

Abstract
The first example of C-alkylation of 1,2-dihydropyridines with alkyl triflates and Michael acceptors was developed to introduce quaternary carbon centers with high regio- and diastereoselectivity. Hydride or carbon nucleophile addition to the resultant iminium ion also proceeded with high diastereoselectivity. Carbon nucleophile addition results in an unprecedented level of substitution to provide piperidine rings with adjacent tetrasubstituted carbons.
Keywords: piperidine, alkylation, tetrasubstituted carbon, diastereoselective, dihydropyridine
Piperidines are valuable and prevalent substructures in biologically active natural products and in medicines that have been enormously important in managing disease.[1-3] As entry to this important class of compounds, a number of powerful methods for the synthesis of 1,2-dihydropyridines have been developed.[4,5] In this context, we previously disclosed an approach to rapidly assemble highly substituted 1,2-dihydropyridines by a Rh(I)-catalyzed C–H alkenylation/electrocyclization cascade from readily available α,β-unsaturated imines and alkynes.[6] We have further recently described that these 1,2-dihydropyridine products are versatile intermediates for the preparation of highly substituted tetrahydropyridines by regio- and diastereoselective protonation followed by hydride or carbon nucleophile addition (Scheme 1a).[7-9] Herein, we demonstrate that C-alkylation of 1,2-dihydropyridines, which to our knowledge has not previously been reported, can be achieved with alkyl triflates and Michael acceptors with high regio and diastereoselectivity to provide quaternary carbon centers within the piperidine core (Scheme 1b).[10-12] Moreover, reduction or carbon nucleophile addition to the resultant iminium ion can also be accomplished with high diastereoselectivity. In the case of carbon nucleophile addition, an unprecedented level of substitution is achieved to provide piperidine rings with adjacent tetrasubstituted carbons.
Scheme 1.
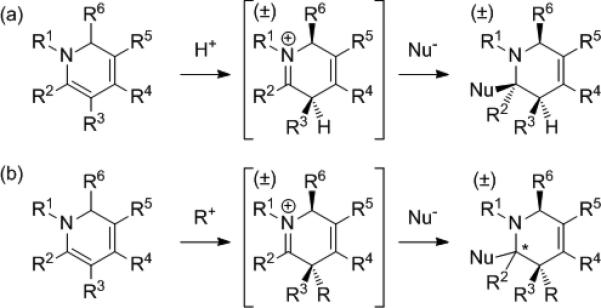
a) Previously reported 1,2-dihydropyridine protonation and nucleophilic addition sequence. b) Transformation developed in this study: Reaction with carbon electrophiles (R+) followed by addition of hydride or carbon nucleophiles.
We initially explored the addition of methyl iodide and dimethyl sulfate to dihydropyridine 1 (R = Me) (Table 1). However, methylation was not observed using a variety of solvents such as THF, toluene and CH2Cl2 even with heating to 60 °C in a sealed reaction vessel (data not shown). We consequently evaluated the considerably more reactive methyl triflate, which resulted in complete conversion to the methylated iminium product at rt (entry 1). Successful alkylation with ethyl triflate demonstrated that less reactive alkylating agents also couple efficiently (entry 2). Moreover, ethylation established that the reaction proceeds with very high diastereoselectivity. To further test the generality of the alkylation protocol, the reactivity of dihydropyridine 1 (R = H) was investigated because it results in a more highly destabilized aldiminium product (entry 3). While generally complete and stereoselective alkylation was observed in toluene with an 14–18 h reaction time, at shorter reaction times CH2Cl2 as solvent proved to be more effective (entry 4–7). Given the instability of benzyl triflates, benzyl chloride and bromide were instead evaluated, but no alkylation was observed for either reagent (entries 8 and 9). Lewis acid-mediated activation of benzyl bromide was therefore investigated, with ZrCl4 providing the benzylated product in >95% conversion within 2 h at –78 °C to rt and with good diastereoselectivity (entry 10).
Table 1.
Optimization of dihydropyridine alkylation conditions.

| ||||||
|---|---|---|---|---|---|---|
| entry | R | R’X | solvent | time | dr [a] | conv[b] |
| 1 | Me | MeOTf | toluene | 16 h | - | >95% |
| 2 | Me | EtOTf | toluene | 14 h | >95% | >95% |
| 3 | H | MeOTf | toluene | 18 h | - | >95% |
| 4 | Me | MeOTf | CH2Cl2 | 1.5 h | - | >95% |
| 5 | Me | EtOTf | CH2Cl2 | 1.5 h | >95% | >95% |
| 6 | H | MeOTf | CH2Cl2 | 1.5 h | - | >55% |
| 7 | H | EtOTf | CH2Cl2 | 1.5 h | >95% | >95% |
| 8 | Me | BnCl | CH2Cl2 | 16 h | nd | <5% |
| 9 | Me | BnBr | CH2Cl2 | 16 h | nd | <5% |
| 10[c] | Me | BnBr | CH2Cl2 | 2 h | >90% | >95% |
Where applicable, the reaction dr was determined by 1H NMR analysis.
