

Abstract
Motivation: Metaproteomic analysis allows studying the interplay of organisms or functional groups and has become increasingly popular also for diagnostic purposes. However, difficulties arise owing to the high sequence similarity between related organisms. Further, the state of conservation of proteins between species can be correlated with their expression level, which can lead to significant bias in results and interpretation. These challenges are similar but not identical to the challenges arising in the analysis of metagenomic samples and require specific solutions.
Results: We introduce Pipasic (peptide intensity-weighted proteome abundance similarity correction) as a tool that corrects identification and spectral counting-based quantification results using peptide similarity estimation and expression level weighting within a non-negative lasso framework. Pipasic has distinct advantages over approaches only regarding unique peptides or aggregating results to the lowest common ancestor, as demonstrated on examples of viral diagnostics and an acid mine drainage dataset.
Availability and implementation: Pipasic source code is freely available from https://sourceforge.net/projects/pipasic/.
Contact: RenardB@rki.de
Supplementary information: Supplementary data are available at Bioinformatics online
1 INTRODUCTION
In contrast to classical proteomic approaches, metaproteomics and environmental proteomics studies aim at deciphering the interplay of different organisms contained within an environmental sample (Muth et al., 2013). Throughout the past years, this idea has seen increasing application primarily in three fields: aqueous ecosystems, terrestrial systems and eukaryotic host microbiomes (Hettich et al., 2013). In addition, metaproteomic approaches have become of interest also for clinical diagnostics, e.g. for characterizing the state of an infection (Fouts et al., 2012) or for identifying and strain-level typing of bacteria (Karlsson et al., 2012).
Similar to metagenomic approaches (Wooley et al., 2010), the analysis of environmental samples and the interplay of organisms offer an enormous potential to further the characterization and understanding of these systems. At the same time, challenges in metaproteomics are manifold and relate to all steps of the analysis. Particularly, this includes the handling of the resulting large and complex datasets of spectra derived from mass spectrometry (MS) experiments and their meaningful comparison with reference proteomes of organisms. It can by no means be generally assumed that this set of references—in particular for bacteria or viruses—is complete or representative for the given sample (Lindner et al., 2013). Depending on the sample of interest, the number of organisms of interest may vary significantly from tens to thousands and more. In all cases, it is non-trivial to identify the correct origin of a spectrum and thereby to allow either the identification of organisms or the quantification of either organisms or key biological processes.
While many goals and strategies correlate for metagenomic and metaproteomic approaches, several distinct differences are noteworthy. In metaproteomic approaches, expression levels are analyzed and thus quantitative measures differ even for proteins from a single organism. This can be highly insightful for functional analyses (Muth et al., 2013), but poses an additional challenge for data analysis. Further, while the method is designed to be unbiased, it cannot be assumed that all proteins are extracted and captured by MS in a metaproteomics experiment. However, as it is an orthogonal technique to metagenomics, metaproteomic and metagenomic approaches have differing error profiles and can jointly provide a much deeper insight than each method on its own (Hettich et al., 2013), even in cases such as the quantification of strains when metagenomics is usually preferable owing to lower demands on material and longer sequences. It should be noted that metaproteomic approaches require the availability of reference proteomes or genomes and cannot assemble them from a given sample as in metagenome protocols [e.g. Lai et al. (2012)].
While numerous tools have been introduced for metagenomic data analysis (see Teeling and Glockner (2012) for an overview), only comparatively few tools exist with focus on the specific difficulties arising in the analysis of metaproteomic data. These cover a broad field ranging from specialized approaches for visualization (Mehlan et al., 2013) to the scalability of database search algorithms (Jagtap et al., 2013, 2012b) and to metaproteogenomics and the difficulty of identifying a suitable reference database (Rooijers et al., 2011; Seifert et al., 2013).
One key difficulty that is hindering metaproteomic data analysis is the ambiguity of peptide identifications (Hettich et al., 2013; Muth et al., 2013; Seifert et al., 2013). Even more pronounced than in classical bottom-up proteomic approaches (Nesvizhskii, 2010), one spectrum can not only match to several peptides occurring in multiple proteins of the same organism, but may match to proteins in different organisms. This is particularly common for closely related organisms with sufficient sequence similarity and for well-conserved proteins. Consequently, this problem hinders the correct identification and quantification of the species present in a sample. While also common in metagenomics (Lindner and Renard, 2013), it is even more challenging in proteomics because peptides are commonly shorter than sequencing reads and thereby less likely to be unique. Furthermore, expression levels are not necessarily uncorrelated to the state of conservation of a protein and thereby constitute a potentially large bias when disregarded.
