

Abstract
Tissue damage resulting from the extracellular production of HOCl (hypochlorous acid) by the MPO (myeloperoxidase)-hydrogen peroxide-chloride system of activated phagocytes is implicated as a key event in the progression of a number of human inflammatory diseases. Consequently, there is considerable interest in the development of therapeutically useful MPO inhibitors. Nitroxides are well established antioxidant compounds of low toxicity that can attenuate oxidative damage in animal models of inflammatory disease. They are believed to exert protective effects principally by acting as superoxide dismutase mimetics or radical scavengers. However, we show here that nitroxides can also potently inhibit MPO-mediated HOCl production, with the nitroxide 4-aminoTEMPO inhibiting HOCl production by MPO and by neutrophils with IC50 values of approx. 1 and 6 μM respectively. Structure–activity relationships were determined for a range of aliphatic and aromatic nitroxides, and inhibition of oxidative damage to two biologically-important protein targets (albumin and perlecan) are demonstrated. Inhibition was shown to involve one-electron oxidation of the nitroxides by the compound I form of MPO and accumulation of compound II. Haem destruction was also observed with some nitroxides. Inhibition of neutrophil HOCl production by nitroxides was antagonized by neutrophil-derived superoxide, with this attributed to superoxide-mediated reduction of compound II. This effect was marginal with 4-aminoTEMPO, probably due to the efficient superoxide dismutase-mimetic activity of this nitroxide. Overall, these data indicate that nitroxides have considerable promise as therapeutic agents for the inhibition of MPO-mediated damage in inflammatory diseases.
Keywords: hypochlorous acid, myeloperoxidase, neutrophil, nitroxide, protein oxidation, superoxide
INTRODUCTION
The haem peroxidase enzyme MPO (myeloperoxidase) is released intraphagosomally and extracellularly by activated neutrophils, monocytes and some macrophages, where it catalyses the production of the potent oxidant HOCl (hypochlorous acid) from H2O2 and Cl− ions [1]. Production of the related oxidants HOBr (hypobromous acid) and HOSCN (hypothiocyanous acid) from Br− and SCN− is also significant at physiological concentrations of these ions [2,3]. Intraphagosomal HOCl production appears to play a beneficial role in immune defense by mediating bacterial cell killing, however tissue damage due to extracellular production of HOCl is implicated as a key pathogenic event in a number of human inflammatory diseases including atherosclerosis, asthma, rheumatoid arthritis, cystic fibrosis, kidney disease and some cancers [4-6]. For example, elevated systemic levels of MPO can predict cardiovascular disease incidence and risk, and MPO accumulates in atherosclerotic lesions at all stages of their development along with specific biomarkers of HOCl-mediated protein oxidation [6]. The levels of these biomarkers also correlate with disease progression [6]. HOCl is believed to contribute to atherosclerotic disease initiation and development in a number of ways, including the generation of highly atherogenic oxidized forms of low-density lipoproteins that are taken up by macrophages in an unregulated manner to give lipid-laden (foam) cells [7], the impairment of the reverse-cholesterol properties of (anti-atherogenic) high-density lipoproteins [8] and the promotion of extracellular matrix degradation [9]. Similar evidence is available for a role for MPO-derived HOCl in other diseases [6].
In the light of the substantial body of evidence that MPO-derived HOCl is involved in the progression of inflammatory diseases, there is considerable interest in the development of therapeutically useful MPO inhibitors [5,10,11]. A promising approach has been the development of inhibitors that act as peroxidase substrates for MPO, which either divert its catalytic activity from HOCl production, or generate metabolites that mediate irreversible haem modification [6,10,12,13], however compounds that can inhibit MPO in vivo at non-toxic doses have yet to be identified.
Nitroxides are stable free radicals that exert a range of antioxidant effects in vitro and have considerable potential as therapeutic inhibitors of oxidative damage in vivo due to their low toxicity [14]. Nitroxides can protect against oxidative damage in animal models of inflammation [14], though the mechanisms responsible are poorly understood. In vitro studies have established that nitroxides can: (i) act as SOD (superoxide dismutase)-mimetics; (ii) reversibly/irreversibly scavenge radicals (e.g. NO2•, thiyl- and carbon-centred radicals); and (iii) reduce the oxy-ferryl (FeIV = O) centre of haem proteins [15]. However, the potential for these compounds to exert protection via inhibition of the enzymatic activity of MPO and other peroxidase enzymes has been largely overlooked. Nitroxides can suppress protein nitration by a MPO-H2O2-NO2− system [16] and prevent cellular damage by a MPO-H2O2-phenol system [16,17], but these activities have been ascribed to their actions as radical scavengers (e.g. R2N-O• + NO2• → R2N+=O + NO2−). The piperidine nitroxide 4-hydroxyTEMPO (TEMPOL; for nitroxide full names see Table 1) was recently shown to be a substrate for compound I and compound II of MPO [16], but the importance of such reactions in modulating the enzymatic activity of MPO under physiologically relevant conditions, and the effects of nitroxides on MPO-mediated HOCl production, are unknown.
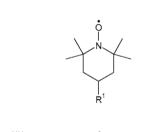
Table 1. Structures of nitroxides (R2N-O•) and related hydroxylamines (R2N-OH) and amines (R2N-H) examined in this study.