The reaction conversion was established by 1H NMR analysis.
With ZrCl4 as a Lewis acid additive, –78 °C to rt.
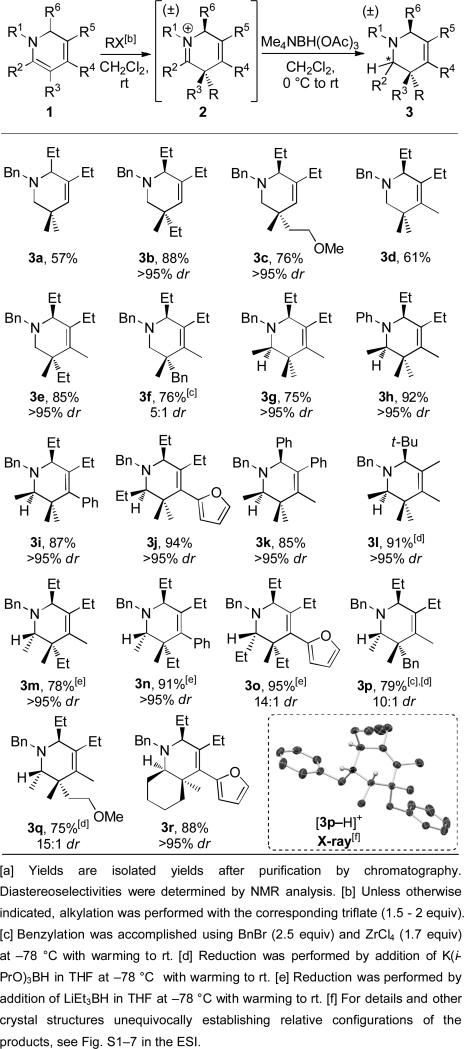
With the feasibility of the key alkylation step established, we next focused on the preparation of the desired tetrahydropyridines 3 by reduction of the reactive iminium ion 2 (Table 2). We first explored the alkylation/reduction sequence for 1,2-dihydropyridines 1 unsubstituted at the R2 position (R2 = H), because this avoids introducing a stereocenter in the reduction step. After alkylation, Me4NBH(OAc)3 served as a mild and inexpensive reducing agent for the synthesis of tetrahydropyridines 3a to 3f in good to high overall yields. Notably, in the key alkylation step, methyl (3a and 3d), ethyl (3b and 3e), functionalized 2-methoxyethyl (3c), and benzyl (3f) groups were all successfully incorporated. The overall yields for the methylation products 3a and 3d were somewhat lower than for the other electrophiles due to competitive methylation at nitrogen when R2 = H. However, for all of the other alkylating agents, which are less reactive than methyl triflate, no alkylation at nitrogen was detected and high diastereoselectivities were observed. For ethylation (3b and 3e) and 2-methoxyethylation (3c), only a single diastereomer was obtained. Alkylation consistently occurs opposite to the R6 group as rigorously established by X-ray structural analysis of 3e as well as for multiple other alkylation products (3g, 3m, 3n, 3p, 6d and 6g, vide infra).[13] The Lewis acid mediated benzylation provided a more modest 5:1 ratio (3f).
Table 2.
One-pot dihydropyridine alkylation and reduction.[a]
|
We next investigated the alkylation/reduction sequence for 1,2-dihydropyridines substituted at the R2 position, which introduces added complexity because a stereocenter is generated in the reduction step (3g to 3r, Table 2). The sequence was first performed by methylating dihydropyridines 1 where R3 = Me such that a stereocenter is only introduced in the reduction step (3g – 3l). In all cases only a single diastereomer was produced as determined by 1H NMR analysis. Hydride addition opposite the R6 group was rigorously established for 3g by X-ray structural analysis and is consistent with the face selectivity observed for reduction in our previously reported protonation/reduction sequence.[7] The methylation/reduction sequence proceeded in excellent overall yields for a range of substituents with diverse steric and electronic properties (75-94%). At the R1 position on nitrogen both the N-benzyl (3g) and the comparatively deactivating N-phenyl (3h) group provided high yields. The R4 position could be substituted with alkyl (3g, h, k and l), phenyl (3i) and furanyl (3j) groups, while alkyl and phenyl groups could be placed at both the R5 and R6 positions (3g versus 3k). A 91% yield was observed when the tert-butyl group was placed at the R6 position (3l), although reduction with K(i-PrO)3BH at -78 °C instead of Me4NBH(OAc)3 at 0 °C was necessary to achieve high selectivity.