Currently, two major ideas are used to address this difficulty: either the analysis is based primarily on unique peptides that are specific for a single organism (Karlsson et al., 2012; Lo et al., 2007; Rooijers et al., 2011) or the phylogenetic resolution is reduced. This can be achieved by limiting the analysis to a set of well-chosen representative species that have no significant overlap (Chourey et al., 2013) or by dynamically allocating results to the lowest common ancestor that allows a distinction (Huson et al., 2007; Jagtap et al., 2012a; Schneider et al., 2011). When disregarding shared peptides and focusing on unique peptides, it is feasible to identify the species present in an organism as long as the coverage is high enough to observe a sufficient number of these peptides with sufficient confidence. This can be a challenge in metaproteomic experiments, which commonly have low coverage for individual species (Hettich et al., 2013), and resulting difficulties in the reliability of peptide identifications (Renard et al., 2010) can lead to false conclusions. Furthermore, quantitative information derived exclusively from distinct unique peptides is not necessarily representative for the presence of organisms or functional groups. When using representatives or lowest common ancestor, the resolution of the approach is reduced and it may no longer be possible to distinguish strains or related species.
Within this contribution, we introduce Pipasic (peptide intensity-weighted proteome abundance similarity correction) as a tool for metaproteomic data analysis, which overcomes the limitations of these two strategies. Pipasic uses all peptide identifications available, not only unique peptides, and generates a strain-specific quantitative output without resorting to a lower phylogenetic resolution. This is possible because Pipasic builds on a similarity correction approach from metagenomics (Lindner and Renard, 2013), which implicitly weights and corrects observed abundances based on the experiment-specific expected similarity between reference genomes. Further, Pipasic avoids potential bias by estimating the similarity only for expressed proteins, which may correlate with the state of conservation of proteins.
We evaluate Pipasic in two settings: a diagnostic setting where we distinguish two closely related cowpox virus strains at varying concentrations and a metaproteomic dataset from an acid mine drainage (AMD) environment. We compare Pipasic to a MEGAN-based (Huson et al., 2007) analysis as a commonly used lowest common ancestor approach (Jagtap et al., 2012a) and analyze the impact of the expression level correction and unique peptides.
2 METHODS
Pipasic is a method for estimating corrected proteome abundances in a metaproteomic dataset and builds on the GASiC approach (Lindner and Renard, 2013), extending it to the specific features of metaproteomic data. The overall workflow is outlined in Figure 1. As input, Pipasic takes a metaproteomic dataset containing a set of tandem mass spectra data and a set of N reference proteomes of organisms or functional groups that are expected to be potentially contained within the sample. The goal then is to quantify the contribution of these references to the spectral data at hand. The first step is the identification of the metaproteomic spectra with the peptides in the reference proteomes. The number of identified spectra per proteome is the naïve observed abundance estimate. The second step calculates a matrix containing the pairwise similarities of the reference proteomes, reflecting only those proteins expressed in the sample according to their expression level. The results of the first two steps are then used to estimate the corrected proteome abundances.
Fig. 1.

Method overview. Pipasic involves three main steps: (i) peptide identification: here metaproteomic peptide spectra are identified by a database search. The number of matches to the proteomes is the observed abundances. (ii) Similarity estimation: the similarities between the reference proteomes are calculated and stored in a similarity matrix. This incorporates the adjustment to only regard expressed proteins and to weight them according to their expression level. (iii) Similarity correction: the observed abundances are corrected using the similarity matrix yielding corrected abundances
There are two main differences to the genomics-based GASiC approach: first, expression levels are analyzed in metaproteomics. Compared with the homogeneous coverage in metagenomic whole genome sequencing, each protein—even within a single organism—will have a different expression level. These expression levels directly influence the similarity estimation. Second, the number of spectra is typically lower than the number of reads in metagenomics. This requires the proteomes to have sufficiently high numbers of matching spectra that the probabilistic correction step works correctly. Therefore, abundance estimates may be distorted if the coverage is low or the proteome only contains few proteins.
2.1 Peptide identification
The peptide spectra in the metaproteomic dataset are searched separately against each reference proteome using an appropriate database search algorithm. The choice of peptide search tool is not restricted; we tested searches with InsPecT (Tanner et al., 2005), Sequest/Tide (Diament and Noble, 2011) and BICEPS (Renard et al., 2012). It is crucial for Pipasic that matches to all reference proteomes are reported instead of a subset of best hits as commonly done by many search engines. We generally run the peptide identification and false discovery rate (FDR) computation separately for each reference proteome to be more independent of database size and quality effects (Jeong et al., 2012) and further to allow a more fine-grained probabilistic weighting of peptide identifications against presence of a species. However, Pipasic can also be run with a joint peptide identification and FDR computation for all reference proteomes.
To ensure specificity, we apply a standard decoy database strategy (Bradshaw et al., 2006) using a reverse database. A FDR is calculated for each identification; identifications below a user-defined FDR threshold are discarded. For each proteome , the number of FDR-controlled identifications is called the observed proteome abundance. Normalizing the observed abundances of all proteomes to one yields relative observed abundances ri.
