

Abstract
We have synthesized and characterized four octahedral polypyridyl d6 metal complexes bearing the 5,6-epoxy-5,6-dihydro-[1,10]phenanthroline ligand (L1) as cysteine specific labeling reagents. The proposed synthetic pathways allow the preparation of the metal complexes containing Re(I), Ru(II), Os(II) and Ir(III) while preserving the epoxide functionality. The complexes were characterized by 1H and 13C NMR, mass spectrometry, UV-visible and luminescence spectroscopies as well as cyclic voltammetry. As proof of concept, a set of non-native single cysteine P450 BM3 heme domain mutants previously developed in our laboratory was used to study the labeling reaction. We demonstrate that the proposed labels can selectively react, often in high yield, with cysteine residues of the protein via the nucleophilic thiol ring opening of the epoxide moiety. In addition, under basic conditions, subsequent loss of a water molecule led to the aromatization of the phenanthroline ring on the protein-bound label compounds, as observed by mass spectrometry and luminescence measurements.
Keywords: biomolecule labeling; sulfhydryl specific; polypyridyl d6 metal complexes; 5,6-epoxy-5,6-dihydro-[1,10]phenanthroline
1. Introduction
The detection of proteins and other biological macromolecules using luminescent materials has enabled important advances in the life sciences and continues to be a major area of investigation both in vitro and in vivo. A wide variety of luminescent materials are already available including organic molecules,[1] inorganic complexes,[2, 3] biological fluorophores,[4, 5] and emerging nanomaterials such as quantum dots.[6] Of these candidates, octahedral polypyridyl d6 metal complexes containing Re(I), Ru(II), Os(II), Ir(III) have been of interest due to their photostability, tunability, large Stokes’ shifts, and long-lived excited states.[7] Bioconjugation of d6 metal complexes to proteins permits their luminescence detection,[3] but also the study of protein structure-function relationships and protein folding dynamics,[8] and has enabled the study of photoinduced electron transfer in proteins.[9, 10] Over the last few decades, the extensive work on photoinduced electron transfer has also led to the use of some of these metal complexes to drive enzymatic processes upon light-excitation.[11–13]
In general, covalent attachment of the luminescent labels is often desired in bioconjugation over non-covalent interactions, as those are much weaker and more sensitive to pH and salt concentration.[14] The covalent attachment has taken advantage of the reactivity of amino acid residues such as histidine, lysine, and cysteine.[15, 16] Among them, the selective covalent attachment to cysteine residues is advantageous due to the nucleophilicity of their side chain and their lower natural abundance.[17] Such properties have motivated the development of sulfhydryl-specific labeling reagents, which have been primarily achieved via the introduction of reactive maleimide,[18, 19] bromoalkyl,[10] and iodoacetamide[20] substituents onto the luminescent compounds. Our own laboratory has taken advantage of the iodoacetamide functionality in Ru(II)-diimine complexes to covalently attach these photosensitizers to cysteine residues of P450 BM3 heme domain enzymes and generate efficient light-activated biocatalysts capable of selective C-H functionalization upon visible light irradiation.[12, 13, 21, 22] However, the introduction of the aforementioned reactive functionality onto polypyridyl ligands (e.g. bipyridine or phenanthroline) has often required several synthetic steps in the ligand preparation and have limited their general applications.
Herein, we report a facile and versatile methodology for the thiol-specific labeling of proteins with various d6 metal complexes in high yield (65–90%). This approach takes advantage of the epoxide functionality on the 5,6-epoxy-5,6-dihydro-[1,10]phenanthroline ligand (L1) and its susceptibility to ring opening by strong nucleophiles such as the cysteine sulfyhydryl group.[23–26] We synthesized and characterized four metal complexes (1–4) containing the L1 ligand and the d6 metals Re(I), Ru(II), Os(II) and Ir(III), respectively (See Scheme 1). The use of these complexes in labeling a set of P450 BM3 mutants with different non-native single cysteine residues was then investigated as proof of concept for the methodology.
Scheme 1.
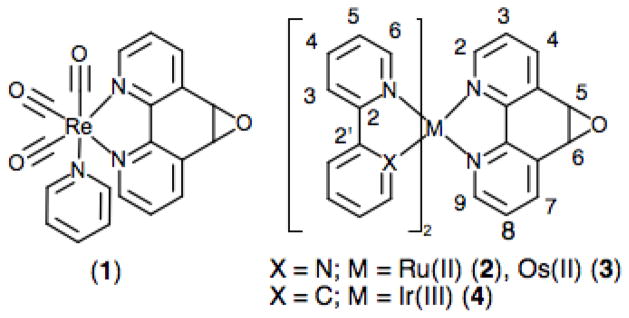
Schematic representation of the d6 metal complexes 1–4 bearing the L1 ligand.
