

Abstract
Nanoparticle (NP)-mediated drug delivery typically relies on cargo release to occur passively or in response to environmental stimuli. Here we present a delivery method based on light-activated disruption of intracellular vesicles after internalization of biofunctionalized mesoporous silica nanoparticles loaded with cargo. This method combines the power of targeted delivery with the spatiotemporal control of light activation. As an example, we delivered a cell-impermeable fluorescent compound exclusively to the cytosol of multidrug resistant cancer cells in a mixed population.
Keywords: Mesoporous silica, nanoparticles, P-glycoprotein, drug delivery, photodynamic therapy, endosome
The potential of nanomaterials for drug delivery has been extensively explored in recent years.1–4 Such efforts have been driven to a large extent by the need to reduce side effects in anticancer drugs (such as doxorubicin’s severe cardiotoxicity) via tissue-specific targeting.5 Beyond enhanced targetability, NP-encapsulation of drugs may also provide protection against premature degradation and enable efficient delivery of substances with poor inherent solubility or membrane permeability. Important determinants of the success of NP-mediated drug delivery are biocompatibility, circulation time, immunogenicity, specific targeting, timing of cargo release and, ideally, access to intracellular compartments. Careful tuning of NP size, especially in the 10–100 nm range, enables control of circulation time,6,7 as well as passive tumor accumulation due to the “enhanced permeability and retention” effect.8 Particle size also affects NP cellular uptake.9 A highly desirable feature of NP-based delivery platforms is precise temporal control of compound release. This can be regulated by incorporating release mechanisms triggered by environmental stimuli such as pH,10 temperature,11 or enzymatic reactions.12 Another important way to control payload delivery for membrane-impermeable drugs is to regulate access to the cytosol, which usually requires escape from the endolysosomal compartment where particles are clustered following receptor-mediated endocytosis. Here we present a method for precise spatial and temporal control over cytosolic delivery of compounds that would otherwise be cell-impermeable by incorporating a light-triggered endosomal escape mechanism within a cell targetable mesoporous silica carrier.
Among potential nanocarriers, mesoporous silicate materials13–15 have demonstrated great potential for biomedical applications due to their biocompatibility, ease of functionalization, and large surface-to-volume ratio.16 They have been successfully used to deliver drugs,17,18 proteins19 and nucleic acids.20,21 Most recent efforts have been focused on their biological targeting in order to achieve site-specific delivery.16,22 Size control in the sub-100 nm range is desirable to ensure efficacy of the NPs and to facilitate access to sterically hindered tissues. This remains, however, a challenge for mesoporous silica nanoparticles (MSN). Most MSN reported for biomedical applications have inherent sizes larger than 150–200 nm, and while various synthetic procedures have successfully enabled production of MSN with grain sizes down to ~20–30 nm, these particles are usually prone to aggregation in solution, leading to dispersions with large hydrodynamic diameters (>200–300 nm) following surfactant removal. Accurate size control and preservation of a small hydrodynamic size during the various purification/functionalization steps can lead to major improvements in targetability. Here we present a novel synthetic procedure that affords excellent size control and MSN homogeneity with hydrodynamic diameters ranging from 30 to 200 nm that are preserved during the extraction and biofunctionalization steps, resulting in high targeting efficiency to cell surface receptors. In addition, we have embedded a photosensitizer in the targetable nanocarriers to promote endosomal escape of their cargo and access to the cytosol upon exposure to light.
The use of light as an activation mechanism for drug delivery has received increased attention23 due to its advantages in spatial and temporal control of compound release, especially for applications in ophthalmology.24 Berg and coworkers used photosensitizers to mediate endosome rupture for improved cellular delivery of nucleic acids.25 Harnessing this mechanism, Kataoka and co-workers reported light-mediated gene delivery to the conjunctival tissue of rats.26 By successfully combining this approach with the versatile properties of MSN we were able to achieve cytosolic release of a model cell impermeable compound with highly precise temporal and spatial control.