| Nitroxide | Side chains | Structure |
|---|---|---|
| Piperidine nitroxides | ||
| TEMPOa | R1 = H |
|
| 4-HydroxyTEMPOb | R1 = OH | |
| 4-AminoTEMPOc | R1 = NH3+ | |
| 4-TrimethylammoniumTEMPOd (I− salt) | R1 = N+Me3 | |
| 4-CarboxyTEMPOe | R1 = CO2− | |
| PEG-TEMPOf | R1 = [O-CH2CH2]n | |
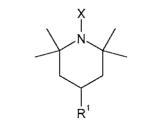
| Piperidine hydroxylamines and amines | ||
| TEMPOHg | R1 = H, X = OH |
|
| TMPh | R1 = H, X = H | |
| 4-AminoTEMPOHi | R1 = NH3+, X = OH | |
| 4-AminoTMPj | R1 = NH3+, X = H | |
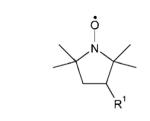
| Pyrrolidine nitroxides | ||
| 3-AminoPROXYLk | R1 = NH3+ |
|
| 3-AminomethylPROXYLl | R1 = CH2 NH3+ | |
| 3-CarbamoylPROXYLm | R1 = C(O)NH2 | |
| Isoindoline nitroxides | ||
| 5-AminoTMIOn | R1 = NH3+, R2 = H |

|
| 5-TrimethylammoniumTMIOo (I− salt) | R1 = N+Me3, R2 = H | |
| 5-NitroTMIOp | R1 = NO2, R2 = H | |
| 5-CarboxyTMIOq | R1 = CO2−, R2 = H | |
| 5,6-DicarboxyTMIOr | R1 = CO2−, R2 = CO2− |
2,2,6,6-tetramethylpiperidin-1-yloxyl
4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yloxyl/TEMPOL
4-amino-2,2,6,6-tetramethylpiperidin-1-yloxyl/TEMPAMINE
4-trimethylammonio-2,2,6,6-tetramethylpiperidin-1-yloxyl iodide/CAT-1
4-carboxy-2,2,6,6-tetramethylpiperidin-1-yloxyl
poly(ethylene glycol)-bis-TEMPO
2,2,6,6-tetramethylpiperidine hydroxylamine, TEMPO hydroxylamine
2,2,6,6-tetramethylpiperidine
4-amino-2,2,6,6-tetramethylpiperidine hydroxylamine, 4-aminoTEMPO hydroxylamine
4-amino-2,2,6,6-tetramethylpiperidine
3-amino-2,2,5,5-tetramethylpyrrolidin-1-yloxyl
3-aminomethyl-2,2,5,5-tetramethylpyrrolidin-1-yloxyl
3-carbamoyl-2,2,5,5-tetramethylpyrrolidin-1-yloxyl
5-amino-1,1,3,3-tetramethylisoindolin-2-yloxyl
5-trimethylammonio-1,1,3,3-tetramethylisoindolin-2-yloxyl iodide
5-nitro-1,1,3,3-tetramethylisoindolin-2-yloxyl
5-carboxy-1,1,3,3-tetra-methylisoindolin-2-yloxyl
5,6-dicarboxy-1,1,3,3-tetramethylisoindolin-2-yloxyl.
In the present study, we demonstrate that nitroxides can potently inhibit HOCl production and protein oxidation by an isolated MPO-H2O2-Cl− system, and HOCl production by human neutrophils, in the presence of physiological concentrations of Cl−. This inhibition is shown to involve one-electron oxidation of nitroxides by MPO compound I (Scheme 1, reaction B) and accumulation of compound II, thereby arresting the catalytic cycles of MPO; haem destruction was also observed with some nitroxides. With neutrophils, cellular production of superoxide (O2•−) antagonizes inhibition, which is attributed to reduction of compound II to the native enzyme (Scheme 1, reaction D); however, nitroxides may counteract this effect via their SOD-mimetic activity (Scheme 1, reaction E). Overall, the results from the present study identify a novel mechanism by which nitroxides may prevent MPO-mediated tissue damage during inflammation and indicate that these compounds have considerable promise as therapeutic agents in inflammatory diseases that involve MPO-derived oxidants.
Scheme 1. HOCl production by MPO and its proposed modulation by nitroxides.

Reaction of H2O2 with native MPO yields compound I, which oxidizes Cl− to yield HOCl and regenerate native MPO (reaction A) (the halogenation cycle). Competing reactions of compound I with nitroxides (R2N-O•) divert the catalytic activity of MPO from HOCl production to the peroxidase cycle and yield compound II and oxoammonium cations (R2N+=O) (reaction B). Reduction of compound II by nitroxides (reaction C) is slow and accumulation of compound II limits HOCl production. Rapid reduction of compound II by O2•− (reaction D), which is generated by activated neutrophils and other cells, stimulates HOCl production by regenerating native MPO, but nitroxides may prevent this process via their SOD-mimetic activity (reaction E).
EXPERIMENTAL
Materials
Nitroxides and related hydroxylamines and amines were obtained from commercial suppliers (Sigma, Invitrogen) or synthesized by published methods (see Table 1 for compound structures). 5-CarboxyTMIO, 5,6-dicarboxyTMIO, 5-aminoTMIO, 5-trimethylammoniumTMIO and 5-nitroTMIO were synthesized as described previously [18,19] and their identities and purity (>98%) were verified by MS and HPLC respectively. TEMPOH and 4-aminoTEMPOH were prepared by catalytic reduction of the parent nitroxides with H2/platinum oxide (IV) in methanol for 2 h and obtained by flash evaporation as described previously [20]; conversion was >99.5% as assessed by EPR spectroscopy. MPO, isolated from human polymorphonuclear leukocytes (purity index A430/A280 = 0.84), was from Planta Natural Products. SOD (from bovine erythrocytes) and catalase (from bovine liver) were from Sigma. Perlecan was immunopurified from medium conditioned by human coronary arterial endothelial cells as described previously [21]. HSA (human serum albumin, fatty acid-free) was from Sigma. All other reagents were analytical grade or better. Solutions were prepared using water filtered through a four-stage MilliQ system and chelex-treated buffers (0.1 M phosphate buffer or PBS, pH 7.4). H2O2 and HOCl solutions were prepared immediately before use and their concentrations were determined spectrophotometrically at pH 12 (H2O2, ε240 = 43.6 M−1 · cm−1; HOCl, ε292 = 350 M−1 · cm−1).
MPO-H2O2-Cl− systems
All reactions involving the MPO-H2O2-Cl− system contained MPO (10–500 nM), Cl− (100 mM), H2O2 (10–50 μM) and Met (methionine) (250 μM). Reactions were performed at pH 7.4 and 25°C and were initiated by the addition of H2O2 or MPO.
HOCl production by the MPO-H2O2-Cl− system
HOCl production by the MPO-H2O2-Cl− system was determined by measuring the conversion of Met to Met sulfoxide [22]. Reactions were stopped after 30 min by addition of catalase (100 μg/ml; 448 units/ml) and Met sulfoxide quantified, relative to standards, by HPLC with fluorescence detection (λex = 340 nm, λem = 440 nm) after pre-column derivatization with OPA (o-phthaldialdehyde) [23].
