

Abstract
MicroRNAs (miRs) are small non-protein-coding RNAs that bind to specific mRNAs and inhibit translation or promote mRNA degradation. Recent reports, including ours, indicated that miR-33a located within the intron of sterol regulatory element-binding protein (SREBP) 2 controls cholesterol homeostasis and can be a possible therapeutic target for treating atherosclerosis. Primates, but not rodents, express miR-33b from an intron of SREBF1. Therefore, humanized mice, in which a miR-33b transgene is inserted within a Srebf1 intron, are required to address its function in vivo. We successfully established miR-33b knock-in (KI) mice and found that protein levels of known miR-33a target genes, such as ABCA1, ABCG1, and SREBP-1, were reduced compared with those in wild-type mice. As a consequence, macrophages from the miR-33b KI mice had a reduced cholesterol efflux capacity via apoA-I and HDL-C. Moreover, HDL-C levels were reduced by almost 35% even in miR-33b KI hetero mice compared with the control mice. These results indicate that miR-33b may account for lower HDL-C levels in humans than those in mice and that miR-33b is possibly utilized for a feedback mechanism to regulate its host gene SREBF1. Our mice will also aid in elucidating the roles of miR-33a/b in different genetic disease models.
Sterol regulatory element-binding proteins (SREBPs) comprise a subclass of basic helix-loop-helix leucine zipper transcription factors conserved from yeasts to humans and regulate the expression of genes required for maintaining cellular lipid homeostasis1. Mammals possess two SREBP genes, SREBP-1 and SREBP-2 (known as SREBF1 and SREBF2, respectively) that express three major SREBP proteins. Two SREBP-1 isoforms, SREBP-1a and SREBP-1c, primarily regulate fatty acid metabolism, and SREBP-2 is the main regulator of cholesterol metabolism, although there is some functional overlap among the three SREBP isoforms2,3,4.
MicroRNAs (miRs) are small non-protein-coding RNAs that bind to specific mRNAs and inhibit translation or promote mRNA degradation5. Recent advances in the understanding of miR biology revealed that the genetic loci encoding for the transcription factors SREBP-1 and SREBP-2 also encode for the miRs miR-33b and miR-33a, respectively. Recent reports, including ours, indicated that miR-33a controls ABCA1 expression and reduces HDL-C levels6,7,8 and that miR-33a deficiency ameliorates atherosclerosis in mice9,10,11. However, in rodents, a part of miR-33b is lacking from a Srebf1 intron (Supplementary Fig. 1a), and it is impossible to determine the precise coordinate mechanisms of miR-33a and miR-33b; the expression of these miR-33s is expected to depend on their corresponding host genes. Of note, SREBP-1 and SREBP-2 are differentially regulated by hormones, dietary challenges, or statin treatment, and the amounts and functions of miR-33a and miR-33b would be greatly affected under these conditions.
miR-33a and miR-33b are identical in their seed sequences, and thus have been predicted to repress the same set of genes with similar specificities. Antisense oligonucleotides against miR-33a are believed to simultaneously target miR-33a and miR-33b. However, there remains a 2-nucleotide mismatch after the seed sequence between miR-33a and miR-33b (Supplementary Fig. 1a), and whether this difference results in differential targeting remains to be established. Moreover, some of the previously established miR-33a target genes were not dysregulated in our miR-33a-deficient mice. Therefore, humanized mice, in which a miR-33b transgene is inserted within a Srebf1 intron, are required to address its function in vivo.
We successfully established miR-33b knock-in (KI) mice for the same intron as in humans. The protein levels of known miR-33a target genes, such as ABCA1, ABCG1, and SREBP-1, were reduced under basal conditions. An LXR agonist, which induces Srebf1 expression, enhanced miR-33b production. In vitro experiments indicated that macrophages from the miR-33b KI mice had a reduced cholesterol efflux capacity via apoA-I and HDL-C. Moreover, HDL-C levels were reduced by almost 35% even in miR-33b KI hetero mice compared with the control mice.
The feasibility of genetic manipulation is one of the many advantages of using mice as a model organism. However, the lack of miR-33b in mouse Srebf1 has raised an important concern regarding the direct translation of data from rodent models to human physiology and metabolic disorders. Our mice will aid in answering these questions and will be useful for assessing the risks and benefits of long-term alterations in miR-33s in different disease models. These mice might also be useful for screening of the drugs that alter the levels of miR-33a and miR-33b.
Results
miR-33b is co-expressed with SREBF1 in the human cell line HepG2
It is assumed that a miR located within an intron of a gene is expressed along with its host gene and exerts its specific function12. Because miR-33b is located in a SREBF1 intron in humans (Supplementary Fig. S1a), we stimulated human cell line HepG2 with the LXR agonist T0901317 and determined miR-33b and miR-33a expression along with the expression of the host genes SREBF1 and SREBF2. As shown in Fig. 1a and b, miR-33b expression seemed to tag along behind SREBF1 expression. In contrast, miR-33a and SREBF2 expression was not affected by LXR stimulation (Fig. 1c and d).
Figure 1. miR-33b is co-expressed with SREBF1 in HepG2 cells.
