

Abstract
Motivation: Identifying regulatory elements is a fundamental problem in the field of gene transcription. Motif discovery—the task of identifying the sequence preference of transcription factor proteins, which bind to these elements—is an important step in this challenge. MEME is a popular motif discovery algorithm. Unfortunately, MEME’s running time scales poorly with the size of the dataset. Experiments such as ChIP-Seq and DNase-Seq are providing a rich amount of information on the binding preference of transcription factors. MEME cannot discover motifs in data from these experiments in a practical amount of time without a compromising strategy such as discarding a majority of the sequences.
Results: We present EXTREME, a motif discovery algorithm designed to find DNA-binding motifs in ChIP-Seq and DNase-Seq data. Unlike MEME, which uses the expectation-maximization algorithm for motif discovery, EXTREME uses the online expectation-maximization algorithm to discover motifs. EXTREME can discover motifs in large datasets in a practical amount of time without discarding any sequences. Using EXTREME on ChIP-Seq and DNase-Seq data, we discover many motifs, including some novel and infrequent motifs that can only be discovered by using the entire dataset. Conservation analysis of one of these novel infrequent motifs confirms that it is evolutionarily conserved and possibly functional.
Availability and implementation: All source code is available at the Github repository http://github.com/uci-cbcl/EXTREME.
Contact: xhx@ics.uci.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Transcription factors (TFs) are proteins that play an important role in transcriptional regulation by promoting or blocking the recruitment of RNA polymerase II. They can bind specifically to recognition sequences on the genome or to other TFs in a complex. High-throughput assays generate a rich amount of information on the sequence preference of TFs. ChIP-Seq (Johnson et al., 2007) can provide the genome-wide binding sites of a single TF. DNase-Seq, which sequences open chromatin regions in the genome, can provide single nucleotide resolution for the binding sites of many TFs (Hesselberth et al., 2009). When sequenced deep enough, binding sites appear as dips, or footprints (FPs), in the DNase-Seq signal. FPs only identify the locations of the TF binding sites; they do not identify the proteins that are bound there. These assays can provide functional information for thousands to millions of base pair regions in the genome.
The task of identifying the sequence preference of a TF is called motif discovery. Motif discovery algorithms can be classified as either search-based or probabilistic. Search-based algorithms infer motifs as consensus sequences. Probabilistic algorithms infer motifs as position frequency matrices (PFMs), which specify the frequency of nucleotides for each position in the binding site.
While PFMs provide more information about a TF’s binding specificity than consensus sequences, inferring PFMs is not always practical. Probabilistic motif discovery programs usually use algorithms such as expectation-maximization (EM) (Dempster et al., 1977) for inference. These algorithms scale poorly with dataset size. Search-based algorithms are therefore preferred for large datasets. DREME (Bailey, 2011) is an example of a search-based algorithm designed for large datasets.
MEME is a popular probabilistic motif discovery program (Bailey and Elkan, 1994). It uses the EM algorithm to infer PFMs. Since its inception in 1994, it has gone through several versions. However, MEME scales poorly with large datasets. One strategy to improve MEME’s performance is to discard many of the sequences. This is the strategy used by MEME-ChIP (Machanick and Bailey, 2011). However, discarding sequences can decrease the chance of discovering motifs corresponding to infrequent cofactors. Another strategy, as used in STEME, applies suffix trees to accelerate MEME (Reid and Wernisch, 2011). However, STEME is only practical for finding motifs of up to width 8 on large datasets because its efficiency tails off quickly as the motif width increases. Other strategies for accelerating MEME involve specialized hardware such as parallel pattern matching chips on PCI cards (Sandve et al., 2006). However, these implementations require hardware not available to most researchers.
To overcome these issues, we propose an online implementation of the MEME algorithm that we have named EXTREME. The online EM algorithm sticks closely to the original EM algorithm (hereafter referred to as the batch EM algorithm) (Cappé and Moulines, 2009). Normally, the online EM algorithm is designed for cases where not all data can be stored at once. Although most computers have enough memory to store entire sequence datasets at once, the online EM algorithm is still advantageous for motif discovery because, for large sample sizes, the online EM algorithm is more efficient, from a computational point of view, than the batch EM algorithm. We show that many of the features of the original MEME algorithm can be adapted to the online methodology. Furthermore, we show that EXTREME can achieve similar results to MEME in a fraction of the execution time. We also show that using the entire dataset is necessary to discover infrequent motifs, which is not always practical to do with MEME. To the best of our knowledge, this is the first application of the online EM algorithm to motif discovery.