HOCl production by human neutrophils
Human neutrophils were isolated from the blood of healthy consenting donors [obtained in accordance with the Declaration of Helsinki (2000) and with approval of the Sydney South West Area Health Service Ethics Review Committee] by density centrifugation with PolymorphPrep™ (Axis-Shield) and hypotonic lysis of red blood cells using PharmLyse™ Lysis Buffer (BD Biosciences). Neutrophils (2 × 106 cells/ml) were suspended in PBS containing CaCl2 (1 mM), MgCl2 (0.5 mM), glucose (1 mg/ml) and Met (250 μM) and pre-incubated for 10 min with nitroxides and related compounds (5–100 μM), SOD (20 μg/ml; 324 units/ml), azide (100 μM) or catalase (50 μg/ml; 224 units/ml) before stimulation by PMA (100 ng/ml). Standard incubations were performed at pH 7.4 and 25°C and were stopped after 30 min by the addition of catalase (50 μg/ml; 224 units/ml); where indicated, incubations were also performed at 37 °C. The cells were pelleted by centrifugation and the generation of Met sulfoxide from Met was quantified in the supernatant after filtration (10 kDa cut-off) as described above.
Visible spectroscopy
The haem absorbance of MPO was monitored using a Perkin-Elmer Lambda 40 UV-vis spectrometer. Solutions were scanned in the region 220–700 nm (960 nm · min−1).
EPR spectroscopy
Nitroxides were quantified by EPR spectroscopy using a standard flattened aqueous solution cell in a Bruker EMX X-band spectrometer equipped with 100 kHz modulation and a cylindrical ER4103TM cavity. EPR spectrometer settings were: gain, 1 × 105; modulation amplitude 0.05 mT; time constant, 40.96 ms; scan time, 30 s; resolution 1024 points; centre field, 384 mT; field scan 6.0 mT; power, 10 mW; and frequency, 9.76 GHz.
H2O2 consumption and O2 evolution
H2O2 and O2 were quantified simultaneously in N2-purged reaction systems within a stirred chamber (NOCHM-4) using H2O2- and O2-specific electrodes (ISO-HPO-2 and ISO-OXY-2) interfaced to an Apollo 4000 Free Radical Analyzer (World Precision Instruments).
Quantification of protein oxidation by ELISA
Perlecan and HSA were adsorbed onto 96-well high-binding polystyrene microtitre plates (4.7 μg/ml protein, 50 μl; approx. 10 nM perlecan and 70 nM HSA). After washing, wells were pre-incubated with MPO (10 nM), Cl− (100 mM) and nitroxides (10 μM) for 5 min and reactions were initiated by addition of H2O2 (10 μM) (total reaction volume: 50 μl). After 30 min reaction time, residual oxidant was quenched with Met (10 mM, 100 μl, 10 min). Wells were blocked with 0.1% casein, then probed with monoclonal antibody CSI-076 (Abcam) (3.3 μg/ml; against perlecan protein core domain I) or monoclonal antibody 2D10G9 (supernatant dilution 1:25; against HOCl-modified protein [24]). Detection of immune complexes was performed using a biotinylated anti-mouse antibody (Chemicon) with streptavidin-conjugated alkaline phosphatase (Amersham) and p-nitrophenylphosphate (Sigma).
Quantification of protein oxidation by amino acid analysis
HSA (2.9 μM) was incubated with MPO (100 nM), Cl− (100 mM) and nitroxides (10 μM) for 5 min and reactions were initiated by the addition of H2O2 (50 μM). After 30 min reaction, proteins were precipitated, dried and subjected to methanesulfonic acid hydrolysis before quantification of amino acids in hydrolysates, relative to authentic standards, by HPLC with fluorescence detection (λex = 340 nm, λem = 440 nm) after pre-column derivatization with OPA [23].
Statistical analyses
Statistical analyses were carried out using one-way ANOVA with Newman Keul’s post-hoc testing, unless otherwise stated, with P < 0.05 taken as significant. IC50 values were determined by fitting a rectangular hyperbola to dose–response curves using non-linear regression. All statistical analyses were performed using GraphPad Prism 4 (GraphPad Software, San Diego, CA, USA, www.graphpad.com).
RESULTS
Inhibition of HOCl production by the MPO-H2O2-Cl− system
HOCl production by the MPO-H2O2-Cl− system (100 nM MPO, 50 μM H2O2, 100 mM Cl−) was quantified by determining the yields, by HPLC, of Met sulfoxide [R-S(O)-CH3] arising from oxidation of the thioether-containing amino acid Met (R-S-CH3, 250 μM) by HOCl. The reaction of HOCl with Met is rapid and, under the conditions employed, the yield of Met sulfoxide is proportional to the concentration of HOCl generated [22]. A range of cyclic aliphatic (piperidine, pyrrolidine) and aromatic (isoindoline) nitroxides and related hydroxylamines and secondary amines (Table 1) were screened for their ability to inhibit HOCl production using this isolated enzyme system. At a concentration of 10 μM, a number of nitroxides inhibited the production of HOCl by approx. 50% or more (Figure 1A). None of the compounds examined significantly inhibited the oxidation of Met to Met sulfoxide by reagent HOCl (50 μM), confirming that these effects derived from enzyme inhibition and not direct scavenging of HOCl. Whereas the nitroxides TEMPO and 4-aminoTEMPO exerted marked inhibition at this concentration, their hydroxylamine derivatives (R2N-OH; TEMPOH and 4-aminoTEMPOH) and related secondary amines (R2N-H; TMP and 4-aminoTMP) were ineffective (Figure 1A). Positively charged and neutral nitroxides exerted much greater inhibitory effects than negatively charged nitroxides, e.g. 4-aminoTEMPO > 4-hydroxyTEMPO >>4-carboxyTEMPO and also 5-aminoTEMPO ~ 5-nitroTMIO >>5-carboxyTMIO ~5,6-dicarboxyTMIO. With 4-trimethylammoniumTEMPO and 5-trimethylammoniumTMIO, which were obtained as I− salts (R-N+Me3I−), MPO-mediated production of hypoiodous acid (HOI) from I− [25] may account for some Met sulfoxide production. Unlike free TEMPO, the corresponding polymer-linked material PEG-TEMPO was a poor inhibitor. IC50 values were determined from dose–response curves for the two most potent inhibitors, 4-aminoTEMPO (IC50 = 1.2 μM) and 3-aminoPROXYL (IC50 = 2.7 μM) (Figures 1B and 1C).