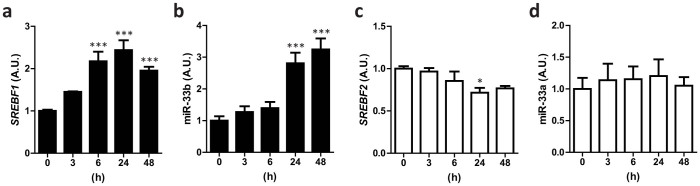
HepG2 cells were treated with T0901317 (10 μM) for the indicated time. The relative expressions of SREBF1 (a), miR-33b (b), SREBF2 (c), and miR-33a (d) are shown (n = 6–9). Values are mean ± s.e.m. *p < 0.05, ***p < 0.001 compared with 0 h.
Generation of miR-33b KI mice
Because miR-33b is located in SREBF1 intron 16 in humans and there are high homologies in exons 16 and 17 between human and mouse (82.6% of nucleotides and 79.7% of amino acids, Supplementary Fig. S1b), we introduced the human miR-33b sequence into intron 16 of mouse Srebf1. We isolated and amplified the region that encoded for the complete pre-miR sequence of human miR-33b and adjacent sequence, which enabled the introduction of miR-33b into intron 16 of mouse Srebf1 (Fig. 2a). Supplementary Figure S2a and Figure 2b show the results of Southern blotting analysis of genomic DNA from ES cells and tail genomic DNA from F1 mice that were successfully targeted by a KI vector, respectively. PCR analysis indicated the specific patterns for wild-type (WT), KI+/−, and KI+/+ mice (Fig. 2c). This miR-33b KI strategy did not alter Srebf1 intron 16 splicing, as confirmed by PCR (Fig. 2d) and sequencing (Fig. 2e). The expression levels of miR-33b in miR-33b KI+/− mice were almost half of those in miR-33b KI+/+ mice (Fig. 2f). We also measured the levels of miR-33b, miR-33a, Srebf1, and Srebf2 in WT and KI mice in both the liver and the peritoneal macrophages (Supplementary Figure S2b–d and S3a–d). Srebf1 levels were similar among these mice (Supplementary Figure S2c and S3c). Although there was no difference in miR-33a levels in macrophages (Supplementary Figure S3b), miR-33a levels were increased in proportion of the expression levels of miR-33b in the liver (Supplementary Figure S2b). The miR-33b KI+/+ mice were born with the expected Mendelian ratios, were viable, fertile, and did not exhibit any obvious abnormalities in size, shape, or structure up to 8 weeks of age. Relative tissue expression pattern of miR-33b was similar to that of Srebf1 (Supplementary Fig. S2e and S2f).
Figure 2. Generation of miR-33b knock-in (KI) mice.

(a). Strategy used to generate miR-33b KI mice. (b). Southern blotting of mouse tail genomic DNA. Representative images are shown. (c). PCR analysis of mouse tail genomic DNA. Representative images are shown. (d). RT-PCR analysis of Srebf1 expression in the livers of 8-week-old mice. Sense primer was designed for exon 13, and antisense primer was designed for exon17. Note that there was no other band except for that of the correct size. Representative images are shown. (e). Sequencing alignment at the joint between exons 16 and 17 of Srebf1 in the indicated mice. (f). Relative expression of miR-33b in the livers of 8-week-old mice (n = 6). N.D., not determined. Values are mean ± s.e.m.
miR-33b is upregulated after inducing Srebf1 expression
We next sought to confirm whether miR-33b expression was affected by endogenous changes in Srebf1 expression by the LXR agonist T090131713. When primary hepatocytes from the miR-33b KI+/+ mice were stimulated with T0901317, Srebf1 and miR-33b mRNA levels were significantly increased in parallel, although this increase was faster for Serbf1 than for miR-33b (Fig. 3a and b). To check this effect in vivo, T0901317 was suspended in 0.5% carboxymethylcellulose and administrated to 8-week-old male miR-33b KI+/+ mice at a dose of 25 mg/kg for 3 days. The average liver weight of the T0901317-treated mice was 1.5-fold greater than that of the control mice (Supplementary Fig. S4a). Srebf1 and miR-33b expression levels in the liver were also significantly increased in parallel (Fig. 3c and d). The average liver weight and Srebf1 expression level in the liver of T0901317-treated WT mice were shown in Supplementary Figure S4b and S4c. These results indicate that miR-33b was co-expressed with Srebf1 in the livers of the T0901317-treated miR-33b KI mice.
Figure 3. miR-33b is co-expressed with Srebf1 in miR-33b KI mice.
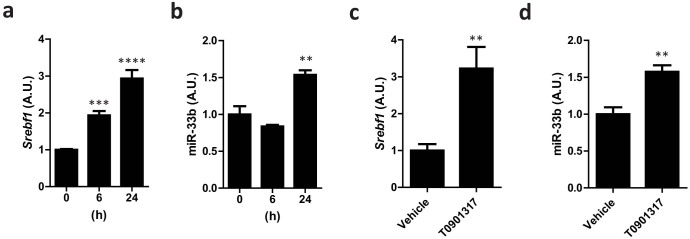
(a). Relative Srebf1 expression levels in primary hepatocytes from miR-33b KI+/+ mice treated with T0901317 (10 μM) for the indicated time. Values are mean ± s.e.m (n = 6). **p < 0.01 ***p < 0.001 by one-way analysis of variance. (b). Relative miR-33b expression levels in primary hepatocytes from miR-33b KI+/+ mice treated with T0901317 (10 μM) for the indicated time. Values are mean ± s.e.m (n = 6). **p < 0.01 by one-way analysis of variance. (c). Relative Srebf1 expression levels in the livers of 8-week-old male miR-33b KI+/+ mice treated with T0901317 (25 mg/kg) for 3 days. Values are means ± s.e.m (n = 6). **p < 0.01 compared with the vehicle. (d). Relative miR-33b expression levels in the livers of 8-week-old male miR-33b KI+/+ mice treated with T0901317 (25 mg/kg) for 3 days. Values are mean ± s.e.m (n = 6). *p < 0.05 compared with the vehicle.