For products 3m to 3r, two stereocenters are introduced, the first in the alkylation step and the second in the reduction step. High overall yields and good stereselectivities were observed for methylation (3r), ethylation (3m to 3o), benzylation (3p) and introduction of the 2-methoxyethyl group (3q). Moreover, different substituents at the R2 through R4 positions were tolerated, including a bicyclic dihydropyridine that resulted in the cis-fused product 3r. However, to achieve high reduction stereoselectivity for many of these substrates, it was necessary to use more reactive hydride reagents at -78 °C. In particular, for 3m, 3n and 3o, reduction with LiBEt3H was optimal, and for 3l, 3p and 3q, reduction with K(i-PrO)3BH proved to be the most effective. The relative configurations of the two newly formed stereocenters were rigorously determined by X-ray structural analysis for the ethylation/reduction products 3m and 3n and the benzylation/reduction product 3p.[13] These X-ray structures establish that upon introducing alkyl groups R larger than a methyl group, steric interactions that result from the newly formed and adjacent quaternary stereocenter control the face selectivity of reduction rather than the more distant R6 substituent.
Michael additions to α,β-unsaturated esters were explored to introduce the ester group as a versatile functionality for further elaboration (Scheme 2). Direct addition of the dihydropyridine to methyl acrylate did not proceed, but after evaluating a variety of Lewis acids, we established that TMSOTf results in high conversion to the silyl enol ether intermediate 4 as an approximately 1:1 E/Z isomer mixture. To obtain tetrahydropyridine 5a, reduction with Me4NBH(OAc)3 was most effective, while for 5b LiBHEt3 provided the highest diastereoselectivity and overall yield.
Scheme 2.
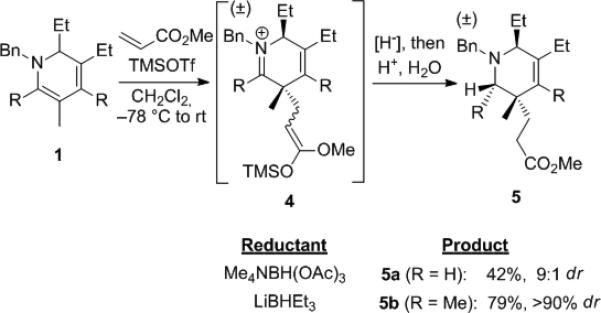
Michael addition of dihydropyridines to methyl acrylate.
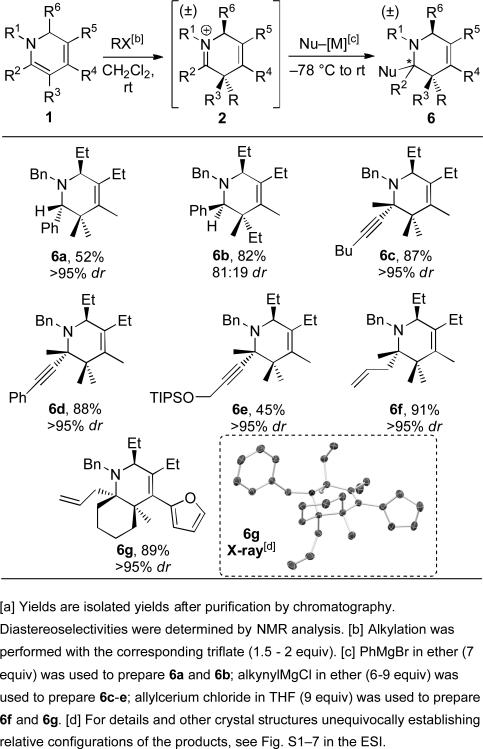
We next investigated the addition of carbon nucleophiles rather than hydride (Table 3). We first evaluated additions to 1,2-dihydropyridines 1 unsubstituted at the R2 position because the iminium intermediate that is generated upon alkylation lacks α-hydrogens and therefore cannot undergo competitive α-deprotonation. For these substrates phenyl Grignard adds cleanly to give 6a and 6b, with the yield for 6a being somewhat lower due to competitive N-methylation of the dihydropyridine in the alkylation step (see previous discussion for 3a and 3d). While high diastereoselectivity was observed for 6a, an attenuated 81:19 selectivity was observed for 6b.