Figure 1. Effect of nitroxides and related compounds on HOCl production by the MPO-H2O2-Cl− system.

Reactions contained MPO (100 nM), Cl− (100 mM), Met (250 μM) and nitroxides and related compounds (0.25–10 μM) and were initiated by the addition of H2O2 (50 μM). In control reactions, nitroxides and related compounds were omitted. Met sulfoxide, a product of the reaction of Met with HOCl, was quantified by HPLC after 30 min. Data are means ± S.E.M., n ≥ 3 independent experiments. IC50 values were determined by fitting a rectangular hyperbola to the dose–response curves using non-linear regression. (A) Effect of nitroxides and related compounds (10 μM). (B) Inhibition by 4-aminoTEMPO (0.25–10 μM). (C) Inhibition by 3-aminoPROXYL (0.25–10 μM).
Role of accumulation of MPO compound II and haem destruction in enzyme inhibition
The ability of nitroxides to alter the distribution of redox intermediates during catalysis, and thereby inhibit the enzymatic activity of MPO (cf. Scheme 1), was monitored by examination of the characteristic haem optical absorbances of the native enzyme (λmax = 430 nm) and compound II (absorption maxima at 453 nm and 625 nm); compound I, which has a short lifetime (t1/2 ~ 100 ms) [26], was not examined. In the absence of nitroxides (500 nM MPO, 50 μM H2O2, 100 mM Cl−, 250 μM Met), where the halogenation cycle (i.e. HOCl production) predominates, the native enzyme was the predominant haem species present (Figure 2A). In contrast, in the presence of positively-charged and neutral nitroxides (50 μM), the major haem species present was MPO compound II, which is inactive in HOCl production (Figures 2B and 2C and Supplementary Figures S1B-S1H). These results demonstrate that nitroxides undergo one-electron oxidation by compound I, yielding oxoammonium cations and converting compound I into compound II (Scheme 1, reaction B), in the presence of physiological concentrations (100 mM) of Cl− (Scheme 1, reaction A). Negatively-charged nitroxides and the polymer-linked nitroxide PEG-TEMPO also promoted compound II accumulation, but much less efficiently (Figure 2D and Supplementary Figures S1K and S1L). With the trimethylammonium-substituted nitroxides (R-N+Me3I−) (Supplementary Figures S1F and S1J), subsequent regeneration of native MPO from compound II is ascribed to I−-mediated reduction of compound II [27]. The compound II substrates ascorbate [27] and paracetamol (acetaminophen) [28] rapidly reversed compound II accumulation induced by 4-aminoTEMPO (not shown) and, as expected, stimulated HOCl production in this system (Supplementary Figure S2). In addition to promoting compound II accumulation, the aromatic nitroxides 5-aminoTMIO and 5-nitroTMIO (Supplementary Figures S1G and S1H) also induced a general loss of haem absorbance, indicating that their metabolites mediate irreversible haem destruction. None of the observed changes in haem absorbance occurred in the absence of H2O2.
Figure 2. Promotion of compound II accumulation by nitroxides.

Reactions contained MPO (500 nM), Cl− (100 mM), Met (250 μM) and nitroxides (50 μM) and were initiated by the addition of H2O2 (50 μM). The wavelengths of absorption maxima for the haem absorbance of native MPO (430 nm) and compound II (453 and 625 nm) are indicated by dotted lines. Results shown are visible spectra recorded at (a) 0.5 min, and (b) 10.5 min after initiation of reactions. (A) No nitroxide. (B) 4-AminoTEMPO. (C) 4-HydroxyTEMPO. (D) 4-CarboxyTEMPO. Results are representative of multiple independent experiments. Results for other nitroxides are shown in Supplementary Figure S1.
4-AminoTEMPO, 4-hydroxyTEMPO and 4-carboxyTEMPO (50 μM) stimulated the decay of pre-formed compound II (500 nM MPO reacted with 50 μM H2O2, then 45 units/ml catalase to quench excess H2O2) to native MPO with half-lives of 2–8 min (Supplementary Figure S3). These results demonstrate that a range of structurally-diverse nitroxides undergo relatively slow one-electron oxidation by compound II (k2 for 4-hydroxyTEMPO is 2.6 × 104 M−1 · s−1 [16]), thereby accounting for their ability to promote compound II accumulation during MPO turnover.
All of the nitroxides examined decreased the rate of H2O2 consumption by isolated MPO (Figure 3 and Supplementary Figure S4), with the magnitude of this effect reflecting the inhibition of HOCl production (Figure 1A), consistent with these processes being interlinked. As 4-aminoTEMPO, 4-hydroxyTEMPO and 4-carboxyTEMPO displayed similar reactivity toward compound II (Supplementary Figure S3), the marked variation in their ability to inhibit H2O2 consumption (Figure 3) can be attributed to differences in the rate of their reaction with compound I (Scheme 1, reaction B).
Figure 3. Nitroxide-mediated inhibition of H2O2 consumption by the MPO-H2O2-Cl− system.

Reactions contained H2O2 (50 μM), Cl− (100 mM), Met (250 μM) and nitroxides (1–500 μM) and were initiated by the addition of MPO (20 nM, indicated by black arrowheads). (A) Inhibition by 4-aminoTEMPO; (a) control (no nitroxide), (b) 1 μM, (c) 10 μM, (d) 50 μM and (e) 500 μM nitroxide. (B) Inhibition by 4-hydroxyTEMPO; legend as for (A). (C) Inhibition by 4-carboxyTEMPO; legend as for (A). Results are representative of multiple independent experiments. Results for other nitroxides are given in Supplementary Figure S4.