2. Experimental
2.1. Material and reagents
All reagents used in this work were of analytical grade and purchased from Thermo Fisher Scientific and Sigma-Aldrich. The complexes Ru(bpy)2Cl2,[27] Os(bpy)2Cl2,[28] Re(CO)3Cl(L1),[29] and [Ir(ppy)2Cl]2[30] (bpy = 2,2′-bipyridine and ppy = 2-phenylpyridine) were prepared following reported procedures. The ligand 5,6-epoxy-5,6-dihydro-[1,10]phenanthroline was purchased from Sigma-Aldrich and could alternatively be synthesized from 1,10-phenanthroline and bleach.[26] 5-(2-aminoethanesulfanyl)-1,10-phenanthroline (L2) was synthesized as reported earlier.[25]
2.2. Instruments and methods
Accurate electrospray ionization mass spectrometry (ESI-MS) data was obtained from an Agilent 6520 Quadrupole Time-of-Flight LC/MS instrument. 1H and 13C NMR were recorded on a Varian 400 MHz NMR spectrometer. UV-visible (UV-vis) spectra were recorded on a Cary 60 UV-vis spectrophotometer and luminescence data on a Varian Cary Eclipse fluorescence spectrophotometer. Quantum yields in acetonitrile solution at 25 °C were determined using Ru(bpy)32+ as a reference (ϕr = 0.061, λex = 455 nm) using the equation:
where I is the integral of the emission spectrum, A is the absorbance at the excitation wavelength, Ru is the reference Ru(bpy)32+ and M denotes the metal complexes 1–4.
2.3. Cyclic Voltammetry
Cyclic voltammograms were obtained on a Pine Wavenow potentiostat instrument in a single-compartment three-electrode cell with a glassy carbon working electrode, Ag/AgCl reference electrode, and platinum wire auxillary electrode. The supporting electrolyte was 0.1 M tetrabutylammonium hexafluorophosphate and dry acetonitrile was used as the solvent. Solutions were degassed with N2 prior to measurements and the voltage was swept at 100 mV/s. Potentials were calibrated using the ferrocene/ferrocenium (Fc/Fc+) couple as a reference and were reported vs. Fc/Fc+ with a measured E1/2(Fc/Fc+) of 0.55 V vs. Ag/AgCl.
2.4. Synthesis of the metal complexes 1–4
[Re(L1)(CO)3(py)](PF6) (1)
Re(L1)(CO)3Cl (32.3 mg, 0.06 mmol) and AgBF4 (12.3 mg, 0.06 mmol) were stirred at 50 °C in anhydrous dimethylformamide (DMF) for 3 hours in the dark. Then, 1 mL of dry pyridine (py) was added and the reaction continued for 3 hrs. The solvent was then removed under vacuum and the desired product isolated upon the addition of saturated aqueous KPF6. The pale yellow solid was filtered and washed with water and diethylether. Yield: 33.7 mg (96 %). ESI-MS m/z = 546.0466 [M+] (calculated: 546.0463). 1H NMR (400 MHz, acetone-d6) δ ppm 5.04 (L1-H5,6, s, 2H), 7.47 (py-H3, t,2H), 7.98 (py-H4, t, 1H), 8.13 (L1-H3,8, d, 2H), 8.59 (py-H2, d, 2H), 8.83 (L1-H2,9, d, 2H), 9.54 (L1-H4,7, d, 2H). 13C NMR δ ppm 55.0, 55.2 (Cepoxide)
[Ru(bpy)2(L1)](PF6)2 (2)
Compound 2 was prepared following a reported procedure.[31] Briefly, 31.0 mg (0.06 mmol) of cis-Ru(bpy)2Cl2 and 14.0 mg of L1 (0.07 mmol) were refluxed in EtOH/water (75/25) for 3 hours in the dark. After removal of the solvent and addition of a saturated solution of KPF6, the desired product (41 mg, 75% yield) was obtained as an orange powder by vacuum filtration. ESI-MS m/z = 305.0539 [M2+] (calculated: 305.0527) 1H NMR (400 MHz, acetone-d6) δ ppm 5.11 (L1-H5,6, s, 2H), 7.40 (bpy-H5,t, 1H), 7.46–7.53 (bpy-H5,5′,5′, m, 3H), 7.57 (L1-H8, dd, 1H), 7.64 (L1-H3, dd, 1H), 7.85 (L1-H9, d, 1H), 8.01–8.09 (bpy-H6,6,6′,6′, m, 4 H), 8.14 (L1-H2, d, 1H), 8.17–8.22 (bpy-H4,4,4′,4′, m, 4H), 8.55 (L1-H7, d, 1H), 8.57 (L1-H4, d, 1H), 8.87–8.81 (H3,3,3′,3′, m, 4H). 13C NMR δ ppm 55.3, 55.4 (Cepoxide).
