes Ewing sarcoma cell migration and invasion
Given its well-established role as a mediator of metastasis in numerous other cancers, we next investigated whether CXCR4 might also contribute to an invasive phenotype in Ewing sarcoma. First, we assessed whether Ewing sarcoma cells demonstrate chemotactic migration towards SDF-1α. As expected, given the very low frequency of CXCR4-positive cells, neither TC-71 nor A673 cells migrated towards SDF-1α (data not shown). In contrast, the CXCR4-high cell lines, CHLA-25 and TC-32 both demonstrated substantial and rapid migration towards SDF-1α (Figure 4A & B). Moreover, exposure of the cells to AMD3100, a small molecule inhibitor of CXCR4, significantly inhibited this chemotactic migration (Figure 4A & B). To further validate these findings, we induced stable knockdown of CXCR4 in both CHLA-25 and TC-32 cell lines using two different short hairpin RNA constructs (Figure 4C & D). Consistent with pharmacologic inhibitor studies, knockdown of CXCR4 significantly impaired the migration of both CHLA-25 and TC-32 cells toward SDF-1α (Figure 4E & F).
Figure 4. CXCR4 promotes chemotactic migration of Ewing sarcoma cells.

A & B. Migration of CHLA-25 (A) and TC32 (B) cells towards SDF-1α (100ng/mL) was measured using real-time cell analysis (xCELLigence CIM-Plate 16) in the presence and absence of the CXCR4 inhibitor AMD3100. AMD3100 significantly inhibited chemotaxis.
C & D. Knockdown of CXCR4 was effectively achieved in CHLA-25 (C) and TC32 cells (D) using lentiviral transduction of 2 different shRNA sequences directed against CXCR4 (sh1 and sh2). Control cells were transduced with an inert non-silencing shRNA vector (shNS). Successful knockdown was confirmed by quantitative RT-PCR (left panels) and flow cytometry (right panels
E & F. Migration of CHLA-25 (E) and TC32 (F) cells towards SDF-1α (100ng/mL) was inhibited following knockdown of CXCR4.
In all panels, graphs represent mean ± SEM of three independent experiments with four replicates per condition. **, P < 0.01, ***, P < 0.001 and ****, P < 0.0001 as compared to controls.
Invasion of cancer cells through basement membranes comprised of extra-cellular matrix proteins is a critical step in the metastatic cascade [23]. To model this process in vitro, we used Matrigel™, a gelatinous protein mixture mimicking extracellular components found in tumors [24]. Both CHLA-25 and TC-32 cells invaded through the Matrigel™ layer toward SDF-1α, and invasion was abrogated by both AMD3100 (Figure 5A & B) and by CXCR4 knockdown (Figure 5C & D). In contrast, SDF-1α had no effect on the invasive potential of A673 cells (data not shown). Thus, CXCR4-positive Ewing sarcoma cells are stimulated to migrate and invade towards SDF-1α but modulation of the CXCR4/SDF-1α axis by pharmacologic or genetic means can profoundly inhibit this response.
Figure 5. CXCR4 promotes invasion of Ewing sarcoma cells.

A & B. Invasion of CHLA-25 (A) and TC32 (B) cells towards SDF-1α (100ng/mL) through a Matrigel™ layer was monitored by real time cell assays as in Figure 4. AMD3100 inhibited migration of both cell lines.
C & D. Knockdown of CXCR4, as in Figure 4, resulted in significant inhibition of invasion of CHLA-25 (C) and TC32 (D) cells.
Graphs represent mean ± SEM of three independent experiments with four replicates per condition. **, P < 0.01 and ***, P < 0.001 as compared to controls.