4-AminoTEMPO (2.5 μM) was not consumed during these reactions (500 nM MPO, 50 μM H2O2, 100 mM Cl−) as determined by EPR spectroscopy (results not shown), indicating that the nitroxide is rapidly regenerated from its oxoammonium cation metabolite. H2O2 is known to reduce oxoammonium cations to nitroxides, yielding O2•− (R2N+=O + H2O2 → + R2N-O• + HO2•− + H+) and ultimately, O2 [29]. This process appears to be significant during inhibition of MPO-mediated HOCl production by 4-aminoTEMPO and 4-hydroxyTEMPO (50 μM), with these nitroxides stimulating the production of approx. 10 μM O2 (Supplementary Figure S5) from approx. 20 μM H2O2 (Figure 3; determined simultaneously) after 15 min reaction. Lower levels of O2 evolution were observed with 4-carboxyTEMPO (Supplementary Figure S5), presumably reflecting its slower metabolism by compound I.
Nitroxides inhibit HOCl-dependent protein oxidation by isolated MPO
The extracellular matrix proteoglycan perlecan and the plasma protein albumin bind MPO and are implicated as important targets for MPO-derived oxidants in vivo (M. D. Rees, unpublished work) [9,30]. The ability of nitroxides to inhibit modification of human endothelial cell-derived perlecan and HSA by the MPO-H2O2-Cl− system was quantified by an ELISA-based assay using monoclonal antibody CSI-076, which recognises perlecan protein core domain I, and monoclonal antibody 2D10G9, which recognises HOCl-modified protein. The MPO-H2O2-Cl− system (10 nM MPO, 10 μM H2O2, 100 mM Cl−) induced a marked loss of recognition of the perlecan protein core by CSI-076 and, in both cases, the generation of HOCl-modified epitopes recognised by 2D10G9 (Figure 4). Inclusion of 4-aminoTEMPO and 4-hydroxyTEMPO at a concentration of 10 μM strongly inhibited these modifications, whereas 4-carboxyTEMPO was ineffective (Figure 4), in accordance with their activities against HOCl production (Figure 1). 4-AminoTEMPO (10 μM) also afforded protection against the loss of Met and histidine residues upon exposure of HSA (2.9 μM; 20 μM Met residues, 86 μM histidine residues) to the MPO-H2O2-Cl− system (100 nM MPO, 50 μM H2O2, 100 mM Cl−) (Supplementary Figure S6).
Figure 4. Nitroxide-mediated inhibition of protein oxidation by the MPO-H2O2-Cl− system.

Modification of surface-adsorbed perlecan (from 10 nM solution) and HSA (from 70 nM solution) by the MPO-H2O2-Cl− system (10 nM MPO, 100 mM Cl− and 10 μM H2O2) in the presence of nitroxides (10 μM) was quantified after 30 min reaction by ELISA by measuring loss of perlecan recognition by monoclonal antibody CSI-076 recognising perlecan domain I (black bars) and gain in recognition by monoclonal antibody 2D10G9 recognising HOCl-modified protein (perlecan, white bars; HSA, grey bars). Results are means ± S.E.M., n = 3 independent experiments. Modification of HSA was also quantified by amino acid analysis (Supplementary Figure S6).
Nitroxides inhibit HOCl production by human neutrophils
Stimulated neutrophils release MPO extracellularly from intracellular granules and produce O2•− by a cell-surface NADPH oxidase complex. Dismutation of O2•− yields H2O2, which is subsequently utilized by MPO to generate HOCl. HOCl production by PMA-stimulated neutrophils was quantified by measuring the conversion of Met into Met sulfoxide [22]. The stimulated cells (2 × 106 cells/ml) generated approx. 40 μM and approx. 80 μM Met sulfoxide after 30 min incubation at 25°C and 37°C respectively. Inclusion of catalase (224 units/ml) or the haem poison azide (100 μM) quenched Met sulfoxide production, consistent with this process being dependent on MPO-derived HOCl. Nitroxides inhibited neutrophil-mediated HOCl production at a concentration of 10 μM (Figure 5A), with the pattern of inhibition generally reflecting their activities against isolated MPO (Figure 1A), although the overall extents of inhibition were lower. Notably, 4-aminoTEMPO inhibited HOCl production by neutrophils far more potently than 3-aminoPROXYL (IC50 = 6.3 μM compared with IC50 = 46 μM) (Figures 5B and 5C), although these nitroxides displayed similar activities with isolated MPO (IC50 = 1.2 μM compared with IC50 = 2.7 μM) (Figures 1B and 1C). The hydroxylamines 4-aminoTEMPOH and TEMPOH, which were poor inhibitors of isolated MPO (Figure 1), displayed significant inhibition against neutrophil-mediated HOCl production (Figure 5A). Stimulated neutrophils were shown by EPR spectroscopy to oxidize the hydroxylamines to their parent nitroxides (approx. 2 μM nitroxide from 10 μM hydroxylamine after 15 min incubation; not shown), rationalizing this inhibition.
Figure 5. Effect of nitroxides and related compounds on HOCl production by human neutrophils, and modulation by SOD.

Neutrophils (2 × 106 cells/ml) in PBS containing glucose (1 mg/ml) were pre-incubated with Met (250 μM) and nitroxides and related compounds (5–100 μM), with or without SOD (324 units/ml), for 10 min before stimulation of the cells with PMA (100 ng/ml); cells were also stimulated in the presence of azide (100 μM) or catalase (224 units/ml). In control systems, cells were stimulated in the absence of nitroxides and related compounds, azide, catalase or SOD. Met sulfoxide was quantified by HPLC after 30 min. Results are means ± S.E.M., n = 3 independent experiments. IC50 values were determined by fitting a rectangular hyperbola to the dose-response curves using non-linear regression. (A) Effect of nitroxides and related compounds (10 μM). (B) Inhibition by 4-aminoTEMPO (5–100 μM) in the absence (solid squares, solid curve) and presence (open circles, dashed curve) of SOD; equivalent extents of inhibition were observed at 37°C in the presence (IC50 approx. 1 μM) and absence (IC50 approx. 5 μM) of SOD (results not shown). (C) Inhibition by 3-aminoPROXYL (5-100 μM) in the absence (solid squares, solid curve) and presence (open circles, dashed curve) of SOD.
Neutrophil-generated O2•− can antagonize MPO inhibitors that promote compound II accumulation by rapidly reducing compound II to the native enzyme (Scheme 1, reaction D) [31]. Inclusion of exogenous SOD (324 units/ml) increased the inhibitory activity of nitroxides against neutrophil-mediated HOCl production to the levels observed with the isolated MPO-H2O2-Cl− system (Figures 5B and 5C and Supplementary Figure S7), confirming that O2•− antagonizes nitroxide-mediated inhibition of MPO, with the magnitude of this effect being dependent on nitroxide structure, e.g. 4-aminoTEMPO <<3-aminoPROXYL (Figures 5B and 5C).