Table 3.
One-pot dihydropyridine alkylation and carbon nucleophile addition.[a]
|
The remaining carbon nucleophile addition products (6c to 6g) were prepared from 1,2-dihydropyridines with carbon substituents at R2, which results in adjacent tetrasubstituted stereocenters being introduced in the alkylation/nucleophilic addition sequence. Each of these substrates has the potential for competitive α-deprotonation in the carbon nucleophile addition step. For this reason, less basic and more covalent organometallic reagents were more effective. Phenyl, alkyl and silyloxymethyl substituted alkynyl Grignard reagents all added diastereoselectively and in good yields (6a to 6e).[14] Allyl cerium reagents also added in high yields and with excellent diastereoselectivities (6f and 6g),[15] including for the preparation of cis-fused bicyclic piperidine 6g with tetrasubstituted carbons at the two bridgehead positions. Finally, X-ray structural analysis of 6d and 6g[13] establishes that when the alkylation step introduces a stereocenter the face selectivity for nucleophilic attack is controlled by steric interactions at this adjacent quaternary site consistent with that previously observed in the alkylation/reduction sequence (vide supra).
In conclusion, a 1,2-dihydropyridine alkylation/nucleophile addition sequence has been developed that proceeds in good yields and with high levels of regio- and diastereoselectivity to provide piperidine derivatives with unprecedented levels of substitution, including products with adjacent tetrasubstituted ring carbons. Because the 1,2-dihydropyridine starting materials are prepared in one step from simple precursors by a tandem C-H alkenylation/electrocyclization reaction cascade, the described alkylation/nucleophile addition sequence provides rapid access to densely substituted piperidines that would be extremely difficult to prepare by alternative methods.
Supplementary Material
Footnotes
This work was supported by the NIH Grant GM069559 (to J.A.E.). R.G.B. acknowledges funding from the DOE, DE-AC02-05CH11231.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx
Contributor Information
Simon Duttwyler, Department of Chemistry Yale University 225 Prospect St., New Haven, CT 06520 (USA).
Shuming Chen, Department of Chemistry Yale University 225 Prospect St., New Haven, CT 06520 (USA).
Colin Lu, Department of Chemistry Yale University 225 Prospect St., New Haven, CT 06520 (USA).
Brandon Q. Mercado, Department of Chemistry Yale University 225 Prospect St., New Haven, CT 06520 (USA)
Robert G. Bergman, Department of Chemistry University of California, Berkeley Berkeley, CA 94720-1416 (USA) and Lawrence Berkeley National Laboratory 1 Cyclotron Road, Berkeley, CA 94720 (USA)
Jonathan A. Ellman, Department of Chemistry Yale University 225 Prospect St., New Haven, CT 06520 (USA).
References
- 1.For an analysis of the prevalence of piperidines in drugs, see: Roughley SD, Jordan AM. J. Med. Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y.
- 2.The structural features of piperidines are consistent with renewed emphasis on the development of drug candidates with increased saturation and nonplanar display of functionality, see: Lovering F, Bikker J, Humblet C. J. Med. Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. Walters WP, Green J, Weiss JR, Murcko MA. J. Med. Chem. 2011;54:6405–6416. doi: 10.1021/jm200504p. Ritchie TJ, Macdonald SJF. Drug. Discov. Today. 2009;14:1011–1020. doi: 10.1016/j.drudis.2009.07.014.
- 3.For general reviews on piperidines, see: Michael JP. Nat. Prod. Rep. 2008;25:139–165. doi: 10.1039/b612166g. Buffat MGP. Tetrahedron. 2004;60:1701–1729. Mitchinson A, Nadin A. J. Chem. Soc. Perkin Trans. 1. 2000:2862–2892. Laschat S, Dickner T. Synthesis. 2000:1781–1813.
- 4.For reviews on the synthesis and applications of 1,2-dihydropyridines, see: Bull JA, Mousseau JJ, Pelletier G, Charette AB. Chem. Rev. 2012;112:2642–2713. doi: 10.1021/cr200251d. Silva EMP, Varandas PAMM, Silva AMS. Synthesis. 2013;45:3053–3089. Tanaka K, Fukase K, Katsumura S. Synlett. 2011:2115–2139.