Rac1 and Cdc42 mediate CXCR4-dependent migration and invasion
The mechanisms by which the CXCR4/SDF-1α axis contributes to tumor growth and metastasis are pleiotropic, and cell type and context dependent [17]. Activation of the mitogen-activated protein kinase (MAPK) and phosphoinositide-3 kinase (PI3K) cascades are both observed downstream of CXCR4 activation [17]. In addition, studies of breast and liver cancer have shown that the small GTPases, Rho, Rac1 and Cdc42 are activated in these tumors following SDF-1α engagement of CXCR4, and that Rho-GTPase signaling is, at least in part, responsible for mediating the invasive/metastatic phenotype [25, 26]. Interestingly, recent studies of Ewing sarcoma have also implicated Rac1 as a key mediator of tumor metastasis [27]. To begin to address the mechanisms by which CXCR4 promotes the invasive cellular phenotype in Ewing sarcoma, we assessed the effects of SDF-1α treatment on the MAPK and PI3K pathways by evaluating phosphorylation of ERK and AKT. As shown, SDF-1α treatment for 24 hours, which promoted cell migration and invasion, had no significant impact on activation of either kinase in CHLA-25 or TC32 cells (Figure 6A). Next we investigated whether SDF-1α-dependent chemotactic migration and invasion were dependent on Rac1 and/or Cdc42. Exposure of Ewing sarcoma cells to either NSC 23766 or ML 141, small molecule inhibitors of Rac1 and Cdc42, respectively, resulted in significant inhibition of both migration (Figure 6B & C, D) and invasion (Figure 6E). In particular, inhibition of Rac1 nearly completely abrogated the chemotactic invasion of CXCR4-positive Ewing sarcoma cells. To determine if Rac1 activation is induced by SDF-1α, TC-32 cells were FACS-sorted on the basis of CXCR4 and Rac1 activity measured in the different populations in the presence or absence of SDF-1α. As shown, CXCR4-high cells displayed higher Rac1 activity than CXCR4-low cells, even in unstimulated conditions (Figure 6F). Exposure to SDF-1α potentiated Rac1 activity in both cell populations but activation of Rac1 was reproducibly most pronounced in SDF-1α–stimulated CXCR4-high cells. Together these studies demonstrate that the invasive cellular phenotype imparted to CXCR4-positive Ewing sarcoma cells following SDF-1α engagement is, at least in part, mediated by downstream activation of Rac1 and Cdc42 Rho-GTPases, in particular Rac1.
Figure 6. CXCR4-mediated chemotaxis is dependent on Rac1 and Cdc42.

A. Western blot of CHLA-25 and TC32 cells shows no significant induction of either PERK (left) or P-AKT (right) following 24 hr. exposure of serum starved cells (SFM) to SDF-1α (100ng/mL).
B & C. End-point analysis of cell migration towards SDF-1α in the presence or absence of Rac1 (NSC 23766) or Cdc42 (ML141) inhibitors was performed as described in Materials and Methods using transwell assays and crystal violet staining. Inhibition of Rac1 and Cdc42 both impeded CXCR4-dependent cell migration.
D & E. Pharmacologic inhibition of Rac1 (NSC 23766) and Cdc42 (ML141) inhibits CXCR4-dependent migration (D) and invasion (E) of CHLA-25 and TC32 cells. Summary histograms show mean ± SEM of three independent experiments with four replicates per condition. *, P < 0.05, **, P < 0.01 and ***, P < 0.001 as compared to controls.
F. Rac1 activity was measured in TC32 cells sorted on the basis of CXCR4. Absorbance values are normalized to control condition (0% in CXCR4-low) and summary histograms show mean ± SEM of two independent sorts with three replicates per condition. *, P < 0.05
CXCR4-dependent migration is increased in Ewing sarcoma cells that are exposed to multiple stresses
Cells in the center of rapidly growing tumors are subjected to a diminished blood supply and must simultaneously endure conditions of both growth factor and oxygen deprivation. Given our findings that CXCR4 and CXCR4-dependent migration are induced by each of these stresses independently we next investigated whether chemotactic migration of Ewing sarcoma cells would be further enhanced in cells that were simultaneously exposed to both serum starvation and hypoxia. As predicted, migration of serum-starved (and thus CXCR4-upregulated) CHLA-25 and TC32 cells towards SDF-1α was increased under hypoxic as compared to normoxic conditions (Figure 7A). Together these studies suggest an additive role of micro-environmental stresses in promoting CXCR4-mediated Ewing sarcoma cell migration.
Figure 7. Hypothetical model of stress-induced, CXCR4-dependent invasion and metastasis.

A. Migration of CHLA-25, and TC32 cells towards SDF-1α (100ng/mL) was measured using real-time cell analysis (xCELLigence CIM-Plate 16) in normoxic (21% O2) or hypoxic conditions (1% O2). Chemotactic migration of each cell line was further increased in hypoxia relative to normoxia.