[Os(bpy)2(L1)](PF6)2 (3)
This complex was synthesized following the same procedure described for 2. A solution of EtOH/water (75/25) containing 10.0 mg of L1 (0.05 mmol) and 26.0 mg of Os(bpy)2Cl2 (0.04 mmol) was refluxed for 3 hours in the dark. The desired product was isolated as a green solid after addition of a saturated KPF6 solution. Yield: 55% ESI-MS m/z = 350.0811 [M2+] (calculated: 350.0812); 1H NMR (400 MHz, acetone-d6) δ ppm 5.10 (L1-H5,6 s, 2H), 7.39 (bpy-H5, t, 1H) 7.47–7.53 (bpy-H5,5′,5′, m, 3H), 7.55 (L1-H8, dd, 1H), 7.63 (L1-H3, dd, 1H), 7.87 (L1-H9, d, 1H), 7.90–7.98 (bpy-H6,6,6′, m, 3H), 7.98–8.05 (bpy-H6,4,4,4′,4′, m, 5H), 8.10 (L1-H2, d, 1H), 8.36 (L1-H4,7, d, 2H) 8.74–8.84 (bpy-H3,3,3′,3′, m, 4 H); 13C NMR δ ppm 55.2, 55.4 (Cepoxide).
[Ir(ppy)2(L1)](PF6) (4)
Ir(ppy)2Cl2 (127.7 mg, 0.22 mmol) was refluxed with a small excess of L1 (57.2 mg, 0.29 mmol) in 20 mL of 1:1 DCM/MeOH in the dark for 3 hrs. The reaction mixture was then concentrated by rotary evaporation and the desired product was precipitated by the addition of a saturated aqueous KPF6 solution. A pale yellow solid was collected by vacuum filtration and washed with water and diethyl ether and dried under vacuum. Yield: 187.0 mg (93%). ESI-MS m/z = 697.1580 [M+] (calculated: 697.1578) 1H NMR (400 MHz, acetone-d6) δ ppm 5.10 (L1-H5,6, d, 2H), 6.33 (ppy-H6′,6′, t, 2H), 6.91 (ppy-H5′,5′, t, 2H), 6.98–7.09 (ppy-H3′,5,5, m, 3H), 7.15 (ppy-H3′, t, 1H), 7.66 (ppy-H6, d, 1H), 7.77–7.97 (ppy-H3,3,4′,4′,6, L1-H3,8, m, 7H), 8.05 (L1-H2, d, 1H), 8.14 (L1-H9, d, 1H), 8.21 (ppy-H4,4, t, 2H), 8.64 (L1-H4,7, d, 2H); 13C NMR δ ppm 55.0,55.2 (Cepoxide).
[Ru(bpy)2(L2)](PF6)2 (2a)
The complex 2a was prepared from two different synthetic routes.
Method A: By reacting compound 2 with a slight excess of 2-aminoethanethiol, dihydrochloride (cysteamine) under the same labeling reaction conditions used for the label protein (see below).
Method B: The L2 ligand was synthesized from L1 and 2-aminoethanethiol, dihydrochloride in sodium ethoxide/ethanol at 50 °C according to reported procedure.[25] Once isolated as a yellow oil, L2 was reacted with cis-Ru(bpy)2Cl2 in water/ethanol for three hours and isolated as the PF6 salt. The addition of a saturated aqueous KPF6 solution produced a red powder that was vacuum filtered and washed with water, chloroform, and diethyl ether. ESI-MS m/z = 334.5631 [M2+] (calculated: 334.5623); 1H NMR (400 MHz, DMSO-d6) δ ppm 3.00 (CH2, t, 2H), 3.29 (CH2, t, 2H), 7.33 (L1-H3,8, dd, 2H), 7.51–7.63 (bpy-H5,5,5′,5′, m, 4H), 7.75–7.93 (bpy-H6,6,6′,6′, m, 4H), 7.97–8.24 (bpy-H4,4,4′,4′, L1-H2,9, m, 6H), 8.29 (L1-H6, s, 1H), 8.58–8.67 (L1-H7, d, 1H), 8.75–8.92 (bpy-H3,3,3′,3′, L1-H4, m, 5H)
2.5. Protein labeling using complexes 1–4
A DMF solution of the metal complexes 1–4 was added to single cysteine residue of the P450 BM3 heme domain mutants in 100 mM Tris buffer pH = 8.2 and the reaction was gently stirred for 3 hours following reaction conditions developed for the iodoacetamide derivatives.[12] After completion, centrifugal filtration was used to concentrate the reaction mixture and a desalting column connected to an AKTA Purifier allowed for the removal of excess label. Subsequently, unlabeled protein was separated from the labeled enzyme by anion exchange chromatography using a HiTrap Q column with a stepwise elution gradient.[12] Completion of the reaction could be assessed by ESI-MS. Typical yields for labeling reactions range from 65 to 90% depending on the mutants and labels used. After mass spectral deconvolution, the hybrid enzymes have experimental mass values as follows, consistent with calculated values in parentheses: 54093.2 (54093.1) for K97C-1, 54093.3 (54093.2) for Q397C-1 and Q109C-1; 54157.2 (54157.2) for K97C-2, 54157.3 (54157.2) for Q397C-2 and Q109C-2; 54247.3 (54247.2) for K97C-3, 54247.4 (54247.3) for Q397C-3 and Q109C-3; 54244.3 (54244.2) for K97C-4, 54244.2 (54244.3) for Q397C-4 and Q109C-4. Addition of small aliquots of potassium carbonate (up to 25 molar eq.) leads to a loss of a water molecule and the aromatization of the ligand on the protein-bound label compounds as detected by mass spectrometry after deconvolution. No significant protein degradation has been observed. The hybrid enzymes with the aromatized metal complexes have experimental mass values as follows, consistent with calculated values in parentheses: 54075.2 (54075.1) for K97C-1a, 54075.3 (54075.2) for Q397C-1a and Q109C-1a; 54139.1 (54139.2) for K97C-2a, 54139.3 (54139.2) for Q397C-2a and Q109C-2a; 54229.2 (54229.2) for K97C-3a, 54229.3 (54229.3) for Q397C-3a and Q109C-3a; 54226.1 (54226.2) for K97C-4a, 54226.2 (54226.3) for Q397C-4a and Q109C-4a.