As a model system relevant for cancer applications, we targeted functionalized NPs to cancer cells expressing P-glycoprotein, a transporter protein responsible for multidrug resistance in many tumors.27,28 As a model drug, we delivered the cell impermeable fluorescent dye Alexa546. By exploiting the photoactive properties of this compound, the timing of cargo release could be precisely controlled by light-stimulated, ROS-mediated endosomal disruption (Figure 1). Our experimental setup (laser scanning confocal microscopy) allowed observation of single vesicle disruption events. This technique offers a combination of features allowing unprecedented control over cytosolic access of the encapsulated drug in the irradiated cells, while preserving unexposed cells. In addition, light-stimulation of the internalized NPs enabled cytosolic delivery of coendocytosed proteins and other test macromolecules (dextran). To the best of our knowledge, this is the first report of successful cytosolic delivery of a cell impermeable compound using NP-mediated targeting in combination with a light-activated endosomal escape mechanism. We believe these NPs hold great potential for accurate spatiotemporal control of drug delivery and may expand the pharmacological arsenal against multidrug resistance to cell-impermeable compounds.
FIGURE 1.

Schematic of light-activated and targeted cytosolic delivery of membrane-impermeable compounds. (a) Antibody-functionalized nanoparticles are loaded with a model compound (the fluorescent dye Alexa546 in our experiments) and targeted to cells expressing P-gp-GFP (GFP bound to the P-glycoprotein transporter). After nanoparticles endocytosis (b), the cargo is released in the endosome (c). Exposure to light at the dye’s excitation wavelength (546 nm) promotes ROS-mediated membrane damage (d) with cytosolic delivery of Alexa546 exclusively in the P-gp expressing cells.
To obtain accurate size control of mesoporous silica nanoparticles and improve targeting efficiency, we developed a modified synthesis procedure based on the co-condensation of tetraethyl orthosilicate (TEOS) and mercaptopropyltrimethoxysilane (MPTMS)17 by introducing a secondary surfactant (Pluronic F-127) into the reaction. Imai and co-workers previously reported the addition of a secondary surfactant to reduce the size of mesoporous silica NPs.29,30 Their synthetic route, based on acidic prehydrolysis of the silicate precursors before base-catalyzed condensation, yielded highly porous NPs as small as 20 nm in diameter. However, we found that this method was not suited for production of well-dispersed colloidal NPs required for biomedical applications, as irreversible agglomeration was observed during synthesis. Nanoparticles obtained by introducing a secondary surfactant in a base-catalyzed synthesis reaction yielded well-dispersed NPs of homogeneous size (Figure 2) and porosity, although the mesopore network in each particle appears to be less organized than in the absence of a secondary surfactant. Full details of the experimental procedure are provided in the Supporting Information. The graph in Figure 2 demonstrates the ability to tune the size of the resulting NPs as a function of secondary surfactant concentration (Pluronic F-127), as measured by dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern). Increasing amounts of the nonionic secondary surfactant limit the growth of the NPs, resulting in dispersions with decreasing average hydrodynamic sizes, displaying excellent reproducibility and low polydispersity.
FIGURE 2.

Size control of mesoporous silica nanoparticles. (a) Transmission electron micrograph of nanoparticles synthesized with 2.5 mg/mL Pluronic F-127 as a secondary surfactant; overlay: size distribution histogram of the sample, obtained by dynamic light scattering. (b) Higher-magnification transmission electron micrograph of the same NPs, showing a mesoporous structure without long-range order. (c) Relationship between the amount of secondary surfactant in the reaction mixture and nanoparticle size (hydrodynamic diameter). Each data point is the average of 3 batches synthesized independently.
This synthetic route could be extended to co-condensation of other organosilicate precursors beside MPTMS, such as aminopropyltriethoxysilane (APTES). In addition, a variety of composite particles could be synthesized by introducing external particles (such as gold nanospheres and nanorods, magnetic NPs, and quantum dots) into the reaction mixture and varying the amount and type of secondary surfactant, to yield core–shell nanostructures (Supporting Information Methods and Figure S1). Purification of the particles from templating and secondary surfactants proved challenging (see Methods in Supporting Information), especially for small sizes; particles tend to aggregate easily at diameters smaller than ~50 nm. For those smaller and delicate NPs, centrifugation was difficult and aggregation-prone. A procedure based on dialysis followed by membrane filtration produced excellent results, preserving the small hydrodynamic size of the dispersed NPs throughout the extraction and washing steps.