2.2 Proteome similarity estimation
The similarity of two reference proteins can be computed in various ways, e.g. based on mismatch statistics or alignment scores. However, for the application to metaproteomics, the quantity of interest is the similarity that may lead to an ambiguous spectra-to-species assignment. The equivalent similarity estimation step in the metagenomic GASiC method involves the simulation of short reads for each genome, which are then mapped to all other genomes. This carefully reflects the risk of incorrectly assigning a read. One could estimate proteome similarities in the same way by simulating spectra for each proteome and identifying them among all other proteomes. However, simulating a significant number of spectra using a simulation method such as the MSSimulator (Bielow et al., 2011) is particularly time-consuming and practically infeasible.
2.2.1 String comparison
As a significantly faster alternative, we thus regard the reference proteomes as sets of protein sequences, i.e. sets of strings. Because the proteins in the experiment are typically digested into tryptic peptides before the spectra are acquired, we perform an in silico digestion of the reference proteomes, yielding a list of short peptide strings for each proteome. For a proteome, we search all short peptide strings in all other proteomes using exact string matching. To account for the amino acids with indistinguishable masses, we replace all occurrences of I by L and Q by K. We do not regard any ambiguity arising from variable modifications (such as oxidized M) because the analysis on the sequence level cannot incorporate the knowledge whether the potential modification indeed occurs. The fraction of tryptic peptides in proteome that can be found in another proteome is denoted the unweighted similarity . Thus, we obtain the—unweighted—similarity matrix .
2.2.2 Weighting by the expression level
A reference proteome often contains proteins that were not expressed or measured in the experiment. This may either result from the fact that not all proteins are expressed or that expression levels span several orders of magnitude and may be below the detection limit or from biases in sample preparation or MS acquisition (Hettich et al., 2013). The similarity of the expressed proteins may strongly differ from the overall similarity because proteins of key cellular functions may be better conserved as well as higher expressed than other proteins of an organism. Thus, we reflect these particular effects in the similarity estimation by introducing weights for all peptides. The weight wp for the tryptic peptide p in proteome is calculated as follows:
Assign a preliminary weight to each peptide p: add to for each spectrum that was identified with p, where Np is the number of peptides the spectrum can be identified with.
For each protein , set the peptide weights , to the average preliminary peptide weight: .
Normalize the sum of all weights to one: .
The matrix entry aij of the weighted similarity matrix A is calculated by summing over the weights of the peptides in that were found in :
2.3 Similarity correction
The similarity correction step corrects the relative observed abundance ri of proteome by estimating the true abundance ci. This step is mathematically identical to the GASiC correction step: the relative observed abundance ri of proteome is assumed to be a mixture of the true abundances cj weighted with the similarity matrix entry aij and can thus be written as
In matrix notation, we can write this equation more briefly as
where and . Directly solving the linear system of equations for c may lead to numerically unstable results. Furthermore, we require the estimated abundances to be ≥ 0 and the sum over all abundances to be ≤ 1. Thus, we formulate the solution for c as a non-negative lasso (Efron et al., 2004; Renard et al., 2008) problem:
In our implementation, we solve this problem with the COBYLA method implemented in SciPy (Oliphant, 2007).
Similarly to the GASiC framework, it is possible to obtain statistically more robust estimates by bootstrapping from the set of spectra and iterating the similarity correction step. Statistical tests for the presence of a proteome as well as error estimates for the obtained abundances can then be computed from the distribution of abundance estimates.
2.4 Technical details
Pipasic is implemented as Python scripts where performance-critical parts are calculated using the scientific computing libraries SciPy and NumPy. Currently, Pipasic is designed to directly work with either InsPecT or Sequest/Tide for peptide identification. Pipasic is freely available from https://sourceforge.net/projects/pipasic/
3 EXPERIMENTS AND RESULTS
To evaluate Pipasic and the impact of the various algorithmic steps, we conducted two experiments. First, we demonstrate the accuracy of the method on a mixture of real datasets of cowpox viruses with a known ground truth. Here, we can identify the benefits of individual steps and compare Pipasic with a metaproteomic analysis based on MEGAN and unique peptides. In a second experiment, we apply Pipasic to a published AMD dataset showing that our method is also able to provide corrected abundance estimates for datasets from a natural environment.
3.1 Performance evaluation
The goal of the first experiment is the quantitative evaluation of the Pipasic method using gold standard ground truth data. In this experiment, we first provide evidence that including the expression information for similarity estimation significantly improves the abundance estimates. Secondly, we demonstrate that Pipasic provides more accurate results with regard to identification and quantification than the analysis with MEGAN and based on unique peptides.