3. Results and discussion
3.1. Synthesis of the metal complexes 1–4
The complexes 1–4 shown in Scheme 1 could be readily synthesized from their chloro precursors following established procedures. Briefly, a slight excess of the ligand L1 was refluxed with the dichloro metal precursors Ru(bpy)2Cl2, Os(bpy)2Cl2, and [Ir(ppy)2Cl]2 to afford 2–4, respectively. The complex 1 was prepared from Re(L1)(CO)3Cl via chloride substitution using AgBF4 and pyridine. All of these complexes could be isolated as PF6 salts in high yield and purity. In some cases when further purification was required, the complexes could be purified by column chromatography on alumina using dichloromethane/methanol (95/5) as co-eluents. It is worth noting that the epoxide ring withstood the synthesis and purification conditions. All of these metal complexes were characterized by ESI-MS, 1H and 13C NMR, UV-vis and luminescence spectroscopies. In addition, their redox properties were also investigated by cyclic voltammetry.
3.2. 1H NMR Spectroscopy
The 1H NMR spectra of 1–4 are shown in Fig. 1 and are consistent with the proposed structures. All spectra exhibit a characteristic resonance at 5.1 ppm corresponding to the aliphatic epoxide protons, which also confirms the integrity of the 5,6-epoxy-5,6-dihydro-[1,10]phenanthroline (L1) ligand in the metal complexes. Regarding the assignment of 1, the coordinating pyridine gives rise to two sets of aromatic protons at 8.6 and 7.5 ppm, respectively, in addition to the lone para hydrogen at 8.0 ppm, while the L1 ligand shows three sets of methine resonances in the aromatic region besides the epoxide singlet at 5.1 ppm. The doubling of the peaks at 9.5 and 5.0 ppm likely indicates a mixture of two fac isomers with the epoxide cis and trans to the axial pyridine, as seen in the parent Re(L1)(CO)3Cl complex [29] (see Fig. S1). The reduced symmetry in the pseudo-octahedral complexes 2, 3, and 4 leads to more complicated spectra with 22 overlapping methine resonances in the aromatic region. Nonetheless, tentative assignments were made by comparison to 1 and based on the wealth of data available on d6 metal complexes. The two doublets and the triplet of the L1 ligand in 1 were used to identify the corresponding protons in 2 and 3. The remaining ancillary bipyridine protons were assigned in the order H3>H4>H6>H5, as recently established.[32] As expected, varying the metal center from Ru(II) to Os(II) results in only minor shifts between the 1H NMR spectra for 2 and 3. The 1H NMR spectrum of complex 4 contains several high-field peaks consistent with the anionic character of the ancillary phenylpyridine (ppy) ligands in the proposed structure.
Fig. 1.

1H NMR spectra of the metal complexes 1–4 in acetone-d6.
3.3. Electronic Spectroscopy
The absorption and emission spectra of the metal complexes 1–4 exhibit typical intraligand (IL) π-π* transitions in the UV region and broad metal-to-ligand charge transfer (MLCT) bands in the visible range (see Fig. S2). Their photophysical properties are summarized in Table 1 with their respective bipyridine analogs. Upon irradiation into the MLCT manifold, all of the complexes exhibit intense luminescence under ambient conditions with quantum yields typical for this class of compounds.[33] Phosphorescence emission from 2 and 3 have been assigned to spin-forbidden 3MLCT states, while a mixture of MLCT and IL π-π* transitions give rise to phosphorescence in 1 and 4. The broad, structureless emission bands are consistent with their MLCT assignment (See Fig. S2).
Table 1.