Graphs represent mean ± SEM of three independent experiments with four replicates per condition. Cell index was normalized to migration in normoxic conditions for each cell line. *, P < 0.05 as compared to controls.
B. A growing tumor begins to deplete its resources, including growth factors and oxygen. Continued tumor growth leads to space constraint at the primary site. Upregulation of CXCR4 in response to these microenvironmental stresses promotes invasion of Ewing sarcoma cells through basement membranes and extracellular matrix and chemotaxis towards SDF-1α rich secondary sites such as lung and bone marrow.
Discussion
In these studies we have shown that expression of CXCR4 is heterogeneous, both in Ewing sarcoma cell lines and primary tumors, and that expression is also highly dynamic. In particular, CXCR4 transcript and protein expression are reversibly increased when cells are exposed to serum deprivation, hypoxia, and confluent growth conditions. All of these stresses are encountered by a growing tumor in vivo as it outstrips its blood supply and expands to abut surrounding adjacent tissues, resulting in growth factor and oxygen deprivation and space constraints. Using both pharmacologic and genetic tools, we have also demonstrated that CXCR4-positive Ewing sarcoma cells display a highly migratory and invasive chemotactic phenotype when exposed to the CXCR4 ligand, SDF-1α/CXLC12. Our finding that Ewing sarcoma cells dynamically regulate CXCR4 leads us to propose a new model of Ewing sarcoma tumor cell invasion in which local microenvironment-induced cell stress results in upregulation of CXCR4, promoting chemotactic migration and invasion of CXCR4-positive Ewing sarcoma cells to distant sites of metastasis. In particular, this model proposes a mechanistic basis for the preferential metastasis of Ewing sarcoma cells to lungs and bone marrow, microenvironments rich in SDF-1α/CXLC12 (Figure 7B).
Studies of Ewing sarcoma tumors and cell lines have previously identified a potential role for the CXCR4/SDF-1α axis in Ewing sarcoma pathogenesis [4, 16, 28, 29]. In particular, interrogation of gene expression databases identified an association between high levels of the CXCR4 transcript and metastatic disease [4]. In addition, concomitant clinical correlative studies in the same study suggested that Ewing sarcoma tumors that express high levels of CXCR4 and a related chemokine receptor, CXCR7, which also binds SDF-1α, are associated with worse overall survival [4]. More recently, an immunohistochemical study of 30 Ewing sarcoma tumors revealed robust CXCR4 staining in approximately one-third of cases, and these investigators also reported an association between CXCR4 expression and poor outcome, although no correlation with metastatic disease was identified [16]. In our own study we also detected robust expression of CXCR4 in approximately one third of cases and an absence of CXCR4-positive cells in another third. However, CXCR4-positive cells were also identified in the remaining third of cases, but tumor cells were found to be heterogeneously positive. Consistent with the study by Berghuis and colleagues [16], the pattern of CXCR4 expression in our tumor cohort did not correlate with the source of the tumor sample. Samples from both primary and recurrent lesions showed equally heterogeneous expression patterns. Together these studies confirm the heterogeneous nature of CXCR4 protein expression in primary Ewing sarcoma tumors and support further investigation of the contribution of CXCR4 signaling to Ewing sarcoma progression. Whether or not high-level expression or an increased frequency of CXCR4-positive cells at the time of diagnosis portends a worse prognosis for patients still requires further investigation. Specifically, given the complexities of prognostic biomarker discovery, it is critical that this question next be addressed prospectively in a large cohort of equivalently treated patients [30]. Moreover, given the highly heterogeneous nature of CXCR4 expression, a single core needle biopsy sample may or may not be representative of CXCR4 expression in other areas of the tumor. Ideally, multiple cores should be assessed when a dynamically regulated and heterogeneous protein like CXCR4 is being evaluated as a potential prognostic biomarker.