2 MATERIALS AND METHODS
2.1 MEME
The original MEME algorithm applies the batch EM algorithm to infer PFMs. Here, we provide a brief overview of MEME’s model and how MEME applies the batch EM algorithm to infer parameters.
2.1.1 MEME’s model
Let 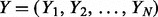 be the dataset of sequences, where N is the number of sequences in the dataset. Each sequence is over the alphabet
be the dataset of sequences, where N is the number of sequences in the dataset. Each sequence is over the alphabet 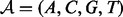 . MEME uses a mixture model that breaks up the dataset into all n (overlapping) subsequences of length W, which it contains. We will refer to this new dataset as
. MEME uses a mixture model that breaks up the dataset into all n (overlapping) subsequences of length W, which it contains. We will refer to this new dataset as 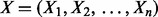 . The mixture model is a two-component model that assumes each subsequence is either an instance of the motif or background. Other variants of MEME place additional constraints. The one occurrence per sequence variant assumes that each sequence contains one instance of the motif. The zero or one occurrence per sequence variant assumes each sequence can have zero or only one occurrence of the motif. These two variants make slight modifications to MEME’s probabilistic model. We will only consider the two-component model.
. The mixture model is a two-component model that assumes each subsequence is either an instance of the motif or background. Other variants of MEME place additional constraints. The one occurrence per sequence variant assumes that each sequence contains one instance of the motif. The zero or one occurrence per sequence variant assumes each sequence can have zero or only one occurrence of the motif. These two variants make slight modifications to MEME’s probabilistic model. We will only consider the two-component model.
The background component is characterized as a zero-order Markov model parameterized by the vector 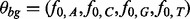 where f0,k is the background frequency of letter k. The motif model is characterized by the PFM
where f0,k is the background frequency of letter k. The motif model is characterized by the PFM 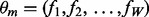 . Each fj = (fj,A, fj,C, fj,G, fj,T) is a parameter of an independent random variable describing a multinomial trial representing the distribution of letters at position j in the motif.
. Each fj = (fj,A, fj,C, fj,G, fj,T) is a parameter of an independent random variable describing a multinomial trial representing the distribution of letters at position j in the motif. 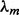 parameterizes the probability that any W-mer is generated by the motif model while
parameterizes the probability that any W-mer is generated by the motif model while 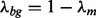 is the probability that any W-mer is generated by the background model.
is the probability that any W-mer is generated by the background model. 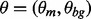 and
and 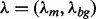 are unknown parameters that are inferred from the known data X. Therefore, the MEME model is
are unknown parameters that are inferred from the known data X. Therefore, the MEME model is
| (1) |
| (2) |
where Zi is a binary latent variable that has a value of 1 if Xi is drawn from the motif model or 0 if Xi is drawn from the background model. Zi’s true value is unknown, but its conditional expected value, defined here as 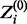 , for a given set of parameters can be calculated as follows:
, for a given set of parameters can be calculated as follows:
| (3) |
To calculate 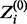 , we need to know the form of
, we need to know the form of 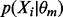 and the form of
and the form of 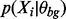 . MEME assumes the distributions of the motif class and background class are
. MEME assumes the distributions of the motif class and background class are
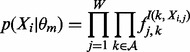 |
(4) |
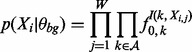 |
(5) |
where Xi,j is the letter in the jth positon of subsequence Xi, and I(k,a) is an indicator function
| (6) |
2.1.2 Batch EM
λ and θ are iteratively improved in the batch EM algorithm. In the E-step, the expected counts of all nucleotides at each position are calculated based on the current guess of the parameters. In the M-step, the parameters are updated based on the values calculated in the E-step. MEME repeats the E and M steps until the change in 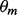 (Euclidean distance) falls below a threshold (default: 10−6). The E and M steps are as follows:
(Euclidean distance) falls below a threshold (default: 10−6). The E and M steps are as follows:
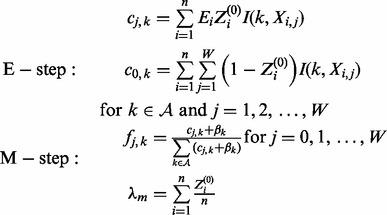 |
To discover multiple motifs, MEME associates an ‘erasing factor’ Ei for each position in the data. The erasing factors vary between 0 and 1 and are set to 1 initially to indicate no erasing has taken place. Each time a motif is discovered, the erasing factors are reduced by a factor representing the probability that the position overlaps an occurrence of that motif. More details concerning how MEME erases are in Bailey and Elkan (1995a). MEME also implements pseudo counts 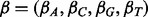 in the M-step to prevent any letter frequency fj,k from becoming 0. This is because if any letter frequency fj,k becomes 0, its value cannot change.