Photophysical and redox properties of the metal complexes 1–4 and their respective bipyridine analogues.
| Complex | λ(ε, M−1 cm− 1)/nm | Λ(excitation)/nm [a] | QYe [b] | E/V [c] | Ref. |
|---|---|---|---|---|---|
| 1 | 257 (12,541) 370 (2,300) |
592(360) | 0.079 | −1.42, −1.53, −1.81 | this work |
| [Re(CO)3(bpy)(py)]+ | 366(2,400) | 558(400) | 0.059 | −1.55, −1.85 | [34] |
| 2 | 242 (23,700) 286 (71,000) 451 (13,300) |
634(458) | 0.068 | 0.92, −1.55, −1.75, −1.92, −2.16 | this work |
| [Ru(bpy)3]2+ | 455(13,642) | 612(455) | 0.061 | 0.89, −1.72, −1.92, −2.16 | this work |
| 3 | 271 (43,400) 290 (51,800) 436 (8,000) 481 (8,500) |
734(480) | 0.028 | 0.48, −1.47, −1.68, −1.85, −2.02 | this work |
| [Os(bpy)3]2+ | 436(10,700) 479(11,100) |
723 | n.d. [d] | 0.35, −1.75 | [35] |
| 4 | 252 (50,500) 376 (7,500) |
617(380) | 0.045 | 0.88, −1.54, −1.77, −2.16 | this work |
| [Ir(ppy)2(bpy)]+ | 376(6,000) | 595(415) | 0.062 | 0.90, −1.77, −2.49, −2.69 | [36] |
measured in acetonitrile
using Ru(bpy)32+ as reference
vs Fc/Fc+
n.d. : not determined.
3.4. Cyclic Voltammetry
We used cyclic voltammetry to investigate the redox potentials of the various d6 metal complexes. Cyclic voltammograms have been obtained for 1–4 and Ru(bpy)32+ as reference in degassed acetonitrile using a standard electrochemical cell. As shown in Fig. 2, each complex exhibits multiple quasi-reversible one-electron ligand-based reduction waves as well as one metal-based oxidation. In addition, an irreversible reduction wave around −1.55 V vs Fc/Fc+ for all complexes is consistent with the reduction of the epoxide moiety.[37]
Fig. 2.
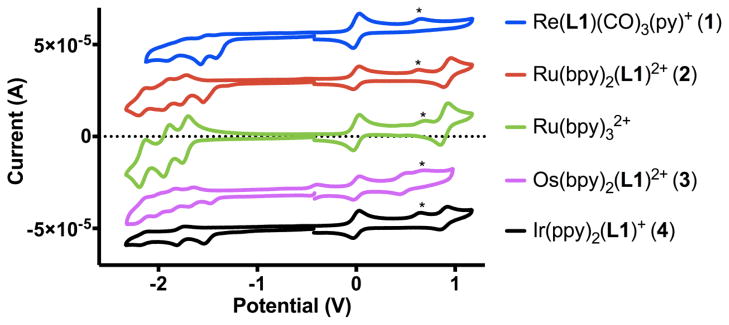
Cyclic Voltammograms for complexes 1–4 and Ru(bpy)32+ in acetonitrile using Fc/Fc+ peak centered at 0 V as reference (*: trace water).
The redox potentials, listed in Table 1, are typical for comparable d6 metal polypyridyl complexes.[38] Three ligand-based reduction waves for 2 and 3 correspond to the successive reduction of the three diimine ligands as also observed for the Ru(bpy)32+ complex. Only two reduction waves are observed for 4 within the voltage range of the solvent. This reflects the more negative reduction potentials for 2-phenylpyridine, owing to the greater electron density on the phenylpyridine rings compared to the bipyridine ligand. The two reduction waves for 1 likely correspond to the double reduction of the diimine ligand L1.
3.5. Thiol-specific protein labeling
In our pursuit of light-driven selective C-H bond functionalization, we have developed a library of hybrid P450 BM3 heme domain enzymes containing covalently attached Ru(II)-polypyridyl photosensitizers.[12, 13] The single labeling of the P450 BM3 enzymes at the engineered cysteine residues using the iodoacetamide functionalized metal complexes was unambiguously confirmed by ESI-MS and UV-vis and luminescence measurements.[13] The position of the covalent attachment was also confirmed by chymotrypsin digest [13] and x-ray crystallography.[21]
A set of previously characterized P450 BM3 heme domains, containing single non-native surface exposed cysteine residues, namely K97C, Q109C and Q397C, was used to investigate the bioconjugation of the metal complexes 1–4. Each protein was subjected to the labeling reaction conditions with the various complexes, and the reaction was followed by mass spectrometry (See Fig. S3). All of the metal complexes could covalently attach to the various P450 BM3 mutants, according to Scheme 2A, via the opening of the epoxy ring and the formation of a β-hydroxy cysteinyl adduct. Modest to high yield could be obtained for the labeling reactions, ranging from 65 to 90% depending on the mutants and labels used (See Fig. S3). As shown in Fig. 3 for the representative K97C protein, a mass spectral shift is observed in the K97C-1 to K97C-4 series consistent with the covalent attachment of the respective labels. In contrast, when a P450 BM3 mutant without any surface cysteine residue [21] is exposed to the same labeling conditions, no covalent adduct could be detected (See Fig. S3), establishing the selectivity of the labeling protocol for sulfhydryl groups vs. the other amino acids present at the protein surface. In addition, the position of the covalent attachment could also unambiguously be confirmed by chymotrypsin digestion of the labeled protein as previously established[13] (See Fig. S4 for K97C-2).