Following surfactant template removal, the particles were functionalized by covalent attachment of streptavidin onto their surface via a heterobifunctional cross-linker with a 5 kDa polyethylene-glycol (PEG) spacer arm. For all diameters, a 5 kDa PEG coat added following streptavidin attachment was essential to ensure stability and agglomeration-free transfer to physiological buffers (Supporting Information Figure S2). Following conjugation and PEG-ylation, unre-acted streptavidin was removed by size-exclusion chromatography. Various polymeric or protein-based coatings (including Pluronic F-127 itself, Synperonic PE-F68, bovine α- and β-casein, and polyethylene-imine) enabled successful transfer to physiological buffers, but resulted in higher nonspecific interactions between NPs and cells or glass coverslips. Particles coated with a mixture of polyethylene-imine (25 kDa) and PEG (PEI–PEG NPs) were also synthesized to mediate high nonspecific cellular uptake due to high electrostatic binding of the NPs to the negatively charged cell surface (see Supporting Information for details).
After synthesis, purification, and functionalization, nanoparticles could be loaded with a variety of molecules up to 12% w/w (Supporting Information Figure S3). For the present study, the cell-impermeable dye, Alexa546 (Invitrogen), was chosen as a model compound for its bright fluorescence. Following surface functionalization, NPs were incubated with an excess of thiol-reactive Alexa546-maleimide to covalently label the free surface thiol groups, as well as saturate the silica surface and the mesopores with hydrolyzed dye. Dye loading in the nanoparticles (~1.6% w/w) was optimized to maximize particle brightness (Supporting Information Figure S3).
To test the ability of our nanoparticles to deliver cargo to the cytosol, we used either surface biotinylation of live LN-229 cells (human glioma, ATCC) in combination with streptavidin-functionalized NPs, or PEI–PEG-coated NPs to mediate cell surface attachment. Both types of particles are efficiently internalized by cells and localize to the endolysosomal compartment after 3 h of incubation at 37 °C (Figure 3). To characterize the internalization pathway, LN-229 cells were transiently transfected with a GFP fusion protein of the lysosomal marker LAMP1,31 incubated at room temperature with PEI–PEG coated NPs (30 min at 20ug/mL), followed by removal of unbound particles and incubation at 37 °C. Cells were then imaged at various time points to monitor NP internalization (Figure 3a). While exclusively present on the cell surface at early time points, the NPs are progressively internalized and fully colocalize with LAMP-1 vesicles for incubations lasting at least 3 h at 37 °C. While all internalized NPs localize to the endolysosomal compartment, some patches of surface bound particles can still be observed after extensive incubation periods in some cells (Figure 3a, bottom row). Similar experiments were also performed using the early endosome marker Rab5 and late endosome marker Rab7.31 The results are summarized in Supporting Information Figure S4. Streptavidin-functionalized NPs were also found to localize to the endolysosomal compartment after overnight incubation with surface-biotinylated LN-229 cells. The internalized NPs were found to colocalize with LysoTracker Blue, a marker of acidic organelles (Figure 3b).
FIGURE 3.

Characterization of the vesicles containing NPs after endocytosis. (a) Confocal micrographs of LN-229 cells transiently transfected with the lysosomal marker, LAMP1-GFP, following incubation with PEI–PEG functionalized NPs. Images show representative cells 10 min (top), 3 h (middle), and 20 h (bottom) after incubation at 37 °C. (b) Confocal micrographs of LN-229 cells after overnight endocytosis of streptavidin functionalized NPs via surface biotinylation. The endolysosomal compartment was stained with LysoTracker Blue. All scale bars are 20 μm.
Following overnight NP endocytosis, cells were exposed to green excitation light (520–550 nm, MWIG3/TRITC filter) for durations ranging from 3 to 120 s. Release into the cytosol was observed immediately after exposure (0.5 mW measured power over the field of view; 500 mW/cm2), for both streptavidin (Figure 4a) and PEI–PEG-coated particles (Figure 4d), as evidenced by a large increase in fluorescence, especially visible across the nucleus for many cells. Vesicle fluorescence was also increased, which can be explained by a reduction of self-quenching for the unreleased cargo as the highly concentrated fluorophore escapes from the lysosomes.32 As detailed in the studies conducted by Berg and co-workers,33–35 this light-induced cytosolic release is due to endosomal membrane damage mediated by reactive oxygen species produced by the photoactive compound (here Alexa546) during illumination. The amount of dye released in the cytosol following light exposure was found to be proportional to the number of NPs internalized by cells (Figure 4b). Dye transfer from endosomes to cytosol is rapid, which is compatible with diffusion kinetics of a small molecule like Alexa546 (Figure 4c), and suggests that the NPs themselves are not required to move into the cytosolic compartment for the effect to occur. When PEI–PEG-coated NPs were incubated overnight with LN-229 cells in the presence of an excess of calcein, exposure of the cells to green light caused cytosolic release of both the NP dye cargo and coendocytosed calcein, as evidenced by strong nuclear accumulation of the compounds (see fluorescence profile plots, Figure 4d).