DISCUSSION
Tissue damage by MPO-derived HOCl is implicated as a key event in the progression of multiple inflammatory diseases including atherosclerosis, rheumatoid arthritis and kidney disease [4-6]. This study demonstrates that nitroxides can inhibit HOCl production by isolated MPO and by human neutrophils at low micromolar concentrations. Nitroxides were also shown to inhibit MPO-mediated oxidation of the extracellular matrix proteoglycan perlecan and the plasma protein albumin, which bind MPO and are implicated as important biological targets for MPO-derived HOCl in vivo (M. D. Rees, unpublished work) [9,30]. Although it is well-established that nitroxides can catalyse O2•− dismutation, scavenge radicals (NO2•, thiyl- and carbon-centred radicals), and mediate reduction of oxy-ferryl (FeIV = O) haem [15], the present study is the first to demonstrate that these compounds can modulate the enzymatic activity of MPO, and prevent HOCl production, under physiologically relevant conditions. As nitroxides exhibit low toxicity [14], the results from the present study indicate that these compounds have considerable promise as therapeutic agents for the prevention of tissue damage by MPO-derived oxidants in a wide spectrum of inflammatory diseases.
It is shown that a range of nitroxides with diverse structures are oxidized rapidly by compound I of MPO (a FeIV = O haem/porphyrin π radical-cation species), in the presence of physiological concentrations of Cl− (a major endogenous substrate), to yield compound II (a FeIV = O species). Nitroxides are poor substrates for compound II and their metabolism by MPO results in the accumulation of compound II and inhibition of H2O2 consumption and, consequently, inhibition of HOCl production. Haem destruction also appears to contribute to the inhibitory activity of some (isoindoline) aromatic nitroxides. The importance of the nitroxide moiety (R2N-O•) is confirmed by the lack of inhibitory activity observed with the corresponding secondary amine compounds (R2NH) and hydroxylamines (R2N-OH), which are reduction products of nitroxides [32].
The efficiency of nitroxide oxidation by compound I, via one-electron transfer to the porphyrin π-cation radical of this redox intermediate (Scheme 1, Reaction B), appears to be a key factor underlying the potency of nitroxides as MPO inhibitors. The results from the present study indicate that this process is profoundly affected by nitroxide structure. Electronic factors are unlikely to underlie these effects, as the one-electron oxidation potentials of all the nitroxides examined (approx. 0.7–1 V) [33,34] are well below the reduction potential of the MPO compound I/compound II couple (1.35 V) [35]. Instead, the topography of the active site appears to play a dominant role in controlling the metabolism of nitroxides by compound I, by determining substrate access and binding. The results from the present study indicate that positively charged and neutral aliphatic nitroxides are metabolized readily by compound I, with nitroxides bearing a free amino group being particularly favoured substrates, whereas structurally-related nitroxides bearing a negatively charged (carboxyl) substituent are poor substrates. Similar structure-activity relationships have been determined for aliphatic thiols [36]. The selectivity against metabolism of negatively charged peroxidase substrates by MPO has been proposed to arise from the presence of a negatively charged Glu residue in the peroxidase substrate channel [36]. We propose that the same residue may promote metabolism of positively charged materials (e.g. those bearing a free amino group) via favourable electrostatic or hydrogen-bonding interactions. Metabolism of aromatic nitroxides appears to be controlled by similar factors, although interactions with an aromatic substrate binding site in the distal cavity [37] could also play a role. The poor inhibitory activity of the polymer-linked nitroxide (PEG-TEMPO) indicates that bulky substituents can limit substrate access and binding. These structure-activity relationships may aid the design of novel MPO inhibitors.
O2•− is shown to antagonize MPO inhibition by nitroxides to varying degrees, with this effect being small in the case of the piperidine nitroxide 4-aminoTEMPO, but greater with the closely related pyrrolidine nitroxide 3-aminoPROXYL. This difference may relate to the differential activities of these nitroxides as SOD-mimetics, as piperidine nitroxides are approx. 100-fold more efficient as SOD-mimetics than pyrrolidine nitroxides [33]. Thus, the potency of 4-aminoTEMPO against neutrophil-mediated HOCl production may arise from a combination of MPO inhibition and efficient removal of O2•− via its SOD-mimetic activity (Scheme 1, reaction E). The latter activity is kinetically viable as the rate constants for reaction of O2•− with 4-aminoTEMPO and related piperidine nitroxides (k2 approx. 1 × 106 M−1 · s−1 at neutral pH [38,39]) are comparable with that for the reaction of O2•− with compound II (k2 = 5 × 106 M−1 · s−1 at neutral pH [40]).
As nitroxides can be regenerated readily from their oxoammonium cation metabolites via one-electron reduction, their inhibitory activity can be catalytic. With isolated MPO, reduction of the oxoammonium cations by H2O2 appears to be a significant process, with this yielding O2 as a product. With neutrophils, cell-derived O2•− may contribute to reduction of the oxoammonium cations as the rate constants for these reactions are high (k2 approx. 1 × 108 M−1 · s−1 [39]). Nitroxides are also generated from hydroxylamines, a major reduction product of nitroxides in vivo [32], in the presence of stimulated neutrophils. Therefore, depletion of nitroxides present at sites of oxidant production via oxidation and reduction reactions should be limited.
MPO-derived HOCl is implicated as a key mediator of inflammatory tissue damage [6], and it will therefore be of considerable interest to investigate the ability of nitroxides to inhibit MPO-mediated HOCl production in vivo. As nitroxides can potently inhibit neutrophil-mediated HOCl production in vitro, it is anticipated that these compounds may inhibit oxidant production by MPO in vivo at pharmacologically achievable concentrations. In this regard, it has been shown that administration of 4-hydroxyTEMPO can reduce levels of 3-nitrotyrosine in animal models of inflammation [41]. This may reflect suppression of NO2− metabolism by MPO in a similar manner to that reported here, however it might reflect direct scavenging of nitrating species (i.e. NO2•) derived from MPO or peroxynitrite [16] or down-regulation of inducible nitric oxide synthase expression [42]. The performance of nitroxides as MPO inhibitors in vivo will reflect a complex range of factors including partitioning between their nitroxide and hydroxylamine forms, their accumulation in and clearance from tissues, as well as their MPO-inhibition and SOD-mimetic activities. It has already been demonstrated that nitroxides can be targeted to intracellular compartments to optimize their antioxidant activity [43], however, our data indicate that nitroxides that target extracellular structures, where MPO-mediated HOCl production occurs, may also be therapeutically useful.