The idea behind this experiment is to mix two pure proteomic MS datasets of highly similar proteomes in predefined ratios. The challenge for the computational method is to correctly estimate the fraction of each proteome in the dataset. To this end, we rely on two datasets containing two different but closely related cowpox virus strains: Krefeld (Kre) and Brighton Red (BR). A more thorough description of these datasets is available in (Doellinger et al., unpublished data). HEp-2 (ATCC-CRL-23) cells were infected with the individual viruses and the viruses were then purified, collected by centrifugation and washed. The viral particles were then dissolved in ammonium bicarbonate. Proteins were digested with trypsin, desalted and fractionated. The peptide fractions were then analyzed with an Easy-nanoLC (Thermo Fisher Scientific) coupled online to an LTQ Orbitrap Discovery mass spectrometer (Thermo Fisher Scientific).
To reduce the number of contaminating spectra, we searched both datasets against the human reference proteome and removed all matches below a 5% FDR. To create the reference proteomes for both viruses, their viral DNA sequences were assembled and genes were identified based on existing NCBI annotations for cowpox viruses. The reference proteomes for both strains were created by translating the identified genes into proteins (Doellinger et al., unpublished data).
We now mixed the remaining spectra to create 11 artificial datasets with mixtures ranging from 100% Kre strain to 100% BR strain by sampling spectra from the original datasets such that each dataset contained 3000 spectra in total. To ensure a balanced spectrum quality in all datasets, we sampled high- and low-quality spectra in a 1:1 ratio from the original datasets where high quality was defined as spectra within the 5% FDR range when searched against the corresponding reference proteome.
3.1.1 Unweighted versus weighted Pipasic
We processed the 11 datasets with Pipasic as described in the Section 2 using InsPecT for peptide identification and both the unweighted and expression-weighted similarity matrix. Figure 2 shows the calculated abundance estimates plotted over the true fraction of Kre spectra on the x-axis, such that the estimates for one dataset lie in a column. The dashed lines represent the observed abundances and emphasize the major challenge with these datasets: high relative abundance values are assigned to both proteomes, as the bulk of the spectra could not be identified uniquely. Although the unweighted correction (dash-dotted lines) clearly improves on the observed abundances, we can still see the discrepancy to the ground truth (solid circles). The expression-weighted correction (solid lines) yields the best approximation of the true abundances. The error bars show the 95% confidence interval of our estimates, which was estimated by 100-fold bootstrapping from our datasets. In particular, the weighted correction estimates zero or low abundances if a proteome was not or almost not contained in the metaproteomic dataset.
Fig. 2.
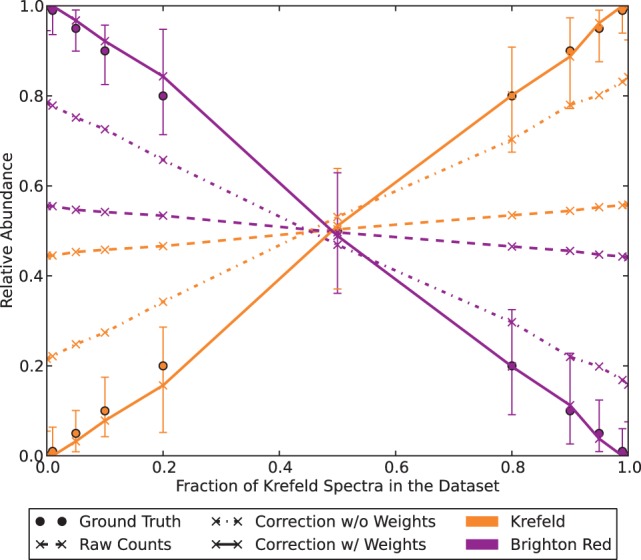
Effect of Pipasic correction: the relative abundances of 11 mixed cowpox virus Kre/BR datasets were corrected with Pipasic without and with expression correction. The observed abundances (dashed lines) are insufficient estimates for the true abundances (solid dots): in the extreme cases of pure Kre or BR datasets the absent virus still receives 45% abundance. The unweighted correction (dash–dotted line) improves on this, but best results are obtained using the expression-weighted similarity matrices (solid line). The error bars indicate the 95% confidence interval after 100-fold bootstrapping
Furthermore, we repeated the experiment using the 20% BR/80% Kre dataset, but successively increased the number of reference proteomes by adding proteomes of other DNA viruses to the database (BR and Kre proteomes were always present). We used up to 20 proteomes (see Supplementary Text) and measured the Pipasic run time and estimation accuracy by calculating the root mean squared error. Both metrics are shown in Figure 3. The most time-consuming step of Pipasic is peptide identification, and therefore, its linear contribution is stronger than the contribution of the similarity matrix calculation with quadratic complexity. We also see that the error of the estimated abundances is low for all considered database sizes and only increases slightly with the number of proteomes.
Fig. 3.