miR-33b KI results in alterations in miR-33a target proteins ABCA1 and SREBP-1
We determined ABCA1, SREBP-1, CPT1a, and AMPKα protein levels in the liver (Fig. 4a and Supplementary Figure S5). As shown in Fig. 4a, Supplementary Figure S5a, and S5b, ABCA1 and SREBP-1 protein levels were lower in the livers of the miR-33b KI mice. However, the protein levels of some of the previously defined miR-33a target genes, such as CPT1a and AMPKα remained unchanged. We also analyzed protein expressions of glucose metabolic genes (Fig. 4b and Supplementary Figure S5c–f). However, no significant change in protein level was observed in PCK1, G6PC, and CREB in the liver of miR-33b KI mice compared with that of control mice. SRC1 was up-regulated in miR-33bKI mice and it was opposite to the results of previous report14.
Figure 4. miR-33b regulates ABCA1 and SREBP-1.
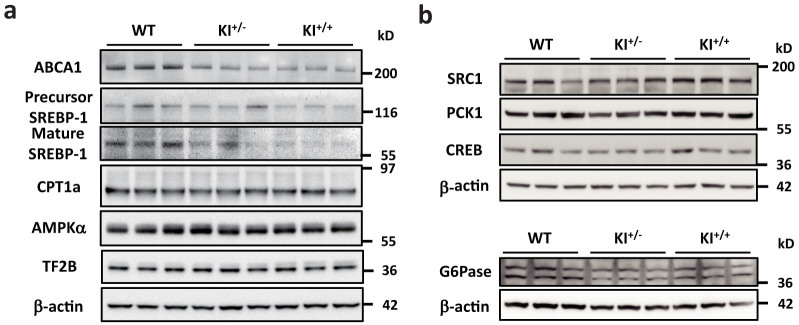
(a). Western blotting analysis for ABCA1, SREBP-1, CPT1a, and AMPKα protein levels in the livers of WT, KI+/−, and KI+/+ mice. Representative images are shown. TF2B and β-actin were used as loading controls. (b). Western blotting analysis for SRC1, PCK1, CREB and G6Pase protein levels in the livers of WT, KI+/−, and KI+/+ mice. Representative images are shown. β-actin were used as loading controls.
miR-33b KI reduces cholesterol efflux in macrophages
To investigate a physiological role of miR-33b in mice, we first compared the functions of peritoneal macrophages from the WT and miR-33b KI+/+ mice. ABCA1 and ABCG1 protein levels were lower in macrophages from the miR-33b KI+/+ mice than from the WT mice (Fig. 5a and Supplementary Figure S3e and S3f), which was compatible with the findings for our miR-33a-deficient mice. We determined apoA-I- and HDL-C-mediated cholesterol efflux from peritoneal macrophages and found that macrophages from the miR-33b KI+/+ mice had lower apoA-I- and HDL-C-mediated cholesterol efflux than those from the WT mice (Fig. 5b).
Figure 5. miR-33b reduces cellular cholesterol efflux and serum HDL-C levels.
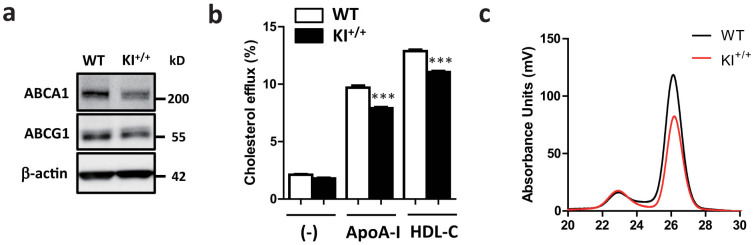
(a). Western blotting for ABCA1 and ABCG1 proteins in peritoneal macrophages from WT and KI+/+ mice. Representative images are shown. β-actin was used as the loading control. (b). Cholesterol efflux to apoA-I and HDL-C in peritoneal macrophages from WT and KI+/+ mice (n = 6 each). Values are mean ± s.e.m. ***p < 0.001 (c). Mean plots of HPLC analysis for serum cholesterol in WT and KI+/+ mice (n = 4 and 5, respectively).
A single miR-33b copy reduces serum HDL levels
Hepatic ABCA1 overexpression increases HDL-C levels15, and liver-specific deletion of ABCA1 results in a substantial decrease in plasma HDL-C levels (approximately 80%) in chow-fed mice16. Moreover, we previously reported that the miR-33a−/− mice had 22%–39% higher serum HDL-C levels than the WT mice8. Thus, we determined the serum HDL-C levels of the WT, miR-33b KI+/−, and miR-33b KI+/+ mice at the age of 8 weeks.