Scheme 2.
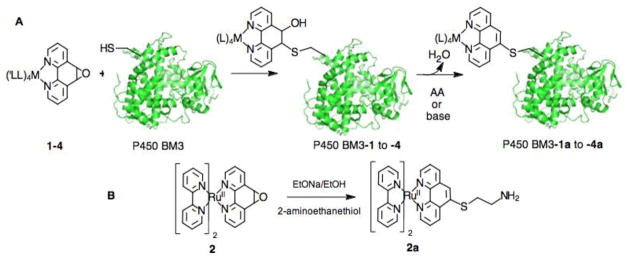
Synthetic routes for A) the covalent attachment of the complexes 1–4 to the various single cysteine residues of P450 BM3 heme domain mutants and B) the fully aromatized model compound 2a.
Fig. 3.

Deconvoluted mass spectra for the K97C protein labeled with various metal complexes (K97C-1 to K97C-4). Dashed lines indicate the labeled proteins (K97C-1a to K97C-4a) with aromatized phenanthroline ring on the protein-bound metal complexes after the addition of a mild base and the loss of a water molecule.
For most of the studied P450 heme domains labeled with the complexes 1–4, only a single mass adduct is observed corresponding to the β-hydroxy cysteinyl adduct (BM3-1 to -4, Scheme 2). While similar β-hydroxy adducts have recently been characterized,[31, 39, 40] the majority of examples investigating the ring opening of the L1 ligand with various nucleophiles have usually observed the full aromatization of the phenanthroline.[25, 26] Similarly, when we investigated the ring opening reaction in the model complex 2 using 2-aminoethanethiol as nucleophile (See Scheme 2B), the sole compound detected and isolated corresponds to the fully aromatized complex, Ru(bpy)2(L2)2+ (2a) with L2 = 5-(2-aminoethanesulfanyl)-1,10-phenanthroline. The same compound 2a could also independently be synthesized from Ru(bpy)2Cl2 and the L2 ligand prepared according to a reported procedure[25] (see experimental section, Fig. S5 and Table S1). The central ring aromatization in the reaction is thought to be promoted by the loss of a water molecule from the ring-opened β-hydroxy sulfanyl intermediate.
Closer examination of the labeling of the Q397C mutant with the Ru(II) and Os(II) complexes (2 and 3) reveals a second peak in the deconvoluted mass with a loss of 18 mass units (see Fig. S6). This peak is attributed to the dehydration of the β-hydroxy cysteinyl intermediate leading to the aromatization of the ligand on the protein-bound label compounds (Q397C-2a and Q397C-3a). We suspected that the aromatization process in the Q397C hybrid enzyme could be base assisted probably by a neighboring basic residue. In the P450 BM3 heme domain crystal structure,[41] a lysine residue at the position 97 is in close proximity to the labeled cysteine Q397C and is a likely candidate responsible for promoting the base-assisted dehydration.
In order to resolve the heterogeneity in the labeling reaction for the Q397C protein, we explored the use of a mild base to promote the complete aromatization. Addition of small aliquots of potassium carbonate to the Q397C-2 and -3 labeled proteins lead to the complete dehydration and full aromatization of the central ring as detected by mass spectrometry (Q397C-2a and -3a) (See experimental section and Fig. S6) without any noticeable protein degradation. While the labeling of the Q397C with the Ir and Re complexes mainly afforded the intermediate ring opened product (Q397C-1 and Q397C-4), complete conversion to the aromatized adduct was similarly obtained upon addition of potassium carbonate. Subsequently, all the previous labeled mutants (BM3-1 to -4) with the intermediate ring opened compound could be driven to the thermodynamically favored aromatized phenanthroline complex by the same approach (BM3-1a to -4a) (Fig. 3, dashed lines). These observations reveal an exquisite control of the properties and reactivities of the 1–4 complexes.
The labeled proteins could be purified following the protocol developed previously [12] and were characterized by UV-vis and luminescence spectroscopies (Fig. 4). CO binding studies with the labeled purified proteins indicate that the labeled proteins are properly folded. In addition, the UV-vis spectra display a Soret band at 418 nm typical for a six-coordinate low-spin Fe(III)-aquo heme thiolate coordination. The MLCT for the Ru(II) and Os(II) complexes could be noted at 460 nm and 475 nm, respectively. In contrast, not much contribution to the heme protein spectra could be detected for the Re and Ir labeled proteins (K97C-1 and -4) likely due to the relatively low absorption coefficient of these complexes compared to the heme Soret band. The emission of the various labeled enzyme matches the emission of the respective d6 metal complexes in the same buffered solution, consistent with the successful covalent label attachment (see Fig. S2).