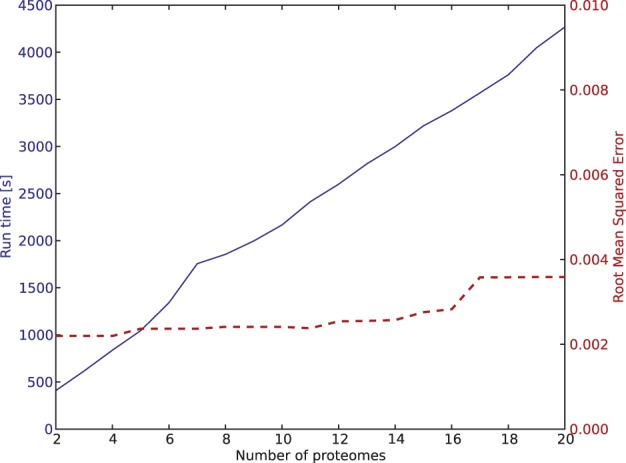
Influence of reference database size on Pipasic prediction accuracy and run time. A dataset containing 20% BR and 80% Kre was searched against a reference proteome database with increasing size. Prediction accuracy was measured using the root mean squared error of the BR and Kre estimates. The run time was measured for the complete Pipasic run, including peptide identification with InsPecT and abundance correction, but without bootstrapping. The error is low for all database sizes and only increases slightly with the database size. The run time increases linearly with the number of reference proteomes, as peptide identification is the most time-consuming step in the pipeline
3.1.2 Comparison to MEGAN
To compare Pipasic with currently used approaches, we also applied MEGAN to the data, which parses the results of a BLAST (Altschul et al., 1990) search against a reference database. An underlying phylogenetic tree allows assigning of shared identifications to lowest common ancestor nodes in the tree, expressing the degree of ambiguity in the results. In this way, MEGAN raises the significance of the unique identifications for the evaluation of the experiment.
We searched all 11 mixed cowpox virus datasets against the Kre and BR reference proteome databases using InsPecT to identify each spectrum with a peptide sequence. Then we searched the peptide sequences with BLASTP in the reference proteomes, such that all identifications could be placed to the correct position in the phylogenetic tree with MEGAN. Figure 4a shows the output of MEGAN for the dataset containing 10% Kre and 90% BR spectra. The size of the circles is log-proportional to the number of assigned spectra, visualizing that the majority of spectra was assigned to the higher level Orthopoxvirus node. The leaves, representing Kre and BR, obtained relatively few spectra: only 8.4% of all matches were unique. In Figure 4b, we plotted for each dataset both the unique matches to each proteome as well as the sum of shared Cowpox virus and unique matches. Because the number of shared matches is much higher than the number of unique matches, the sum of unique and shared (dashed lines) is not informative, as both proteomes obtain close to 50% relative abundance. The number of unique matches (dash–dotted lines) contains more information, and the resulting relative abundances are closer to the ground truth. However, in the case of pure datasets, still a significant number of spectra is matching uniquely to the absent species (about 15%). Here, the Pipasic estimates (solid lines) are much closer to the ground truth.
Fig. 4.
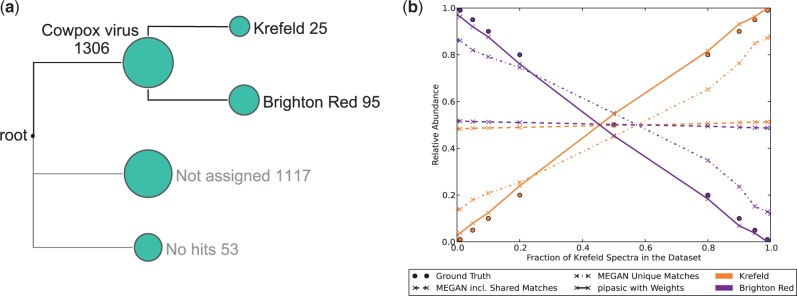
Comparison of Pipasic and MEGAN on the cowpox virus datasets. (a) MEGAN output for the 10% Kre/90% BR dataset. (b) Comparison of MEGAN and Pipasic on all 11 mixed cowpox virus datasets. For MEGAN, the number of unique and shared matches (dashed lines) shows almost no difference between the two proteomes because the number of unique matches is low. The number of unique matches (dash–dotted lines) provides abundances closer to the ground truth, but Pipasic (solid lines) yields the best estimates
3.2 AMD experiment
In the second experiment, we demonstrate the applicability of Pipasic to metaproteomic data originating from a natural environment, which is more complex than our in silico metaproteome. With this experiment, we show that Pipasic automatically corrects abundances of highly similar reference proteomes without affecting the abundances of other unrelated proteomes. For that purpose, we used metaproteomic spectra of an AMD biofilm dataset described in Denef et al. (2010). AMD biofilms are bacterial communities in a highly acidic environment. Thus, AMD communities are not as complex as other microbial communities and their composition is well understood.
We downloaded the metaproteomic spectra of sample 20 run 2 and the corresponding protein database from the authors’ Web site (http://compbio.ornl.gov/biofilm_amd_PIGT/ accessed in March 2013). As the protein database contained sequences for all dominant organisms, we manually divided the database into six reference proteomes: Leptospirillum group II and III (Lepto2 and Lepto3), Ferroplasma acidarmanus Type I and II (Fer1 and Fer2), G-plasma and others, like contaminants and unassigned archaea and bacteria. Then we searched the spectra in the reference proteomes with Tide and counted the number of matching spectra. We applied Pipasic with data weighting on the results to obtain the corrected abundance estimates.