Serum HDL-C levels were significantly decreased in the miR-33b KI+/− and miR-33b KI+/+ mice compared with the WT mice (Table). We also classified and quantified serum lipoproteins using high-performance liquid chromatography (HPLC). Mean plots of the HPLC elution profile of serum from male mice are shown in Fig. 5c, and the lipid profiles are summarized in Supplementary Table S1. These results show that only one copy of miR-33b was sufficient to substantially reduce HDL-C and total cholesterol to the same levels as those in the miR-33b KI+/+ mice. Moreover, the decreased HDL levels mainly comprised very large-, large-, and medium-sized HDLs (mature HDLs) (Fig. 5c and Supplementary Table S1).
Table 1. Serum profiling of WT, KI+/−, and KI+/+ mice.
| WT (n = 4) | KI+/− (n = 4) | KI+/+ (n = 4) | |||
|---|---|---|---|---|---|
| TP (g/dL) | 4.375 ± 0.1109 | 4.275 ± 0.04787 | 4.350 ± 0.05000 | ||
| ALB (g/dL) | 2.950 ± 0.1190 | 2.825 ± 0.1109 | 2.900 ± 0.04082 | ||
| BUN (mg/dL) | 21.75 ± 0.6801 | 20.58 ± 1.248 | 21.58 ± 1.680 | ||
| CRE (mg/dL) | 0.1125 ± 0.002500 | 0.0925 ± 0.004787 | * | 0.0975 ± 0.006292 | |
| Na (mEq/L) | 152.5 ± 0.6455 | 153.5 ± 0.2887 | 153.8 ± 0.4787 | ||
| K (mEq/L) | 3.350 ± 0.05000 | 3.325 ± 0.0750 | 3.350 ± 0.1041 | ||
| Cl (mEq/L) | 110.5 ± 0.6455 | 110.8 ± 0.2500 | 111.0 ± 0.5774 | ||
| Ca (mg/dL) | 8.500 ± 0.1871 | 8.325 ± 0.1109 | 8.350 ± 0.08660 | ||
| IP (mg/dL) | 7.775 ± 0.4589 | 7.225 ± 0.2955 | 7.400 ± 0.4637 | ||
| T-BIL (mg/dL) | 0.0875 ± 0.004787 | 0.0925 ± 0.008539 | 0.0825 ± 0.01109 | ||
| AST (IU/L) | 39.25 ± 1.702 | 33.50 ± 1.658 | 39.25 ± 1.702 | ||
| ALT (IU/L) | 26.50 ± 3.663 | 21.00 ± 2.415 | 22.75 ± 1.702 | ||
| ALP (IU/L) | 505.5 ± 48.55 | 398.5 ± 40.01 | 480.0 ± 29.31 | ||
| LDH (IU/L) | 278.3 ± 77.21 | 243.5 ± 55.30 | 255.0 ± 55.26 | ||
| AMY (IU/L) | 2295 ± 68.22 | 2224 ± 62.39 | 2363 ± 97.02 | ||
| γ-GTP (IU/L) | 3> | 3> | 3> | ||
| T-CHO (mg/dL) | 98.50 ± 5.694 | 66.25 ± 2.287 | ** | 62.00 ± 1.225 | *** |
| TG (mg/dL) | 34.75 ± 2.780 | 32.25 ± 3.065 | 35.25 ± 4.328 | ||
| NEFA (μEq/L) | 471.0 ± 47.36 | 474.8 ± 71.81 | 459.5 ± 55.01 | ||
| LDL-C (mg/dL) | 6.750 ± 0.6292 | 6.750 ± 0.6292 | 6.000 ± 0.0 | ||
| HDL-C (mg/dL) | 57.75 ± 4.171 | 39.25 ± 0.7500 | ** | 37.25 ± 0.6292 | ** |
| GLU (mg/dL) | 216.3 ± 22.98 | 180.5 ± 8.930 | 197.8 ± 11.92 |
Values are mean ± s.e.m. Blood was obtained from chow-fed 8-wk-old male mice after 4 h fasting.
*p < 0.05;
**p < 0.01;
***p < 0.001 compared with WT mice.
Discussion
In the present study, we successfully established humanized mice, in which a miR-33b transgene was inserted within the same intron as that in human SREBF1. The LXR agonist T0901317, which is a well-established Srebf1 expression inducer, enhanced miR-33b production. The protein levels of known miR-33a/b target genes, such as ABCA1, ABCG1, and SREBP-1, were reduced under basal conditions. In vitro experiments indicated that macrophages from the miR-33b KI+/+ mice had a reduced cholesterol efflux capacity via apoA-I and HDL-C. Finally, HDL-C levels were reduced by almost 35% even in the miR-33b KI+/− mice compared with the WT mice without any changes in triglyceride (TG) levels.