Fig. 4.

UV-vis and luminescence spectra for the K97C labeled proteins with metal complexes 1–4.
In addition, the synthesis of complex 2a allows to probe the effect of the introduction of the electron withdrawing cysteamine group and the aromatization of the ring relative to the parent complex 2. The complex 2a shows a 20 mV shift in the first redox potential as well as a blue-shift in the MLCT absorption and emission (see Table S1). Conveniently, the shift in the luminescence can also be used to determine the state of aromatization of the protein-bound label compounds. As shown in Fig. 5, the 20 nm blue-shift observed in the model complexes 2 and 2a is also noticeable in the various labeled proteins with the single adduct (BM3-2) and the full aromatized complex (BM3-2a). This emission shift may find interesting applications in differentiating between aromatized and β-hydroxy cysteinyl labeled biomolecules.
Fig. 5.

Emission spectra of the labeled proteins, K97C-2 and K97C-2a, and the corresponding metal complexes (2 and 2a) reflecting the blue-shift emission due to the aromatization of the phenanthroline ligand and the presence of the thiol group.
4. Conclusion
The set of four d6 metal complexes bearing the L1 ligand could be readily synthesized and characterized. The complexes were selectively conjugated in high yield to cysteine residues of P450 BM3 enzymes leading to a single β-hydroxy cysteinyl adduct that could be driven to the fully aromatized phenanthroline ligand on the protein-bound complexes. The 5,6-epoxy-5,6-dihydro-[1,10]phenanthroline ligand offer new approaches for the labeling of cysteine residues. This methodology can easily be extended to various proteins containing surface exposed sulhydryl residues.
Supplementary Material
Highlights.
Octahedral d6 metal complexes bearing 5,6-epoxy-5,6-dihydro-[1,10]phenanthroline.
Selective nucleophilic epoxy-ring opening by surface-exposed cysteine residues.
Base-assisted dehydration of protein-bound label compounds promoting aromatization.
Acknowledgments
L.C., S.D., A. C. and N.-H. T. would like to thank the National Institute of Health (GM095415) for financial support. This research was also supported by an award from Research Corporation for Science Advancement. L.C. thanks the National Science Foundation (MRI grant 0923573) and San José State University for the use of mass spectrometry facilities in the PROTEIN LAB.
Abbreviations
- bpy
2,2′-bipyridine
- DMF
dimethylformamide
- ESI-MS
electrospray mass spectrometry
- Fc/Fc+
ferrocene/ferrocenium couple
- IL
intraligand
- L1
5,6-epoxy-5,6-dihydro-[1,10]phenanthroline ligand
- L2
5-(2-aminoethanesulfanyl)-1,10-phenanthroline ligand
- MLCT
metal-to-ligand-charge transfer
- ppy
2-phenylpyridine
- py
pyridine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sameiro M, Goncalves T. Chem Rev. 2009;109:190–212. doi: 10.1021/cr0783840. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez-Moreira V, Thorp-Greenwood FL, Coogan MP. Chem Commun. 2010;46:186–202. doi: 10.1039/b917757d. [DOI] [PubMed] [Google Scholar]
- 3.Lo KKW, Choi AWT, Law WHT. Dalton Trans. 2012;41:6021–6047. doi: 10.1039/c2dt11892k. [DOI] [PubMed] [Google Scholar]
- 4.Shcherbakova DM, Subach OM, Verkhusha VV. Angew Chem Int Ed. 2012;51:10724–10738. doi: 10.1002/anie.201200408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis L, Chin JW. Nat Rev Mol Cell Biol. 2012;13:168–182. doi: 10.1038/nrm3286. [DOI] [PubMed] [Google Scholar]
- 6.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat Methods. 2008;5:763–775. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 7.Happ B, Winter A, Hager MD, Schubert US. Chem Soc Rev. 2012;41:2222–2255. doi: 10.1039/c1cs15154a. [DOI] [PubMed] [Google Scholar]
- 8.Bouley Ford ND, Shin DW, Gray HB, Winkler JR. J Phys Chem B. 2013;117:13206–13211. doi: 10.1021/jp403234h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gray HB, Winkler JR. Proc Nat Acad Sci USA. 2005;102:3534–3539. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geren L, Durham B, Millett F. Methods Enzymol. 2009;456:507–520. doi: 10.1016/S0076-6879(08)04428-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roth LE, Tezcan FA. J Am Chem Soc. 2012;134:8416–8419. doi: 10.1021/ja303265m. [DOI] [PubMed] [Google Scholar]