The results of this experiment are shown in Table 1. Here, the effect of the correction is not as pronounced as in the previous experiment owing to the relatively low similarity values (maximum 0.21 compared with 0.92). Lepto3 receives the strongest absolute correction (−359 PSMs) owing to the protein sequence similarities with Lepto2, which receives low relative correction. Fer1 and Fer2 have the highest proteome similarities in this experiment (0.21/0.19); their abundances were reduced in sum by 48.3%. G-plasma has the least similarity to the other proteomes (<0.04) and therefore receives only little correction by 3%. Notably, the correction within the Lepto group is asymmetric: Lepto3 receives stronger relative correction than Lepto2. Two opposing factors contribute to this effect: first, the number of peptide spectrum matches to Lepto2 is more than twice as high as to Lepto3 (Table 1) and, second, the probability to find a Lepto2 spectrum in Lepto3 (Fig. 5) is ∼30% higher than vice versa. Taken together, the difference in abundance dominates the correction in the Lepto group, such that the absolute number of Lepto2 peptides that can also be found in Lepto3 is much higher than vice versa.
Table 1.
AMD dataset abundance estimation
| Proteome | Fer1 | Fer2 | Lepto2 | Lepto3 | G-Plasma | Other |
|---|---|---|---|---|---|---|
| Observed PSMs | 195 | 189 | 4470 | 2014 | 692 | 87 |
| Pipasic estimate | 111 | 88 | 4281 | 1655 | 671 | 32 |
| Relative correction (%) | 43.1 | 53.4 | 4.2 | 17.8 | 3.0 | 63.2 |
Note: The peptide spectrum matches (PSM) were counted for each proteome and subsequently corrected with Pipasic using a weighted similarity matrix. The results show a strong relative correction for the highly similar Fer 1/2 and only small relative correction for Lepto 2/3 and G-Plasma.
Fig. 5.
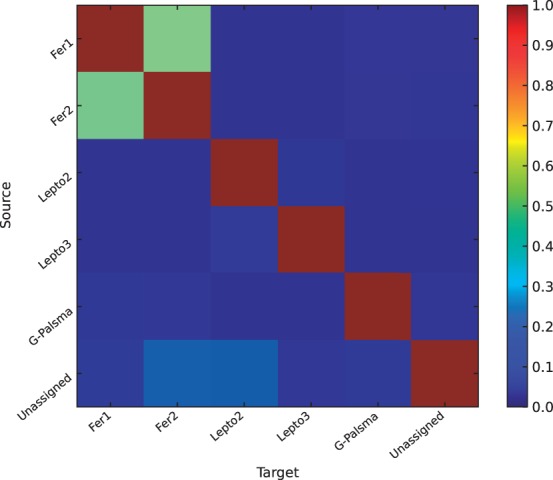
Pipasic similarity matrix with data weighting for the AMD experiment. The matrix entries encode the probability that a peptide in a source proteome can be found in a target proteome, modulated by the metaproteomic data (see Section 2). Here we see that the intra-group matrix coefficients for the Fer and Lepto group are greater than the inter-group coefficients. This means in practice that Pipasic corrects abundances within but not between the two groups. It is noteworthy that the matrix coefficients can be asymmetric, which has the effect that abundance can be shifted from one proteome to another rather than correcting both proteomes equally
This experiment demonstrates that the proposed Pipasic method can handle real metaproteomic data and the calculated estimates are in agreement with the expectation. The two main groups Fer1/2 and Lepto2/3 receive abundance corrections within each group, but not between the groups. This is noteworthy because we did not require any prior information other than the reference proteomes and shows that the similarity estimates reflect the nature of the microbial community.
4 DISCUSSION
The experiments indicate that Pipasic allows the reliable separation of highly similar strains in metaproteomics experiments. It can be used for reliably identifying and quantifying the contributions of organisms and functional units even in cases when—as in the cowpox virus data experiment—92% of all expressed tryptic peptides are identical. In particular, Pipasic allows having a phylogenetic resolution down to the strain level, which is inherently not feasible for lowest common ancestor approaches for highly related species. This is also clearly visible in the comparison with MEGAN on the cowpox virus strain data (Fig. 4).