In contrast to humans and other mammals, rodents lack miR-33b and only have miR-33a in Srebf2. This needs to be kept in mind when attempting to directly translate to humans the previous results that miR-33a inhibition could prevent atherosclerosis in mouse models because of two reasons. First, SREBF1 and SREBF2 are differentially regulated by hormones, dietary challenges, and lipid-lowering agents, including statins17. This indicates that both isoforms of miR-33 participate in regulating the primary risk factors of metabolic syndrome, which accelerate atherosclerosis. Second, miR-33b differs from miR-33a by 2-nucleotides and may have a different target profile, including stronger effects on targets in the SREBP-1-dependent regulation of fatty acid/TG homeostasis and insulin signaling. We found increased miR-33b expression after treatment with LXR agonist in our mice, which indicated that miR-33b was co-expressed with its Srebf1 host gene and enabled us to study the impact of Srebf1-derived miR-33b on cholesterol/lipid homeostasis.
We have not yet succeeded in identifying miR-33b-specific target genes. Even previously reported miR-33b target genes were not reduced in the liver of miR-33b KI mice compared with that of control mice. One of the reasons of such result may be that the previous study was conducted in human cell line and potential binding sites of miR-33b are not conserved at least in PCK1 3′UTR of mice. It is also possible that some compensated mechanisms may have occurred in miR-33b KI mice. However, the protein levels of miR-33a target genes, such as ABCA1, ABCG1, and SREBP-1, were reduced18. Moreover, the protein levels of previously defined miR-33a target genes, which were not dysregulated in miR-33a KO mice, including CPT1a and AMPKα, remained unchanged19,20. Thus, it may be necessary to assess those conditions when Srebf1 expression is strongly affected to establish the importance of the functions of miR-33b. In any event, the numbers of miR-33b transcripts were greater than those of miR-33a transcripts, and this underscores the importance of miR-33b21. Although there were no differences in the levels of miR-33a in macrophages, it is interesting that the levels of miR-33a were increased in proportion to the expression levels of miR-33b in the liver. Because Srebf1 level is higher in the liver than that in macrophages22, it is possible that miR-33b and miR-33a compete for the same target gene binding sites in the liver, and that the degradation of miR-33a is inhibited by miR-33b expression. In addition, there may be other unknown mechanisms.
miRs are known to target long non-coding RNAs whose functions are largely unknown, and interactions between miRs are also possible23. Thus, miR-33b-specific functions should be determined in future experiments.
Rayner et al. recently showed that inhibiting miR-33a and miR-33b in healthy male non-human primates increased circulating HDL-C levels21. More recently, Rottiers et al. reported that miR-33a and miR-33b acted in a redundant manner and that inhibiting both isoforms by an 8-mer LNA-modified anti-miR enhanced HDL-C levels24. Our data demonstrated that miR-33b indeed functions to control HDL-C levels, which highlights the importance of targeting both miR-33 family members simultaneously. It is noteworthy that only one copy of miR-33b (miR-33b KI+/− mice) significantly reduced HDL-C levels to the same levels as those in the miR-33b KI+/+ mice. This explains one of the reasons why human HDL-C levels are lower than those of mice and indicates that it is important to considerably reduce miR-33b levels if pharmacological targeting of miR-33s is used to increase HDL-C levels. In this context, the current LNA-modified anti-miR technique is quite potent for reducing the levels of both miR-33 isoforms and may be useful for anti-atherosclerosis therapy.
In addition to the effects on HDL-C, a study by Rayner et al. showed that miR-33 antagonism reduced very low-density lipoprotein-associated TGs in their cohort of normal male African green monkeys21. However, Rottiers et al. did not find any significant changes in TG levels when using miR-33a/b-targeting LNA-anti-miR treatment24. In our present miR-33b KI study and in previous miR-33a KO experiments8, we did not observe any changes in TG levels, indicating that modulation of miR-33s is unlikely to have a strong effect on TG levels, although species differences and different dietary conditions need to be considered.
In contrast, we found a significant inhibitory effect of miR-33b on SREBP-1. A feedback system of SREBP-2 by cholesterol levels is well known, which maintains appropriate levels of cellular cholesterol. However, a similar mechanism has not been established for SREBP-1. Chronic activation of SREBP-1c in cases of overnutrition can lead to serious obesity-related problems. miR-33b may be utilized for a feedback mechanism to regulate its host gene SREBF1 because insulin induces hepatic SREBP-1c expression and promotes lipogenesis and hepatic TG synthesis (Supplementary Fig. S6).
In the present study, we demonstrated the effect of miR-33b on HDL-C levels in vivo. We assume that inhibiting both miR-33a and miR-33b will have a significant effect on HDL-C levels in clinical settings. However, it is known that one miR can have hundreds of target genes and unexpected side effects may occur due to long-term therapeutic modulation of miR-33 to cure metabolic diseases. Careful observations of miR-33b KI and miR-33a-deficient mice and intercrossing of these mice will enable us to detect miR-33a- and miR-33b-specific target genes and to elucidate the overall functions of miR-33a and miR-33b in vivo. Moreover, our mice will aid in analyzing the roles of miR-33a/b in different genetic disease models and in screening drug candidates that can modulate miR-33a and miR-33b levels and activities.