FIGURE 4.

Light-induced cytosolic release of Alexa546 loaded into mesoporous silica nanoparticles. (a) Confocal micrographs of live LN-229 cells after surface biotinylation-mediated uptake of streptavidin-functionalized particles loaded with Alexa546 (60× water-immersion objective). Images were acquired before (left panels) and immediately after (right panels) exposure to light from a TRITC-filtered mercury lamp. (b) Relationship between the amount of cytosol-released Alexa546 and the amount of endocytosed NPs. Each data point in the scatter plot represents one cell. (c) Time evolution of Alexa546 fluorescence following stimulation. Fluorescence is normalized for each cell to its initial value preceding light exposure. The bars represent SD (n = 57 cells). (d) Confocal micrographs of live LN-229 cells following overnight uptake of PEI–PEG coated NPs in the presence of calcein (0.25 mM). Images were acquired before and after 2 min light exposure as in (a). The profile plots display calcein and Alexa546 fluorescence across a representative cell before and after exposure. Scale bars are 20 μm in all images.
We then investigated whether the NPs could liberate coendocytosed macromolecules and nanoparticles. The NPs successfully mediated cytosolic delivery of a 3 kDa dextran-FITC conjugate (Figure 5). In this experiment, one set of LN-229 cells was incubated with NPs loaded with a releasable cargo dye (Alexa546), while a second set was incubated with NPs carrying a dye covalently bound to their matrix during synthesis (Alexa633; so it could not be released). The two cell populations were then plated together, and incubated in the presence of dextran-FITC. Illumination of this mixed population gave the results summarized in Figure 5. Both types of NPs successfully mediated cytosolic delivery of the FITC-dextran macromolecules, albeit with different kinetics. The lower release kinetics of dextran in the case of the covalently labeled Alexa633-NPs may be explained by a lower excitation efficiency of this fluorophore through the TRITC filter set used for exposure. Covalently labeled Alexa633-NPs allowed successful release of dextran upon illumination; yet, no Alexa633 signal was observed in the cytosol after >30 min following exposure to light. These results indicate that the NPs themselves remained confined inside endosomes even after their rupture. Proteins such as NeutrAvidin could also be delivered to the cell cytosol with slower diffusion kinetics than a dye molecule (Supporting Information Figure S5). However, coendocytosed quantum-dot-streptavidin conjugates remained clustered in the endolysosomal compartment following light activation (Supporting Information Figure S6).
FIGURE 5.
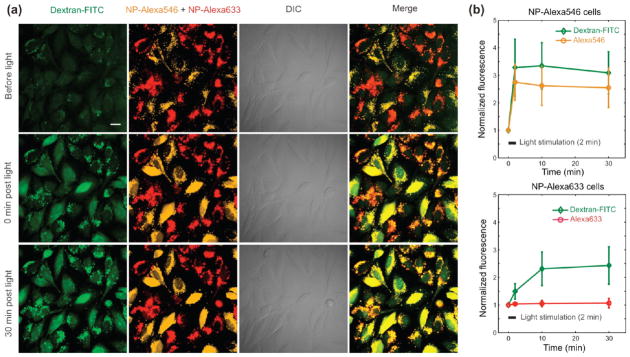
(a) Confocal micrographs of LN-229 cells after NP and dextran-FITC coendocytosis (60× water-immersion objective). Orange NPs were loaded with Alexa546 after synthesis, while red NPs contain covalently bound Alexa633, which cannot be released. The two types of NPs were endocytosed separately and the two populations subsequently mixed and incubated with dextran. Images were acquired before (top row) and at various time points following 2 min light stimulation of the NPs (following rows). Dextran-FITC cytosolic release is observed with both NP types with different kinetics. Scale bars are 20 μm. (b) Time evolution of NP and dextran-FITC average cell fluorescence (normalized to initial fluorescence) following light stimulation, for both Alexa546-NPs loaded cells (left, n = 14 cells) and Alexa633-NPs loaded cells (right, n = 20 cells). The bars represent SD.