Given its reliability, Pipasic is preferable to approaches relying solely on the analysis of unique peptides. Figure 4 indicates the risk of analyzing unique peptides for highly related strains. Even though the overall number of identified peptides per species is above 1000, which is high for a metaproteomic setting, the number of unique peptides remains low owing to the sequence similarity. Thus, only few peptide identifications out of a thousand decide on the identification of a species when relying on unique peptides. The example in Figure 4 highlights the risk: even in cases when the ground truth contains 0% spectra of the Kre strain, MEGAN finds 17 unique peptides; this effect was also observed when using the more conservative OMSSA (Geer et al., 2004) search engine instead of InsPecT. These may incorrectly be interpreted as proof of the presence of the Kre strain. However, given that the original peptide identification search was conducted at a 5% FDR and given the large number of spectra searched, these identifications are incorrect. Because Pipasic leverages the computed similarity and the shared peptides into the analysis, it is less at risk to overvalue these incorrect identifications and correctly reduces the presence of the Kre strain in this example down to a level where it cannot be distinguished from a 0% presence.
This example also indicates that the statistical model behind Pipasic contains and quantifies uncertainty. A bootstrapping strategy in the abundance estimation step allows us to obtain confidence statements for all estimates, and thereby the definition of cutoffs for diagnostic decisions can be supported by statistical statements. The reliability of an estimate obviously primarily depends on data abundance—the number of supporting spectra—as well as on the computed and weighted similarity of the species of interest.
Pipasic computes its similarity correction adjusted to the expression level of proteins. The cowpox virus data experiment clearly indicates the significance of this step for the results. This step highlights a major difference between metaproteomics and metagenomics: although the main idea of the metagenomic method could be applied in metaproteomics, the method itself must be tuned to the underlying difference in the biological data. The expression level correction should also be applicable and helpful in metatranscriptomics settings where also expression level information can be confounded with the state of conservation. Here, we applied the correction only for complete proteomes of species because the number of spectra per species was limited. In large-scale experiments, it should also be feasible to adjust for protein groups separately.
With regard to quantification, Pipasic currently relies exclusively on spectral counts. While we observe positive results for both the expression level correction and the quantification in the cowpox virus experiment, spectral counts have been shown to have limitations with regard to the quantitative range and precision that they cover. Methods combining the intensity of mass spectra with spectral counts, e.g. Dicker et al. (2010), could in principle be integrated into the Pipasic framework and further improve the quantification exactness.
One general difficulty for metaproteomics is that all analyses depend on the completeness of the provided reference proteomes because purely de novo peptide identification approaches are not yet sufficiently reliable (Hettich et al., 2013). Thus, any quantification or identification by Pipasic is also at risk of only reflecting the available reference proteomes. Using an error-tolerant peptide search strategy such as BICEPS (Renard et al., 2012), peptides containing up to two amino acid substitutions can be included and thereby this risk can be reduced.
Pipasic is currently not optimized for large-scale datasets and can become computationally expensive because peptide identifications need to be performed separately against all reference proteomes and all pairwise string comparisons need to be computed and accounted for. To overcome this, a two-step procedure may be helpful to first identify all species having unique identifications with existing methodology and then to run Pipasic on those subsets that are expected to have a high sequence similarity to ensure specificity of results.
5 CONCLUSION
With this contribution, we introduced Pipasic as a tool for identification and quantification in metaproteomics. Pipasic focuses on correcting observed proteome abundances without having to exclusively assign ambiguous peptide spectrum matches to their correct origin among a potentially large number of reference proteomes. Its particular strength is that it computes the peptide level similarity between reference proteomes and thereby can reliably distinguish on the strain level. Further, Pipasic includes the expression level in the analysis and thereby avoids bias resulting from the correlation of conservation and expression in metaproteomics. Pipasic is implemented in Python and freely available as an open-source project.
ACKNOWLEDGEMENTS
The authors would like to acknowledge all members of the Research Group Bioinformatics (NG 4, Robert Koch Institute) for critical discussion and Mathias Kuhring for critical reading of the manuscript.
Funding: B.Y.R. acknowledges financial support by Deutsche Forschungsgemeinschaft (DFG), grant number (RE3474/2-1).
Conflict of Interest: none declared.