- 12.Tran NH, Huynh N, Chavez G, Nguyen A, Dwaraknath S, Nguyen TA, Nguyen M, Cheruzel L. J Inorg Biochem. 2012;115:50–56. doi: 10.1016/j.jinorgbio.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran NH, Nguyen DN, Dwaraknath S, Mahadevan S, Chavez G, Nguyen A, Dao T, Mullen S, Nguyen TA, Cheruzel LE. J Am Chem Soc. 2013;135:14484–14487. doi: 10.1021/ja409337v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalia J, Raines RT. Curr Org Chem. 2010;14:138–147. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendoza VL, Vachet RW. Mass Spectrom Rev. 2009;28:785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sahoo H. Rsc Adv. 2012;2:7017–7029. [Google Scholar]
- 17.Chalker JM, Bernardes GJL, Lin YA, Davis BG. Chem Asian J. 2009;4:630–640. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- 18.Terpetschnig E, Dattelbaum JD, Szmacinski H, Lakowicz JR. Anal Biochem. 1997;251:241–245. doi: 10.1006/abio.1997.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weh J, Duerkop A, Wolfbeis OS. Chembiochem. 2007;8:122–128. doi: 10.1002/cbic.200600316. [DOI] [PubMed] [Google Scholar]
- 20.Castellano FN, Dattelbaum JD, Lakowicz JR. Anal Biochem. 1998;255:165–170. doi: 10.1006/abio.1997.2468. [DOI] [PubMed] [Google Scholar]
- 21.Ener ME, Lee YT, Winkler JR, Gray HB, Cheruzel L. Proc Nat Acad Sci USA. 2010;107:18783–18786. doi: 10.1073/pnas.1012381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran NH, Huynh N, Bui T, Nguyen Y, Huynh P, Cooper ME, Cheruzel LE. Chem Commun. 2011;47:11936–11938. doi: 10.1039/c1cc15124j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoffers E, Kohler L. Tet Asymm. 2009;20:1897–1902. doi: 10.1016/j.tetasy.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shee NK, Adekunle FAO, Das D, Drew MGB, Datta D. Inorg Chim Acta. 2011;375:101–105. [Google Scholar]
- 25.Dotsenko IA, Curtis M, Samoshina NM, Samoshin VV. Tetrahedron. 2011;67:7470–7478. [Google Scholar]
- 26.Shen YB, Sullivan BP. Inorg Chem. 1995;34:6235–6239. [Google Scholar]
- 27.Collin JP, Sauvage JP. Inorg Chem. 1986;25:135–141. [Google Scholar]
- 28.Kober EM, Caspar JV, Sullivan BP, Meyer TJ. Inorg Chem. 1988;27:4587–4598. [Google Scholar]
- 29.Marti AA, Mezei G, Maldonado L, Paralitici G, Raptis RG, Colon JL. Eur J Inorg Chem. 2005:118–124. [Google Scholar]
- 30.Lowry MS, Hudson WR, Pascal RA, Bernhard S. J Am Chem Soc. 2004;126:14129–14135. doi: 10.1021/ja047156+. [DOI] [PubMed] [Google Scholar]
- 31.Wei H, Yin JY, Wang E. Anal Chem. 2008;80:5635–5639. doi: 10.1021/ac8001462. [DOI] [PubMed] [Google Scholar]
- 32.Pazderski L, Pawlak T, Sitkowski J, Kozerski L, Szlyk E. Magn Reson Chem. 2010;48:450–457. doi: 10.1002/mrc.2600. [DOI] [PubMed] [Google Scholar]
- 33.Balzani V, Bergamini G, Campagna S, Puntoriero F. Top Curr Chem. 2007;280:1–36. [Google Scholar]
- 34.Sacksteder L, Zipp AP, Brown EA, Streich J, Demas JN, Degraff BA. Inorg Chem. 1990;29:4335–4340. [Google Scholar]
- 35.Kober EM, Sullivan BP, Dressick WJ, Caspar JV, Meyer TJ. J Am Chem Soc. 1980;102:7383–7385. [Google Scholar]
- 36.Ladouceur S, Fortin D, Zysman-Colman E. Inorg Chem. 2010;49:5625–5641. doi: 10.1021/ic100521t. [DOI] [PubMed] [Google Scholar]
- 37.Boujlel K, Simonet J. Electrochim Acta. 1979;24:481–487. [Google Scholar]
- 38.Hankache J, Niemi M, Lemmetyinen H, Wenger OS. Inorg Chem. 2012;51:6333–6344. doi: 10.1021/ic300558s. [DOI] [PubMed] [Google Scholar]
- 39.Lezhnina MM, Hofmann D, Santiago-Schubel B, Klauth P, Kynast UH. New J Chem. 2012;36:2322–2327. [Google Scholar]
- 40.Wei H, Wang E. Chem Lett. 2007;36:210–211. [Google Scholar]
- 41.Girvan HM, Seward HE, Toogood HS, Cheesman MR, Leys D, Munro AW. J Biol Chem. 2007;282:564–572. doi: 10.1074/jbc.M607949200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.