Berghuis et al. identified a role for CXCR4/SDF-1α in promoting cell proliferation, rather than metastasis [16]. Given the pleiotropic nature and cell context-specific response of CXCR4-dependent signaling, it is not surprising that different experimental designs have uncovered different results and elucidated different functions for the CXCR4/SDF-1α axis in Ewing sarcoma pathogenesis. We have shown that exposure of CXCR4-positive Ewing sarcoma cells to SDF-1α results in robust induction of chemotaxis, and that both migration and invasion are promoted by activation of CXCR4 signaling. In addition, studies with small molecule inhibitors AMD3100, NSC 23766 and ML 141 showed that migration and invasion towards SDF-1α are dependent on CXCR4 and its downstream effectors, Rac1 and Cdc42, respectively. Interestingly, our studies also indicated that the basal activity of Rac1 is higher in CXCR4-positive Ewing sarcoma cells than CXCR4-negative cells, even in the absence of ligand and that Rac1 was maximally activated by SDF-1α in the CXCR4-positive population. Moreover, we have also found that inhibiting Rac1 blocks SDF-1α –independent invasion of serum starved Ewing cells that do not express high levels of CXCR4 (data not shown). In addition, Rac1 was also recently implicated as a key mediator of Ewing sarcoma cell invasion and metastasis downstream of the tyrosine kinase receptor ERBB4 [27]. Thus, activation of Rac1 is implicated in both non-chemotactic and SDF-1α mediated Ewing sarcoma migration and invasion, downstream of and in parallel to CXCR4-dependent signaling, suggesting that this Rho-GTPase may be a critical downstream hub, present at the convergence of multiple Ewing sarcoma metastatic pathways.
The origins of tumor heterogeneity are multi-factorial, and contributing factors include genetic variation, stochastic processes, different microenvironments, and cell plasticity [31]. Indeed, dynamic regulation of metastasis-inducing genes in response to exogenous cues is a hallmark of epithelial cancer cell plasticity, resulting in epithelial-mesenchymal transition (EMT), a critical initiating event in the onset of carcinoma metastasis [32]. Unlike most adult solid tumors, pediatric solid tumors mainly arise from non-epithelial tissues, predominantly neural and mesenchymal lineages, thus obviating a role for EMT. We have discovered that, like EMT genes in epithelial cancers, CXCR4 expression in Ewing sarcoma is highly plastic and this phenotypic plasticity results in functional changes that can contribute to cell invasion and metastatic dissemination. In particular, CXCR4 expression is highly responsive to stresses in the local microenvironment, reverting to its basal state when the stressor is removed. Consistent with this observation, dynamic regulation of CXCR4 has also been observed in neuroblastoma, a neural crest-derived solid tumor [33, 34], demonstrating that plasticity of CXCR4 is not limited to Ewing sarcoma. Interestingly, high levels of CXCR4 have also been identified in tumor- and metastasis-initiating cancer stem cell populations [7, 35, 36] suggesting that dynamic regulation of CXCR4 may contribute to the dynamic regulation of stemness that has been described in highly plastic cancer cell populations [37]. We hypothesize that dynamic regulation of CXCR4 in Ewing sarcoma, as well as other pediatric solid tumors, contributes to cellular heterogeneity and supports the dynamic transition of cells between non-metastatic and metastatic states. Studies are ongoing in our lab to determine the precise molecular mechanisms that underlie the dynamic regulation of CXCR4 expression and to define whether it is under the control of epigenetic, transcriptional, and/or post-transcriptional regulatory pathways.
Current systemic cytotoxic agents have reached the limit of tolerability, and novel approaches to treatment, in particular approaches that prevent metastatic relapse, are desperately needed for Ewing sarcoma and other invasive solid tumors [38]. The CXCR4/SDF-1α axis is a well-established mediator of tumor metastasis, and it offers a potentially attractive therapeutic target for the treatment and prevention of metastatic disease [17]. Our current work, along with recent studies of other sarcomas and neuroblastoma [9, 10, 14, 34, 39], suggest that this axis represents a potential target for metastasis prevention in Ewing sarcoma as well as other aggressive pediatric tumors and should be further investigated in relevant pre-clinical therapeutic models of these cancers. In particular, studies of spontaneous metastasis using orthotopic, patient-derived xenograft models will be most informative and should be pursued for pre-clinical studies of CXCR4-targeted therapies.
Implications.
This study reveals the highly plastic and dynamic nature of CXCR4 expression in Ewing sarcoma and supports a model in which stress-induced up-regulation of CXCR4 contributes to tumor metastasis to lung and bone marrow, which express high levels of SDF-1a.
Acknowledgments
The authors would like to thank Dr. Gary Luker and his laboratory and staff of the UMICH FACS core for their technical assistance. We also thank current and past members of the Lawlor laboratory for helpful discussion.