REFERENCES
- Altschul SF, et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bielow C, et al. MSSimulator: simulation of mass spectrometry data. J. Proteome Res. 2011;10:2922–2929. doi: 10.1021/pr200155f. [DOI] [PubMed] [Google Scholar]
- Bradshaw RA, et al. Reporting protein identification data: the next generation of guidelines. Mol. Cell. Proteomics. 2006;5:787–788. doi: 10.1074/mcp.E600005-MCP200. [DOI] [PubMed] [Google Scholar]
- Chourey K, et al. Environmental proteomics reveals early microbial community responses to biostimulation at a uranium- and nitrate-contaminated site. Proteomics. 2013;13:2921–2930. doi: 10.1002/pmic.201300155. [DOI] [PubMed] [Google Scholar]
- Denef VJ, et al. Proteogenomic basis for ecological divergence of closely related bacteria in natural acidophilic microbial communities. Proc. Natl Acad. Sci. USA. 2010;107:2383–2390. doi: 10.1073/pnas.0907041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diament BJ, Noble WS. Faster SEQUEST searching for peptide identification from tandem mass spectra. J. Proteome Res. 2011;10:3871–3879. doi: 10.1021/pr101196n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicker L, et al. Increased power for the analysis of label-free LC-MS/MS proteomics data by combining spectral counts and peptide peak attributes. Mol. Cell. Proteomics. 2010;9:2704–2718. doi: 10.1074/mcp.M110.002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, et al. Least angle regression. Ann. Stat. 2004;32:407–499. [Google Scholar]
- Fouts DE, et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med. 2012;10:174. doi: 10.1186/1479-5876-10-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, et al. Open mass spectrometry search algorithm. J. Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- Hettich RL, et al. Metaproteomics: harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Anal. Chem. 2013;85:4203–4214. doi: 10.1021/ac303053e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, et al. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap P, et al. Deep metaproteomic analysis of human salivary supernatant. Proteomics. 2012a;12:992–1001. doi: 10.1002/pmic.201100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap P, et al. Workflow for analysis of high mass accuracy salivary data set using MaxQuant and ProteinPilot search algorithm. Proteomics. 2012b;12:1726–1730. doi: 10.1002/pmic.201100097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap P, et al. A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics. 2013;13:1352–1357. doi: 10.1002/pmic.201200352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong K, et al. False discovery rates in spectral identification. BMC Bioinformatics. 2012;13(Suppl. 16):S2. doi: 10.1186/1471-2105-13-S16-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson R, et al. Strain-level typing and identification of bacteria using mass spectrometry-based proteomics. J. Proteome Res. 2012;11:2710–2720. doi: 10.1021/pr2010633. [DOI] [PubMed] [Google Scholar]
- Lai B, et al. A de novo metagenomic assembly program for shotgun DNA reads. Bioinformatics. 2012;28:1455–1462. doi: 10.1093/bioinformatics/bts162. [DOI] [PubMed] [Google Scholar]
- Lindner MS, Renard BY. Metagenomic abundance estimation and diagnostic testing on species level. Nucleic Acids Res. 2013;41:e10. doi: 10.1093/nar/gks803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner MS, et al. Analyzing genome coverage profiles with applications to quality control in metagenomics. Bioinformatics. 2013;29:1260–1267. doi: 10.1093/bioinformatics/btt147. [DOI] [PubMed] [Google Scholar]
- Lo I, et al. Strain-resolved community proteomics reveals recombining genomes of acidophilic bacteria. Nature. 2007;446:537–541. doi: 10.1038/nature05624. [DOI] [PubMed] [Google Scholar]
- Mehlan H, et al. Data visualization in environmental proteomics. Proteomics. 2013;13:2805–2821. doi: 10.1002/pmic.201300167. [DOI] [PubMed] [Google Scholar]
- Muth T, et al. Searching for a needle in a stack of needles: challenges in metaproteomics data analysis. Mol. Biosystems. 2013;9:578–585. doi: 10.1039/c2mb25415h. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics. 2010;73:2092–2123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant TE. Python for scientific computing. Comput. Sci. Eng. 2007;9:10–20. [Google Scholar]
- Renard BY, et al. NITPICK: peak identification for mass spectrometry data. BMC Bioinformatics. 2008;9:355. doi: 10.1186/1471-2105-9-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard BY, et al. Estimating the confidence of peptide identifications without decoy databases. Anal. Chem. 2010;82:4314–4318. doi: 10.1021/ac902892j. [DOI] [PubMed] [Google Scholar]
- Renard BY, et al. Overcoming species boundaries in peptide identification with Bayesian information criterion-driven error-tolerant peptide search (BICEPS) Mol. Cell. Proteomics. 2012 doi: 10.1074/mcp.M111.014167. 11, M111.014167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooijers K, et al. An iterative workflow for mining the human intestinal metaproteome. BMC Genomics. 2011;12:6. doi: 10.1186/1471-2164-12-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, et al. Structure and function of the symbiosis partners of the lung lichen (Lobaria pulmonaria L. Hoffm.) analyzed by metaproteomics. Proteomics. 2011;11:2752–2756. doi: 10.1002/pmic.201000679. [DOI] [PubMed] [Google Scholar]
- Seifert J, et al. Bioinformatic progress and applications in metaproteogenomics for bridging the gap between genomic sequences and metabolic functions in microbial communities. Proteomics. 2013;13:2786–2804. doi: 10.1002/pmic.201200566. [DOI] [PubMed] [Google Scholar]
- Tanner S, et al. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 2005;77:4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- Teeling H, Glockner FO. Current opportunities and challenges in microbial metagenome analysis–a bioinformatic perspective. Brief. Bioinform. 2012;13:728–742. doi: 10.1093/bib/bbs039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooley JC, et al. A primer on metagenomics. PLoS Comput. Biol. 2010;6:e1000667. doi: 10.1371/journal.pcbi.1000667. [DOI] [PMC free article] [PubMed] [Google Scholar]