Methods
Materials
The following antibodies were used: anti-ABCA1 (NB400-105) and anti-ABCG1 (NB400-132) (Novus Biologicals, Littleton, CO, USA); anti-AMPKα (#2532) and anti-CREB (#9197) (Cell Signaling Technology, Beverly, MA, USA); anti-CPT1a (ab128568) and anti-PCK1 (ab70358) (Abcam, Cambridge, UK); anti-β-actin (AC-15; A5441, Sigma-Aldrich, St. Louis, MO, USA); anti-SREBP-1 (sc-13551, sc-8984), anti-SRC1 (sc-8995), anti-G6Pase (sc-27198), and anti-TF2B (sc-225) (Santa Cruz, Biotechnology, California, USA). Anti-mouse, anti-rabbit and anti-goat IgG HRP-linked antibodies were purchased from GE Healthcare (Amersham, UK). Human apoA-I was purchased from Sigma-Aldrich. Human acetylated LDL (acLDL) and human HDL-C were purchased from Biomedical Technologies, Inc. (Stoughton, MA, USA). [1, 2-3H (N)]-Cholesterol was purchased from Perkin Elmer (Boston, MA, USA). T0901317 was purchased from Cayman Chemical (Ann Arbor, MI, USA).
Generation of miR-33b KI mice
A targeting vector was constructed by modifying bacterial artificial chromosome RP24-310C22 (Invitrogen) using a defective prophage λ-Red recombination system25,26. As a selection marker, a neomycin resistance cassette flanked by loxP sites (loxP-PGK-gb2-neo-loxP cassette; Gene Bridges, Germany) was inserted at the adjacent site of the human pre-miR-33b site. The targeting vector was electroporated into C57BL/6 mouse ES cells (DS Pharma Biomedical) using a Nucleofector system (Lonza). Positive clones were selected by incubating cells with 200 μg/ml geneticin (Invitrogen) for 5 days, and homologous recombination was confirmed by Southern blotting. Successfully recombined ES cells were injected into blastocysts from ICR strain mice supplied by Unitech Inc. (Japan), and chimeric mice were bred with C57BL/6 mice to generate F1 mice. F1 mice genotypes were confirmed by Southern blotting. The neomycin resistance cassette was removed from the mouse germ line by breeding heterozygous mice with Ayu-1 Cre KI mice, which expressed Cre recombinase in multiple tissues, including the germ line27. Descendant miR-33b knock-in heterozygous mice without the Ayu-1 Cre allele were crossed with each other to generate the miR-33b KI+/+ mice. All experiments were performed with male C57BL/6 background mice and wild-type littermates were used as a control. All of the experimental protocols were approved by the Ethics Committee for Animal Experiments of Kyoto University and the methods were performed in accordance with the guidelines approved by the ethics committee. Primers used for genotyping were as follows: WT/KI sense, ATGGATTTACCTCAGTTTTAACGAC; WT antisense, CATCACTGAAGCACTGCATCTGC; KI antisense, AAGTGGATCCAGAATTCGTGA; Cre sense, GCTGCCACGACdCAAGTGACAGCAATG; and Cre antisense, GTAGTTATTCGGATCATCAGCTACAC.
Southern blotting
Southern blotting was performed using DIG High Prime DNA Labeling and Detection Starter Kit II (Roche) according to the manufacturer's protocol. Genomic DNA samples were purified and digested with MfeI and AseI for a 5′ probe, EcoRI for a 3′ probe, and NheI for a Neo probe. Primer sequences used to amplify these probes were as follows:
5′ probe sense, CACGGTTGTGAGAAGTCAGTATTC; 5′ probe antisense, CTTTGCAAGCTCCTTGAGAATAAG; 3′ probe sense, AGTAAAATTCTCCTCAATGAACGTG; 3′ probe antisense, CAGTAGGTGACATTGTGATTGATCT; Neo probe sense, GAACAAGATGGATTGCACGCAGGTTCTCCG; and Neo probe antisense, GTAGCCAACGCTATGTCCTGATAG.
Determination of splicing between exons 16 and 17 in Srebf1
We amplified the fragment between Srebf1 exons 13 and 17 using cDNA from the livers of the indicated mice by PCR, and these products were then electrophoresed. Extension time was sufficient to expand the fragment when the correct splicing did not occur. There was no other band except for that of the correct size. Sequencing was performed using a primer for exon 16 and an ABI 3130 genetic analyzer. Primer sequences used were as folows:
Exon 13 sense, CCTAGAGCGAGCGTTGAACT; Exon 17 antisense, CTACCTGGACTGAAGCTGGTG; and Exon 16 sequence primer, AGGGCAGCTCTGTACTCCTTC.
Cell culture
HepG2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Nacalai Tesque, Japan) supplemented with 10% fetal bovine serum (FBS). Mouse primary hepatocytes were obtained from miR-33b KI+/+ mice using a two-step collagenase perfusion method28. In brief, hepatocyte suspensions were obtained by passing a collagenase type II (Gibco BRL, Life Technologies Inc., Rockville, MD, USA)-digested liver sample through a 70-μm cell strainer, followed by centrifugation to isolate mature hepatocytes. Hepatocytes were then resuspended in DMEM supplemented with 10% FBS and seeded on collagen type I-coated dishes (Iwaki Asahi Glass Co. Ltd., Japan) at a density of 7 × 104 cells/ml. After incubation for 24 h, the cells were used for experiments.