Grant Support
The work was supported by grants from the NIH: SARC SPORE 1U01-CA114757 (ERL, RC); NHLBI-5T32HL007622 (LN), and NICHD-5T32HD007513 (LN) and through the University of Michigan’s Cancer Center Support Grant (P30 CA046592) by the use of the following Cancer Center Core(s): Flow cytometry. Additional financial support from the Liddy Shriver Sarcoma Initiative, the Russell G. Adderley Endowment, the Nancy Newton Loeb Fund, the Evan Shapiro Fund to Combat Pediatric Cancer, and Go4 the Goal is gratefully acknowledged.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Balamuth NJ, Womer RB. Ewing’s sarcoma. The Lancet Oncology. 2010;11(2):184–192. doi: 10.1016/S1470-2045(09)70286-4. [DOI] [PubMed] [Google Scholar]
- 2.Gorlick R, et al. Children’s Oncology Group’s 2013 blueprint for research: Bone tumors. Pediatric Blood & Cancer. 2013;60(6):1009–1015. doi: 10.1002/pbc.24429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Domanska UM, et al. A review on CXCR4/CXCL12 axis in oncology: No place to hide. European Journal of Cancer. 2013;49(1):219–230. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Bennani-Baiti IM, et al. Intercohort gene expression co-analysis reveals chemokine receptors as prognostic indicators in Ewing’s sarcoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16(14):3769–78. doi: 10.1158/1078-0432.CCR-10-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cojoc M, et al. Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. Onco Targets Ther. 2013;6:1347–1361. doi: 10.2147/OTT.S36109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller A, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 7.Hermann PC, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Mohle R, et al. Overexpression of the chemokine receptor CXCR4 in B cell chronic lymphocytic leukemia is associated with increased functional response to stromal cell-derived factor-1 (SDF-1) Leukemia. 1999;13(12):1954–9. doi: 10.1038/sj.leu.2401602. [DOI] [PubMed] [Google Scholar]
- 9.Libura J, et al. CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood. 2002;100(7):2597–606. doi: 10.1182/blood-2002-01-0031. [DOI] [PubMed] [Google Scholar]
- 10.Strahm B, et al. The CXCR4-SDF1alpha axis is a critical mediator of rhabdomyosarcoma metastatic signaling induced by bone marrow stroma. Clinical & experimental metastasis. 2008;25(1):1–10. doi: 10.1007/s10585-007-9094-6. [DOI] [PubMed] [Google Scholar]
- 11.Tarnowski M, et al. Regulation of expression of stromal-derived factor-1 receptors: CXCR4 and CXCR7 in human rhabdomyosarcomas. Molecular cancer research: MCR. 2010;8(1):1–14. doi: 10.1158/1541-7786.MCR-09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laverdiere C, et al. Messenger RNA expression levels of CXCR4 correlate with metastatic behavior and outcome in patients with osteosarcoma. Clin Cancer Res. 2005;11(7):2561–7. doi: 10.1158/1078-0432.CCR-04-1089. [DOI] [PubMed] [Google Scholar]
- 13.Lin F, et al. Relationships between levels of CXCR4 and VEGF and blood-borne metastasis and survival in patients with osteosarcoma. Medical oncology. 2011;28(2):649–53. doi: 10.1007/s12032-010-9493-4. [DOI] [PubMed] [Google Scholar]
- 14.Namlos HM, et al. Global gene expression profiling of human osteosarcomas reveals metastasis-associated chemokine pattern. Sarcoma. 2012;2012:639038. doi: 10.1155/2012/639038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawlor ER, et al. Anchorage-independent multi-cellular spheroids as an in vitro model of growth signaling in Ewing tumors. Oncogene. 2002;21(2):307–18. doi: 10.1038/sj.onc.1205053. [DOI] [PubMed] [Google Scholar]
- 16.Berghuis D, et al. The CXCR4-CXCL12 axis in Ewing sarcoma: promotion of tumor growth rather than metastatic disease. Clinical sarcoma research. 2012;2(1):24. doi: 10.1186/2045-3329-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harvey JM, et al. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol. 1999;17(5):1474–81. doi: 10.1200/JCO.1999.17.5.1474. [DOI] [PubMed] [Google Scholar]