Supplementary Material
ACKNOWLEDGEMENT
We thank Dr Antony Ward (University of Sydney) for assistance with the synthesis of hydroxylamines.
FUNDING
Financial support from the Australian Research Council (through the Discovery and Centres of Excellence programs), the National Health and Medical Research Council, and the Austrian Science Fund [FWF (Fonds zur Förderung der wissenschaftlichen Forschung)] [grant numbers P19074-B05 and F3007-B05] is gratefully acknowledged.
Abbreviations used
- HOCl
hypochlorous acid
- HSA
human serum albumin
- Met
methionine
- MPO
myeloperoxidase
- OPA
o-phthaldialdehyde
- SOD
superoxide dismutase
REFERENCES
- 1.Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92:3007–3017. [PubMed] [Google Scholar]
- 2.Senthilmohan R, Kettle AJ. Bromination and chlorination reactions of myeloperoxidase at physiological concentrations of bromide and chloride. Arch. Biochem. Biophys. 2006;445:235–244. doi: 10.1016/j.abb.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 3.van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klebanoff SJ. Myeloperoxidase: friend and foe. J. Leukoc. Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 5.Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 6.Davies MJ, Hawkins CL, Pattison DI, Rees MD. Mammalian heme peroxidases: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008;10:1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 7.Hazell LJ, Stocker R. Oxidation of low-density lipoprotein with hypochlorite causes transformation of the lipoprotein into a high-uptake form for macrophages. Biochem. J. 1993;290:165–172. doi: 10.1042/bj2900165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shao BH, Cavigiolio G, Brot N, Oda MN, Heinecke JW. Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-1. Proc. Natl. Acad. Sci. U.S.A. 2008;105:12224–12229. doi: 10.1073/pnas.0802025105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rees MD, Kennett EC, Whitelock JM, Davies MJ. Oxidative damage to extracellular matrix and its role in human pathologies. Free Radic. Biol. Med. 2008;44:1973–2001. doi: 10.1016/j.freeradbiomed.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 10.Malle E, Furtmuller PG, Sattler W, Obinger C. Myeloperoxidase: a target for new drug development? Br. J. Pharmacol. 2007;152:838–854. doi: 10.1038/sj.bjp.0707358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451:904–913. doi: 10.1038/nature06796. [DOI] [PubMed] [Google Scholar]
- 12.Kettle AJ, Winterbourn CC. Mechanism of inhibition of myeloperoxidase by anti-inflammatory drugs. Biochem. Pharmacol. 1991;41:1485–1492. doi: 10.1016/0006-2952(91)90565-m. [DOI] [PubMed] [Google Scholar]
- 13.Kettle AJ, Gedye CA, Winterbourn CC. Mechanism of inactivation of myeloperoxidase by 4-aminobenzoic acid hydrazide. Biochem. J. 1997;321:503–508. doi: 10.1042/bj3210503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soule BP, Hyodo F, Matsumoto KI, Simone NL, Cook JA, Krishna MC, Mitchell JB. Therapeutic and clinical applications of nitroxide compounds. Antioxid. Redox Signal. 2007;9:1731–1743. doi: 10.1089/ars.2007.1722. [DOI] [PubMed] [Google Scholar]
- 15.Soule BP, Hyodo F, Matsumoto K, Simone NL, Cook JA, Krishna MC, Mitchell JB. The chemistry and biology of nitroxide compounds. Free Radic. Biol. Med. 2007;42:1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaz SM, Augusto O. Inhibition of myeloperoxidase-mediated protein nitration by tempol: kinetics, mechanism, and implications. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8191–8196. doi: 10.1073/pnas.0708211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borisenko GG, Martin I, Zhao Q, Amoscato A, Kagan VE. Nitroxides scavenge myeloperoxidase-catalyzed thiyl radicals in model systems and in cells. J. Am. Chem. Soc. 2004;126:9221–9232. doi: 10.1021/ja0495157. [DOI] [PubMed] [Google Scholar]
- 18.Reid DA, Bottle SE, Micallef AS. The synthesis of water soluble isoindoline nitroxides and a pronitroxide hydroxylamine hydrochloride UV-VIS probe for free radicals. Chem. Commun. 1998;17:1907–1908. [Google Scholar]
- 19.Barrett AGM, Hanson GR, White AJP, Williams DJ, Micallef AS. Synthesis of nitroxide-functionalized phthalocyanines. Tetrahedron. 2007;63:5244–5250. [Google Scholar]
- 20.Mitchell JB, Degraff W, Kaufman D, Krishna MC, Samuni A, Finkelstein E, Ahn MS, Hahn SM, Gamson J, Russo A. Inhibition of oxygen-dependent radiation-induced damage by the nitroxide superoxide dismutase mimic, Tempol. Arch. Biochem. Biophys. 1991;289:62–70. doi: 10.1016/0003-9861(91)90442-l. [DOI] [PubMed] [Google Scholar]
- 21.Whitelock JM, Graham LD, Melrose J, Murdoch AD, Iozzo RV, Underwood PA. Human perlecan immunopurified from different endothelial cell sources has different adhesive properties for vascular cells. Matrix Biol. 1999;18:163–178. doi: 10.1016/s0945-053x(99)00014-1. [DOI] [PubMed] [Google Scholar]
- 22.Meotti FC, Senthilmohan R, Harwood DT, Missau FC, Pizzolatti MG, Kettle AJ. Myricitrin as a substrate and inhibitor of myeloperoxidase: implications for the pharmacological effects of flavonoids. Free Radic. Biol. Med. 2008;44:109–120. doi: 10.1016/j.freeradbiomed.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 23.Pattison DI, Hawkins CL, Davies MJ. Hypochlorous acid-mediated protein oxidation: How important are chloramine transfer reactions and protein tertiary structure? Biochemistry. 2007;46:9853–9864. doi: 10.1021/bi7008294. [DOI] [PubMed] [Google Scholar]