Cholesterol efflux from macrophages
Cellular cholesterol efflux via apoA-I was determined as described previously29. In brief, thioglycollate-elicited mouse peritoneal macrophages were plated in 24-well microplates at a density of 5 × 106 cells/ml. Cells were cultured for 24 h in RPMI 1640 containing 3H-labeled acLDL (1.0 μCi/ml of 3H-cholesterol and 25 μg/ml of acLDL). On the next day, the cells were washed 3 times with RPMI 1640 and incubated for 6 h in RPMI 1640 with or without apoA-I (10 μg/ml) or HDL (100 μg/ml). Cholesterol efflux was expressed as the percentage of radioactivity released from cells in medium relative to the total radioactivity in cells plus medium.
Western blotting
Western blotting was performed using standard procedures as described previously30. A lysis buffer was supplemented with a complete mini protease inhibitor (Roche), ALLN (25 μg/ml), 0.5 mM NaF, and 10 μM Na3VO4 just prior to use. Protein concentrations were determined using a bicinchoninic acid protein assay kit (Bio-Rad). All samples (20 μg of protein) were suspended in lysis buffer, fractionated using NuPAGE 4%–12% Bis-Tris (Invitrogen) gels, and transferred to a Protran nitrocellulose transfer membrane (Whatman). The membrane was blocked using 1× phosphate-buffered saline (PBS) containing 5% non-fat milk for 1 h and incubated with a primary antibody [anti-ABCA1 (1:1000), anti-ABCG1 (1:1000), anti-AMPKα (1:1000), anti-SREBP-1 (1:250), anti-TF2B (1:1000), anti-β-actin (1:3000), anti-CREB (1:1000), anti-PCK1 (1:1000), anti-SRC1 (1:200), anti-G6Pase (1:200) or anti-CPT1a (1:1000)] overnight at 4°C. After washing with PBS–0.05% Tween 20 (0.05% T-PBS), the membrane was incubated with a secondary antibody (anti-rabbit, anti-mouse and anti-goat IgG HRP-linked; 1:2000) for 1 h at 4°C. The membrane was then washed with 0.05% T-PBS and detected with an ECL Western Blotting Detection Reagent (GE Healthcare) using an LAS-1000 system (Fuji Film).
RNA extraction and quantitative RT-PCR (qRT-PCR)
Total RNA was isolated and purified using TriPure Isolation Reagent (Roche). cDNA was synthesized from 1 μg of total RNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche) according to the manufacturer's instructions. For qRT-PCR, specific genes were amplified in 40 cycles using SYBR™ Green PCR Master Mix (Applied Biosystems). Expression was normalized to that of the housekeeping gene β-actin. Gene-specific primers are as follows;
SREBF1 sense, AACAGTCCCACTGGTCGTAGAT; SREBF1 antisense, TGTTGCAGAAAGCGAATGTAGT; SREBF2 sense, AGGAGAACATGGTGCTGA; SREBF2 antisense, TAAAGGAGAGGCACAGGA; ACTB sense, AGGCACTCTTCCAGCCTTCC; ACTB antisense, GCACTGTGTTGGCGTACAGG; Srebf1 sense, TAGAGCATATCCCCCAGGTG; Srebf1 antisense, GGTACGGGCCACAAGAAGTA; Srebf2 sense, GTGGAGCAGTCTCAACGTCA; Srebf2 antisense, TGGTAGGTCTCACCCAGGAG; Actb sense, GATCTGGCACCACACCTTCT; and Actb antisense, GGGGTGTTGAAGGTCTCAAA.
Quantitative PCR for miRs
Total RNA was isolated using the TriPure Isolation Reagent (Roche). miR-33a and miR-33b were measured using TaqMan MicroRNA assay protocols (Applied Biosystems). Products were analyzed using a thermal cycler (ABI Prism® 7900HT sequence detection system). miRs expression of samples were normalized by U6 snRNA expression.
Serum biochemical analysis
After mice were fasted for 4–6 h, blood was obtained from the inferior vena cava of an anesthetized mouse, and serum was separated by centrifugation at 4°C and stored at −80°C. Employing standard methods, biochemical measurements were made using a Hitachi 7180 Auto Analyzer (Nagahama Life Science Laboratory, Nagahama, Japan). Lipoproteins were analyzed by HPLC at Skylight Biotech (Akita, Japan), according to the procedures described previously31.
Statistical analysis
Results are given as mean ± s.e.m. Statistical comparisons were made using unpaired two-tailed Student's t-tests or one-way analysis of variance with the Bonferroni post hoc test, as appropriate. A p value of <0.05 was considered significant.