Incubation of the PEI–PEG coated NPs with bovine aortic endothelial cells (BAEC) resulted in accumulation of the NPs in large lysosomal vesicles, enabling monitoring of single vesicle disruption events following light exposure (Figure 6). After overnight incubation with a low concentration of NPs (20ug/mL), cells were exposed to TRITC filtered light for very short durations (3 s) and monitored under confocal microscopy at fast scanning rates (580 ms/frame) with minimal imaging laser power. Multiple vesicle disruption events could be observed over a period of minutes following light activation. Figure 6a shows dye fluorescence at various time points up to 20 min, demonstrating progressive cytosolic accumulation of the NP cargo with strong nuclear translocation. The first 5 min of this time-course are provided as Supporting Information Movie S1.
FIGURE 6.

Confocal micrographs of BAEC cells exposed to green light after overnight endocytosis of Alexa546-loaded PEI–PEG MSNs. (a) Time course of Alexa546 fluorescence at various time points following 3 s light exposure. (b) DIC images of cells before and 6 min after a 3 s light exposure showing decreased light absorption of the NP-containing lysosomes after dye release (bar diagram, mean absorbance ± SD, n = 30 vesicles). (c) Magnified view of individual vesicles at various time points following a 3 s exposure. Two individual vesicle disruptions can be seen at ~160 and 201 s following exposure. (d) Fluorescence (top row) and absorbance (from DIC transmittance, bottom) profile plots along the lines defined in (c). All scale bars are 20 μm.
Strikingly, observation of the cells in transmitted light showed a purple color in the intracellular vesicles characteristic of the NP-loaded dye, which was strongly attenuated following cargo release. This can be monitored via DIC microscopy (Figure 6b), as light absorption by the vesicles (quantified as the difference between average transmittance for the entire image and average transmittance of a given vesicle) significantly decreases following exposure to light. Figure 6c shows details of NP-loaded lysosomal vesicles following a 3 s exposure to green light (at time 0 s) with successive disruption of two vesicles. Line profile plots of dye fluorescence and absorption (computed from DIC intensity) across these two vesicles are displayed in Figure 6d. The disruption events are characterized by the appearance of a short-lived fluorescence halo surrounding them as dye rapidly diffuses into the cell cytosol, accompanied by a concomitant drop in absorption within the vesicles (which is directly proportional to dye concentration). Multiple endosome disruptions can be observed in Supporting Information Movie S1. When a vesicle breaks in the vicinity of a dendritic process, the diffusing dye is especially noticeable as a fluorescence wave fills up the membrane protrusion, followed by a rapid decrease in signal as the dye diffuses in the entire cell (see Supporting Information Movie S2).
To illustrate the potential of these nanoparticles, we applied our delivery strategy to multidrug resistant (MDR) cells. Almost half of human tumors develop MDR, whereby exposure to a chemotherapeutic agent triggers simultaneous resistance to a wide spectrum of different compounds, even to those to which the cell had never been exposed.27,36 In most cases, MDR results from the expression of membrane transporters that actively extrude cell-permeable cytotoxic compounds. The most well characterized MDR transporter is P-glycoprotein (P-gp), a 170 kDa member of the adenosine triphosphate (ATP)-binding cassette (ABC) superfamily that can extrude a wide spectrum of compounds.37 Such lack of substrate specificity makes this clearance pathway difficult to circumvent. As our method allows cytosolic delivery of cell-impermeable compounds, which are generally not P-gp substrates, we believe our technique is a novel approach to bypass MDR.