- 24.Malle E, Hazell L, Stocker R, Sattler W, Esterbauer H, Waeg G. Immunological detection and measurement of hypochlorite-modified LDL with specific monoclonal antibodies. Arterioscler. Thromb. Vasc. Biol. 1995;15:982–989. doi: 10.1161/01.atv.15.7.982. [DOI] [PubMed] [Google Scholar]
- 25.Furtmuller PG, Burner U, Obinger C. Reaction of myeloperoxidase compound I with chloride, bromide, iodide, and thiocyanate. Biochemistry. 1998;37:17923–17930. doi: 10.1021/bi9818772. [DOI] [PubMed] [Google Scholar]
- 26.Harrison JE, Araiso T, Palcic MM, Dunford HB. Compound I of myeloperoxidase. Biochem. Biophys. Res. Commun. 1980;94:34–40. doi: 10.1016/s0006-291x(80)80183-5. [DOI] [PubMed] [Google Scholar]
- 27.Marquez LA, Dunford HB, Van Wart H. Kinetic studies on the reaction of compound II of myeloperoxidase with ascorbic acid. Role of ascorbic acid in myeloperoxidase function. J. Biol. Chem. 1990;265:5666–5670. [PubMed] [Google Scholar]
- 28.Marquez LA, Dunford HB. Interaction of acetaminophen with myeloperoxidase intermediates - optimum stimulation of enzyme-activity. Arch. Biochem. Biophys. 1993;305:414–420. doi: 10.1006/abbi.1993.1440. [DOI] [PubMed] [Google Scholar]
- 29.Krishna MC, Samuni A, Taira J, Goldstein S, Mitchell JB, Russo A. Stimulation by nitroxides of catalase-like activity of hemeproteins - Kinetics and mechanism. J. Biol. Chem. 1996;271:26018–26025. doi: 10.1074/jbc.271.42.26018. [DOI] [PubMed] [Google Scholar]
- 30.Tiruppathi C, Naqvi T, Wu YB, Vogel SM, Minshall RD, Malik AB. Albumin mediates the transcytosis of myeloperoxidase by means of caveolae in endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7699–7704. doi: 10.1073/pnas.0401712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kettle AJ, Gedye CA, Winterbourn CC. Superoxide is an antagonist of antiinflammatory drugs that inhibit hypochlorous acid production by myeloperoxidase. Biochem. Pharmacol. 1993;45:2003–2010. doi: 10.1016/0006-2952(93)90010-t. [DOI] [PubMed] [Google Scholar]
- 32.Swartz HM, Sentjurc M, Kocherginsky N. Metabolism of nitroxides and their products in cells. In: Kocherginsky N, Swartz HM, editors. Nitroxide Spin Labels. CRC Press; FL: 1995. pp. 113–147. [Google Scholar]
- 33.Goldstein S, Samuni A, Hideg K, Merenyi G. Structure-activity relationship of cyclic nitroxides as SOD mimics and scavengers of nitrogen dioxide and carbonate radicals. J. Phys. Chem. A. 2006;110:3679–3685. doi: 10.1021/jp056869r. [DOI] [PubMed] [Google Scholar]
- 34.Hodgson JL, Namazian M, Bottle SE, Coote ML. One-electron oxidation and reduction potentials of nitroxide antioxidants: A theoretical study. J. Phys. Chem. A. 2007;111:13595–12605. doi: 10.1021/jp074250e. [DOI] [PubMed] [Google Scholar]
- 35.Furtmuller PG, Arnhold J, Jantschko W, Pichler H, Obinger C. Redox properties of the couples compound I/compound II and compound II/native enzyme of human myeloperoxidase. Biochem. Biophys. Res. Commun. 2003;301:551–557. doi: 10.1016/s0006-291x(02)03075-9. [DOI] [PubMed] [Google Scholar]
- 36.Burner U, Jantschko W, Obinger C. Kinetics of oxidation of aliphatic and aromatic thiols by myeloperoxidase compounds I and II. FEBS Lett. 1999;443:290–296. doi: 10.1016/s0014-5793(98)01727-x. [DOI] [PubMed] [Google Scholar]
- 37.Hori H, Fenna RE, Kimura S, Ikeda-Saito M. Aromatic substrate molecules bind at the distal heme pocket of myeloperoxidase. J. Biol. Chem. 1994;269:8388–8392. [PubMed] [Google Scholar]
- 38.Samuni A, Krishna CM, Mitchell JB, Collins CR, Russo A. Superoxide reaction with nitroxides. Free Radic. Res. Commun. 1990;9:241–249. doi: 10.3109/10715769009145682. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein S, Merenyi G, Russo A, Samuni A. The role of oxoammonium cation in the SOD-mimic activity of cyclic nitroxides. J. Am. Chem. Soc. 2003;125:789–795. doi: 10.1021/ja028190w. [DOI] [PubMed] [Google Scholar]
- 40.Kettle AJ, Anderson RF, Hampton MB, Winterbourn CC. Reactions of superoxide with myeloperoxidase. Biochemistry. 2007;46:4888–4897. doi: 10.1021/bi602587k. [DOI] [PubMed] [Google Scholar]
- 41.Thiemermann C. Membrane-permeable radical scavengers (tempol) for shock, ischemia-reperfusion injury, and inflammation. Crit. Care Med. 2003;31:S76–S84. doi: 10.1097/00003246-200301001-00011. [DOI] [PubMed] [Google Scholar]
- 42.Linares E, Giorgio S, Augusto O. Inhibition of in vivo leishmanicidal mechanisms by tempol: nitric oxide down-regulation and oxidant scavenging. Free Radic. Biol. Med. 2008;44:1668–1676. doi: 10.1016/j.freeradbiomed.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 43.Kagan VE, Jiang JF, Bayir H, Stoyanovsky DA. Targeting nitroxides to mitochondria: location, location, location, and… concentration - Highlight Commentary on “Mitochondrial superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide”. Free Radic. Biol. Med. 2007;43:348–350. doi: 10.1016/j.freeradbiomed.2007.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.