Author Contributions
T.H., T.N. and K.O. designed the project; T.H., T.N., O.B., Y.K., T.N., M.N., S.U., M.I., F.N., Y.I., S.K. and N.S. performed experiments; T.H., T.N., N.Y., H.S., T.N., K.H., N.K., M.Y., T.K., T.K. and K.O. analyzed and interpreted data; and T.H., T.N. and K.O. prepared the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Neal G. Copeland (Institute of Molecular and Cell Biology, Singapore) for the defective prophage λ-Red recombination system; Junji Takeda (Osaka University, Osaka, Japan) and Kosuke Yusa (Osaka University, Osaka, Japan) for the plasmid used for the construction of the targeting vector; Ken-ichi Yamamura (Kumamoto University, Kumamoto, Japan) and Kimi Araki (Kumamoto University) for Ayu-1 Cre expressing mice. This work was supported in part by grants from the Japan Society for the Promotion of Science, by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to T. Kimura, T. Kita, K.H., T.H., and K.O.), by a Grant-in-Aid for Scientific Research on Innovative Areas “Crosstalk between transcriptional control and energy pathways, mediated by hub metabolites”(3307) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to K.O.), by Otsuka Pharmaceutical Co., Ltd. - Kyoto University SRP Program (to T. Kimura, T. Kita, M.Y., T.H., and K.O.), by grants from Banyu Life Science Foundation International, Takeda Memorial Foundation, Suzuken Memorial Foundation, Sakakibara Memorial Foundation, Japan Foundation of Applied Enzymology, SENSHIN Medical Research Foundation, Kowa Life Science Foundation, and ONO Medical Research Foundation (to T.H.), and by grants from ONO Medical Research Foundation, the Cell Science Research Foundation, Daiichi-Sankyo Foundation of Life Science, and Takeda Memorial Foundation (to K.O.).
References
- Osborne T. F. & Espenshade P. J. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. Genes Dev 23, 2578–2591 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espenshade P. J. & Hughes A. L. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet 41, 401–427 (2007). [DOI] [PubMed] [Google Scholar]
- Horton J. D., Goldstein J. L. & Brown M. S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109, 1125–1131 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama C. et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 75, 187–197 (1993). [PubMed] [Google Scholar]
- Ono K., Kuwabara Y. & Han J. MicroRNAs and Cardiovascular Diseases. FEBS J (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi-Shoushtari S. H. et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 328, 1566–1569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner K. J. et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328, 1570–1573 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T. et al. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc Natl Acad Sci U S A 107, 17321–17326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner K. J. et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest 121, 2921–2931 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T. et al. MicroRNA-33 Deficiency Reduces the Progression of Atherosclerotic Plaque in ApoE(−/−) Mice. J Am Heart Assoc 1, e003376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T. et al. MicroRNAs and Lipoprotein Metabolism. J Atheroscler Thromb 21, 17–22 (2014). [DOI] [PubMed] [Google Scholar]
- Sowa N. et al. MicroRNA 26b encoded by the intron of small CTD phosphatase (SCP) 1 has an antagonistic effect on its host gene. J Cell Biochem 113, 3455–3465 (2012). [DOI] [PubMed] [Google Scholar]
- DeBose-Boyd R. A., Ou J., Goldstein J. L. & Brown M. S. Expression of sterol regulatory element-binding protein 1c (SREBP-1c) mRNA in rat hepatoma cells requires endogenous LXR ligands. Proc Natl Acad Sci U S A 98, 1477–1482 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez C. M. et al. MicroRNA 33 regulates glucose metabolism. Mol Cell Biol 33, 2891–2902 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso F. et al. Role of the hepatic ABCA1 transporter in modulating intrahepatic cholesterol and plasma HDL cholesterol concentrations. J Lipid Res 44, 296–302 (2003). [DOI] [PubMed] [Google Scholar]
- Timmins J. M. et al. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest 115, 1333–1342 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimano H. SREBPs: physiology and pathophysiology of the SREBP family. FEBS J 276, 616–621 (2009). [DOI] [PubMed] [Google Scholar]
- Horie T. et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat Commun 4, 2883 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin I. et al. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem 285, 33652–33661 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Hernando C. & Moore K. J. MicroRNA modulation of cholesterol homeostasis. Arterioscler Thromb Vasc Biol 31, 2378–2382 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner K. J. et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 478, 404–407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. et al. Genetic deletion of low density lipoprotein receptor impairs sterol-induced mouse macrophage ABCA1 expression. A new SREBP1-dependent mechanism. J Biol Chem 283, 2129–2138 (2008). [DOI] [PubMed] [Google Scholar]
- Memczak S. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338 (2013). [DOI] [PubMed] [Google Scholar]
- Rottiers V. et al. Pharmacological Inhibition of a MicroRNA Family in Nonhuman Primates by a Seed-Targeting 8-Mer AntimiR. Sci Transl Med 5, 212ra162 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland N. G., Jenkins N. A. & Court D. L. Recombineering: a powerful new tool for mouse functional genomics. Nat Rev Genet 2, 769–779 (2001). [DOI] [PubMed] [Google Scholar]
- Horiguchi M. et al. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc Natl Acad Sci U S A 106, 19029–19034 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M. et al. HLF/HIF-2alpha is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J 22, 1134–1146 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seglen P. O. Preparation of isolated rat liver cells. Methods Cell Biol 13, 29–83 (1976). [DOI] [PubMed] [Google Scholar]
- Zhang Z. et al. Expression of cholesteryl ester transfer protein in human atherosclerotic lesions and its implication in reverse cholesterol transport. Atherosclerosis 159, 67–75 (2001). [DOI] [PubMed] [Google Scholar]
- Horie T. et al. Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc Res 87, 656–664 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui S., Kakuuchi H., Okamoto M., Mizukami Y. & Okazaki M. Differential reactivity of two homogeneous LDL-cholesterol methods to LDL and VLDL subfractions, as demonstrated by ultracentrifugation and HPLC. Clin Chem 48, 1946–1954 (2002). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information