To study the targetability of the functionalized NPs to P-gp-expressing cells, we induced stable expression of a P-gp-GFP C-terminal fusion protein in wild type LN-229 cells. We ascertained P-gp-GFP localization and function by antibody staining and efflux of known substrates, such as rhodamine 123, tetra-methyl-rhodamine-esther, and JC-1 (Supporting Information Figure S7). Our streptavidin-NP conjugates were successfully targeted to P-gp expressing cells following antibody staining (Figure 7a). P-gp positive cells showed high levels of NP binding after 20 min of incubation with few nonspecific interactions (Figure 7a, bottom right panel). As noted above, bioconjugation using a long arm (5 kDa) PEG cross-linker was essential to maximize targetability of the NPs. Shorter arm cross-linkers (such as LC-SMCC, Pierce Biotechnology) also allowed covalent grafting of streptavidin, but resulted in reduced labeling efficiency of cell surface receptors, presumably due to lower mobility of the attached streptavidin as well as lower binding site accessibility (Supporting Information Figure S8). The use of size exclusion chromatography for conjugate purification also proved important. This method was superior to repeated centrifugation and resuspension steps by sonication, which were found to significantly decrease bioactivity of the immobilized streptavidin (Supporting Information Figure S8). Moreover, repeated centrifugation steps contributed to the generation of particles aggregates, while column purification did not increase the average hydrodynamic diameter of the NPs.
FIGURE 7.

(a) Antibody-mediated targeting of mesoporous silica nanoparticles to P-gp. Confocal micrographs showing P-gp-GFP-expressing cells after incubation with streptavidin-functionalized, Alexa546 loaded NPs (60× water-immersion objective). The NPs were added following staining with a primary mouse anti-hMDR1 antibody and a biotinylated goat-antimouse secondary antibody. Plates also contained cells lacking P-gp to serve as contemporaneous controls. The bottom right graph displays the amount of NPs bound to individual cells as a function of their P-gp expression level. Each data point in the scatter plot represents one cell. The NPs used here correspond to a hydrodynamic diameter postsynthesis of ~60 nm (see Figure 1). (b) Targeted, light-activated cytosolic release of Alexa546 into cells expressing P-gp-GFP. Streptavidin-functionalized particles (75 nm diameter) were loaded with Alexa546 and targeted to cells expressing P-gp-GFP. Confocal micrographs showing cells after overnight endocytosis of targeted NPs before (left panel) and immediately after (right panel) light exposure. Cells were imaged using a 60× water-immersion objective. The scatter plot shows the variation in dye fluorescence following light exposure as a function of cell P-gp expression level. Each data point represents one cell. Scale bars are 20 μm in all images.
Following overnight endocytosis of the membrane-bound NPs, regions were chosen that included both wild type cells and cells expressing P-gp. Because of the high specificity of NP uptake, illumination of the entire field released dye exclusively in the P-gp-expressing cells (Figure 7b). While the amount of cytosol-released dye does not linearly correlate with P-gp expression, there appears to be a threshold beyond which significant release is observed, as evidenced when plotting the increase in dye fluorescence within individual cells following light exposure as a function of their P-gp expression level (Figure 7b, bottom right panel). The few cells showing a decrease in total cell fluorescence are cells that did not display cargo release and for which the slight reduction in NP fluorescence can be attributed to either photobleaching or a small focus drift during the course of the experiment.
In summary, we have developed a method for producing size-tunable (30–200 nm), highly monodispersed mesoporous silica nanoparticles that can be biofunctionalized and targeted to specific cell surface proteins. These nanoparticles can be loaded with a wide variety of compounds and can mediate cytosolic release of cell-impermeable molecules in P-gp-expressing cells via light-mediated endosomal breakage. This strategy combines the advantages of nanoparticle-mediated targeted delivery with the temporal control of light activation. We believe this approach is promising in expanding the pharmacological arsenal to cell-impermeable compounds to overcome multidrug resistance. In addition, these novel nanoparticles may be useful vectors for highly specific protein and nucleic acid delivery.
Supplementary Material
Acknowledgments
We wish to thank Dr. Markus Delling for the kind gift of the LAMP1-GFP, Rab5-GFP, and Rab7-GFP plasmids, as well as for useful discussion of the manuscript. This work was funded in part by the MIT Deshpande Center for Technological Innovation.