- 18.Don-Salu-Hewage AS, et al. Cysteine (C)-X-C Receptor 4 Undergoes Transportin 1-Dependent Nuclear Localization and Remains Functional at the Nucleus of Metastatic Prostate Cancer Cells. PLoS ONE. 2013;8(2):e57194. doi: 10.1371/journal.pone.0057194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu T-p, et al. The impact of chemokine receptor CXCR4 on breast cancer prognosis: A meta-analysis. Cancer Epidemiology. 2013;37(5):725–731. doi: 10.1016/j.canep.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 20.Orimo A, et al. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell. 2005;121(3):335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, et al. Hypoxic preconditioning advances CXCR4 and CXCR7 expression by activating HIF-1α in MSCs. Biochemical and Biophysical Research Communications. 2010;401(4):509–515. doi: 10.1016/j.bbrc.2010.09.076. [DOI] [PubMed] [Google Scholar]
- 22.Oh YS, et al. Hypoxia induces CXCR4 expression and biological activity in gastric cancer cells through activation of hypoxia-inducible factor-1alpha. Oncol Rep. 2012;28(6):2239–46. doi: 10.3892/or.2012.2063. [DOI] [PubMed] [Google Scholar]
- 23.Valastyan S, Weinberg Robert A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell. 2011;147(2):275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benton G, et al. Multiple uses of basement membrane-like matrix (BME/Matrigel) in vitro and in vivo with cancer cells. International Journal of Cancer. 2011;128(8):1751–1757. doi: 10.1002/ijc.25781. [DOI] [PubMed] [Google Scholar]
- 25.Gassmann P, et al. CXCR4 regulates the early extravasation of metastatic tumor cells in vivo. Neoplasia. 2009;11(7):651–61. doi: 10.1593/neo.09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yagi H, et al. A synthetic biology approach reveals a CXCR4-G13-Rho signaling axis driving transendothelial migration of metastatic breast cancer cells. Sci Signal. 2011;4(191):ra60. doi: 10.1126/scisignal.2002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendoza-Naranjo A, et al. ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO Mol Med. 2013;5(7):1019–34. doi: 10.1002/emmm.201202343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chansky HA, et al. Targeting of EWS/FLI-1 by RNA interference attenuates the tumor phenotype of Ewing’s sarcoma cells in vitro. J Orthop Res. 2004;22(4):910–7. doi: 10.1016/j.orthres.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 29.Jin Z, et al. Wnt5a promotes ewing sarcoma cell migration through upregulating CXCR4 expression. BMC Cancer. 2012;12:480. doi: 10.1186/1471-2407-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shukla N, et al. Biomarkers in Ewing Sarcoma: The Promise and Challenge of Personalized Medicine. A Report from the Children’s Oncology Group. Front Oncol. 2013;3:141. doi: 10.3389/fonc.2013.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saunders NA, et al. Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO Mol Med. 2012;4(8):675–84. doi: 10.1002/emmm.201101131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 33.Carlisle AJ, et al. CXCR4 expression heterogeneity in neuroblastoma cells due to ligand-independent regulation. Mol Cancer. 2009;8:126. doi: 10.1186/1476-4598-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L, et al. Tissue microenvironment modulates CXCR4 expression and tumor metastasis in neuroblastoma. Neoplasia. 2007;9(1):36–46. doi: 10.1593/neo.06670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duda DG, et al. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17(8):2074–80. doi: 10.1158/1078-0432.CCR-10-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikegaki N, et al. Transient treatment with epigenetic modifiers yields stable neuroblastoma stem cells resembling aggressive large-cell neuroblastomas. Proc Natl Acad Sci U S A. 2013;110(15):6097–102. doi: 10.1073/pnas.1118262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta PB, et al. Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell. 2011;146(4):633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 38.Steeg PS. Perspective: The right trials. Nature. 2012;485(7400):S58–9. doi: 10.1038/485S58a. [DOI] [PubMed] [Google Scholar]
- 39.Ma M, et al. Mesenchymal stromal cells may enhance metastasis of neuroblastoma via SDF-1/CXCR4 and SDF-1/CXCR7 signaling. Cancer letters. 2011;312(1):1–10. doi: 10.1016/j.canlet.2011.06.028. [DOI] [PubMed] [Google Scholar]