- 5.For recent methods for the synthesis of 1,2-dihydropyridines, see Mizoguchi H, Oikawa H, Oguri H. Nature Chem. 2013 Advanced online publication, DOI:10.1038/nchem.1798. Chau ST, Lutz JP, Wu K, Doyle AG. Angew. Chem. 2013;125:9323–9326. doi: 10.1002/anie.201303994. Angew. Chem. Int. Ed. 2013;52:9153–9156. doi: 10.1002/anie.201303994. Yamakawa T, Yoshikai N. Org. Lett. 2013;15:196–199. doi: 10.1021/ol303259m. Oshima K, Ohmura T, Suginome M. J. Am. Chem. Soc. 2012;134:3699–3702. doi: 10.1021/ja3002953. Amatore M, Leboeuf D, Malacria M, Gandon V, Aubert C. J. Am. Chem. Soc. 2013;135:4576–4579. doi: 10.1021/ja309301n. Nadeau C, Aly S, Belyk K. J. Am. Chem. Soc. 2011;133:2878–2880. doi: 10.1021/ja111540g. Jarvis SBD, Charette AB. Org. Lett. 2011;13:3830–3833. doi: 10.1021/ol201349k.
- 6.Colby DA, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2008;130:3645–3651. doi: 10.1021/ja7104784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Duttwyler S, Lu C, Rheingold AL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2012;134:4064–4067. doi: 10.1021/ja2119833. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Duttwyler S, Chen S, Takase MK, Wiberg KB, Bergman RG, Ellman JA. Science. 2013;339:678–682. doi: 10.1126/science.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ischay MA, Takase MK, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2013;135:2478–2481. doi: 10.1021/ja312311k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For reviews on tetrahydropyridines, see: Mateeva NN, Winfield LL, Redda KK. Curr. Med. Chem. 2005;12:551–571. doi: 10.2174/0929867310504050551. Felpin FX, Lebreton J. Curr. Org. Synth. 2004;1:83–109.
- 9.For recent methods for the preparation of highly substituted tetrahydropyridines, see: Martin TJ, Rovis T. Angew. Chem. 2013;125:5476–5479. Angew. Chem. Int. Ed. 2013;52:5368–5371. doi: 10.1002/anie.201301741. Shi F, Tan W, Zhou R-Y, Xing G-J, Tu S-J. Adv. Synth. Catal. 2013;355:1605–122. Li X, Zhao Y, Qu H, Mao Z, Lin X. Chem. Commun. 2013;49:1401–1403. doi: 10.1039/c2cc38349g. Lemonnier G, Charette AB. J. Org. Chem. 2012;77:5832–5837. doi: 10.1021/jo300690h. Yang DX, Micalizio GC. J. Am. Chem. Soc. 2012;134:15237–15240. doi: 10.1021/ja306362m. Harrison DP, Iovan DA, Myers WH, Sabat M, Wang SS, Zottig VE, Harman WD. J. Am. Chem. Soc. 2011;133:18378–18387. doi: 10.1021/ja2075086.
- 10.For stereospecific 3-aza-Cope rearrangement of quaternary N-allyl enammonium salts to install quaternary centers in tetahydroisoquinolines, see: McComsey DF, Maryanoff BE. J. Org. Chem. 2000;65:4938–4943. doi: 10.1021/jo000363h.
- 11.For leading references, on diastereoselective alkylations of α-carboxy or cyano-substituted hydrazones to introduce quaternary carbon stereocenters, see: Enders D, Zmaponi A, Raabe G, Runsink J. Synthesis. 1993:725–728. Enders D, Zamponi A, Schafer T, Nubling C, Eichenauer H, Demir AS, Raabe G. Chem. Ber. 1994;127:1707–1721.
- 12.For leading references on diastereolective enolate alkylations for the asymmetric synthesis of α,α-disubstituted α-hydroxy or amino acids, see: Seebach D, Sting AR, Hoffmann M. Angew. Chem. 1996;108:2881–2921. Angew. Chem. Int. Ed. 1996;35:2708–2748. Hugelshofer CL, Mellem KT, Myers AG. Org. Lett. 2013;15:3134–3137. doi: 10.1021/ol401337p.
- 13.Synthetic details, characterization, and molecular structures obtained by X-ray structural analysis of compounds are described in the Supporting Information under CCDC Nos: 974855 (3e), 974821 (3g), 974854 (3m), 974857 (3n), 974819 (3p), 974856 (6d), and 974820 (6g). These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 14.For diastereoselective, sequential addition of Grignard reagents and cyanide to N-methoxy-piperidin-2-ones to install a tetrasubstituted carbon center, see: Jäkel M, Qu J, Schnitzer T, Helmchen G. Chem. Eur. J. 2013;19:16746–16755. doi: 10.1002/chem.201302735.
- 15.We previously observed that allyl cerium reagents minimize competitive iminium ion deprotonation, see ref 7b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.