Footnotes
Supporting Information Available. Materials and Methods, Figures S1–S8, and Movies 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- 1.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 3.Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Seven challenges for nanomedicine. Nat Nanotechnol. 2008;3:242–244. doi: 10.1038/nnano.2008.114. [DOI] [PubMed] [Google Scholar]
- 4.Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 5.Sengupta S, Eavarone D, Capila I, Zhao G, Watson N, Kiziltepe T, Sasisekharan R. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436:568–572. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 6.Boddy AV, Todd R, Sludden J, Griffin M, Robson L, Cassidy J, Bissett D, Bernareggi A, Verrill MW, Calvert AH. A phase I and pharmacokinetic study of paclitaxel poliglumex (XYOTAX), investigating both 3-weekly and 2-weekly schedules. Clin Cancer Res. 2005;11:7834–7840. doi: 10.1158/1078-0432.CCR-05-0803. [DOI] [PubMed] [Google Scholar]
- 7.Sutton D, Nasongkla N, Blanco E, Gao J. Functionalized micellar systems for cancer targeted drug delivery. Pharm Res. 2007;24:1029–1046. doi: 10.1007/s11095-006-9223-y. [DOI] [PubMed] [Google Scholar]
- 8.Matsumura Y, Maeda H. A new concept of macromolecular therapies in cancer chemotherapy: mechanism of tumortropic accumulation of proteins and the antitumor agent SMANCS. Cancer Res. 1986;6:6387–6392. [PubMed] [Google Scholar]
- 9.Jiang W, Kim BYS, Rutka JT, Chan WCW. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 10.Hu Y, Litwin T, Nagaraja AR, Kwong B, Katz J, Watson N, Irvine DJ. Cytosolic delivery of membrane-impermeable molecules in dendritic cells using pH-responsive core-shell nanoparticles. Nano Lett. 2007;7:3056–3064. doi: 10.1021/nl071542i. [DOI] [PubMed] [Google Scholar]
- 11.Lee SH, Choi SH, Kim SH, Park TG. Thermally sensitive cationic polymer nanocapsules for specific cytosolic delivery and efficient gene silencing of siRNA: Swelling induced physical disruption of endosome by cold shock. J Controlled Release. 2008;125:25–32. doi: 10.1016/j.jconrel.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Park C, Kim H, Kim S, Kim C. Enzyme responsive nanocontainers with cyclodextrin gatekeepers and synergistic effects in release of guests. J Am Chem Soc. 2009;131:16614–16615. doi: 10.1021/ja9061085. [DOI] [PubMed] [Google Scholar]
- 13.Yu C, Fan J, Tian B, Stucky GD, Zhao D. Synthesis of mesoporous silica from commercial poly(ethylene oxide)/poly-(butylene oxide) copolymers: toward the rational design of ordered mesoporous materials. J Phys Chem B. 2003;107:13368–13375. [Google Scholar]
- 14.Wang J, Zhang J, Asoo BY, Stucky GD. Structure-selective synthesis of mesostructured/mesoporous silica nanofibers. J Am Chem Soc. 2003;125:13966–13967. doi: 10.1021/ja036967v. [DOI] [PubMed] [Google Scholar]
- 15.Tsung CK, Fan J, Zheng N, Shi Q, Forman AJ, Wang J, Stucky GD. A general route to diverse mesoporous metal oxide submicrospheres with highly crystalline frameworks. Angew Chem, Int Ed. 2008;47:8682–8686. doi: 10.1002/anie.200802487. [DOI] [PubMed] [Google Scholar]
- 16.Slowing II, Vivero-Escoto JL, Wu CW, Lin VSY. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv Drug Delivery Rev. 2008;60:1278–1288. doi: 10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 17.Lai CY, Trewyn BG, Jeftinija DM, Jeftinija K, Xu S, Jeftinija S, Lin VSY. A mesoporous silica nanosphere-based carrier system with chemically removable CdS nanoparticle caps for stimuli-responsive controlled release of neurotransmitters and drug molecules. J Am Chem Soc. 2003;125:4451–4459. doi: 10.1021/ja028650l. [DOI] [PubMed] [Google Scholar]
- 18.Trewyn BG, Whitman CM, Lin VSY. Morphological control of room-temperature ionic liquid templated mesoporous silica nanoparticles for controlled release of antibacterial agents. Nano Lett. 2004;4:2139–2143. [Google Scholar]
- 19.Slowing II, Trewyn BG, Lin VSY. Mesoporous silica nanoparticles for intracellular delivery of membrane-impermeable proteins. J Am Chem Soc. 2007;129:8845–8849. doi: 10.1021/ja0719780. [DOI] [PubMed] [Google Scholar]
- 20.Radu DR, Lai CY, Jeftinija K, Rowe EW, Jeftinija S, Lin VSY. A polyamidoamine dendrimer-capped mesoporous silica nanosphere-based gene transfection reagent. J Am Chem Soc. 2004;126:13216–13217. doi: 10.1021/ja046275m. [DOI] [PubMed] [Google Scholar]
- 21.Slowing II, Trewyn BG, Giri S, Lin VSY. Mesoporous silica nanoparticles for drug delivery and biosensing applications. Adv Funct Mater. 2007;17:1225–1236. [Google Scholar]
- 22.Rosenholm JM, Peuhu E, Eriksson JE, Sahlgren C, Linden M. Targeted intracellular delivery of hydrophobic agents using mesoporous hybrid silica nanoparticles as carrier systems. Nano Lett. 2009;9:3308–3311. doi: 10.1021/nl901589y. [DOI] [PubMed] [Google Scholar]
- 23.Alvarez-Lorenzo C, Bromberg L, Concheiro A. Light-sensitive intelligent drug delivery systems. Photochem Photobiol. 2009;85:848–860. doi: 10.1111/j.1751-1097.2008.00530.x. [DOI] [PubMed] [Google Scholar]
- 24.Christie JG, Kompella UB. Ophthalmic light sensitive nanocarrier systems. Drug Discovery Today. 2008;13:124–134. doi: 10.1016/j.drudis.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Høgset A, Prasmickaite L, Selbo PK, Hellum M, Engesæter BØ, Bonsted A, Berg K. Photochemical internalisation in drug and gene delivery. Adv Drug Delivery Rev. 2004;56:95–115. doi: 10.1016/j.addr.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 26.Nishiyama N, Iriyama A, Jang WD, Miyata K, Itaka K, Inoue Y, Takahashi H, Yanagi Y, Tamaki Y, Koyama H, Kataoka K. Light-induced gene transfer from packaged DNA enveloped in a dendrimeric photosensitizer. Nat Mater. 2005;4:934–941. doi: 10.1038/nmat1524. [DOI] [PubMed] [Google Scholar]
- 27.Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 28.Boumendjel A, Di Pietro A, Dumontet C, Barron D. Recent advances in the discovery of flavonoids and analogs with high-affinity binding to P-glycoprotein responsible for cancer cell multidrug resistance. Med Res Rev. 2002;22:512–529. doi: 10.1002/med.10015. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki K, Ikari K, Imai H. Synthesis of silica nanoparticles having a well-ordered mesostructure using a double surfactant system. J Am Chem Soc. 2004;126:462–463. doi: 10.1021/ja038250d. [DOI] [PubMed] [Google Scholar]
- 30.Ikari K, Suzuki K, Imai H. Structural control of mesoporous silica nanoparticles in a binary surfactant system. Langmuir. 2006;22:802–806. doi: 10.1021/la0525527. [DOI] [PubMed] [Google Scholar]
- 31.Mukherjee S, Ghosh RN, Maxfield FR. Endocytosis. Phys Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- 32.Dacey DM, Peterson BB, Robinson FR, Gamlin PD. Fireworks in the primate retina: in vitro photodynamics reveals diverse LGN-projecting ganglion cell types. Neuron. 2003;37:15–27. doi: 10.1016/s0896-6273(02)01143-1. [DOI] [PubMed] [Google Scholar]
- 33.Høgset A, Prasmickaite L, Selbo PK, Hellum M, Engesæter BØ, Bonsted A, Berg K. Photochemical internalisation in drug and gene delivery. Adv Drug Delivery Rev. 2004;56:95–115. doi: 10.1016/j.addr.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 34.Norum OJ, Selbo PK, Weyergang A, Giercksky KE, Berg K. Photochemical internalization (PCI) in cancer therapy: from bench towards bedside medicine. J Photochem Photobiol, B. 2009;96:83–92. doi: 10.1016/j.jphotobiol.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 35.Berg K, Folini M, Prasmickaite L, Selbo PK, Bonsted A, Engesaeter BO, Zaffaroni N, Weyergang A, Dietze A, Maelandsmo GM, Wagner E, Norum OJ, Hogset A. Photochemical internalization: a new tool for drug delivery. Curr Pharm Biotechnol. 2007;8:362–372. doi: 10.2174/138920107783018354. [DOI] [PubMed] [Google Scholar]
- 36.Seeger MA, van Veen HW. Molecular basis of multidrug transport by ABC transporters. Biochim Biophys Acta. 2009;1794:725–737. doi: 10.1016/j.bbapap.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 37.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo RP, Harrell PM, Trinh YT, Zhang QH, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.