in the M-step to prevent any letter frequency fj,k from becoming 0. This is because if any letter frequency fj,k becomes 0, its value cannot change.
EM performs maximum likelihood estimation to maximize an objective function. The new estimates in the M-step are always guaranteed to increase the value of the objective function. As the E and M steps are repeated, EM algorithms converge to a maximum. For MEME, the objective function is the expected value of the log likelihood of the model parameters θ and λ given the joint distribution of the data X and missing data Z:
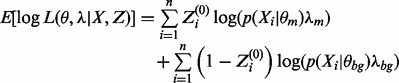 |
(7) |
2.1.3 Seeding
The EM algorithm is sensitive to initial conditions and prone to converging to local maxima. To mitigate this problem, MEME tests many seeds and runs the EM algorithm to convergence from the ‘best’ seed. The exact details of how MEME performs seeding can be found in Bailey and Elkan (1995b).
2.1.4 Scoring the motifs
Motif instances are determined according to Bayesian decision theory. After a motif is discovered, a subsequence Xi is classified as being an occurrence of the motif only if
| (8) |
For each motif discovered, MEME calculates its E-value. This E-value is the number of motifs, with the same width and number of occurrences, that can generate an equal or higher log likelihood ratio if the dataset had been generated according to background model. The log likelihood ratio 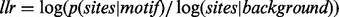 is a measure of how different the sites are from the background model. Calculating the E-value exactly can be time consuming, so it is not computed directly. It is instead heuristically calculated as a function of the total information content and the number of occurrences (Bailey et al., 2010).
is a measure of how different the sites are from the background model. Calculating the E-value exactly can be time consuming, so it is not computed directly. It is instead heuristically calculated as a function of the total information content and the number of occurrences (Bailey et al., 2010).
2.1.5 Time complexity
For each iteration of the batch EM algorithm, the number of operations performed is approximately proportional to W. Each batch EM iteration has a time complexity of O(nW). Although the number of iterations can vary, it is typically proportional to n. Therefore, the algorithm scales quadratically with the size of the dataset and has a time complexity of O(n2W). The seed searching also scales quadratically with the size of the dataset (Bailey and Elkan, 1995b).
2.2 EXTREME
EXTREME shares many similarities with MEME, especially in the implementation. At the center of the EXTREME algorithm is the online EM algorithm. We provide an overview of the online EM algorithm and how EXTREME implements the online EM algorithm to discover motifs.
2.2.1 Online EM
Like the batch EM algorithm, the online EM algorithm also repeatedly iterates between E and M steps, which update the parameters. In contrast to the batch EM algorithm, each iteration of the online EM algorithm operates on only one observation, Xi, instead of the whole dataset X.
Following the instructions in Cappé and Moulines (2009), the E and M steps, as derived from (7), are as follows:
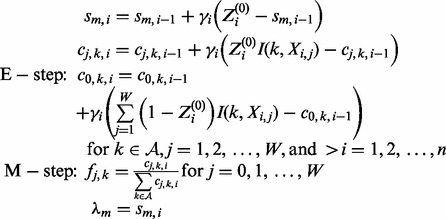 |
The step size is 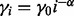 . α and
. α and 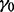 are set to 0.6 and 0.05, respectively. These are by no means the most optimized set of parameters, but they are adequate for accurate motif discovery. As shown in Cappé and Moulines (2009), the online EM algorithm converges to a local maximum of the likelihood function (7) for
are set to 0.6 and 0.05, respectively. These are by no means the most optimized set of parameters, but they are adequate for accurate motif discovery. As shown in Cappé and Moulines (2009), the online EM algorithm converges to a local maximum of the likelihood function (7) for 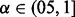 .
.
The E and M steps are repeated until a convergence threshold (default: 10−6) in terms of the symmetrized Kullback–Leibler divergence between the PFM estimates at a user-defined number of intervals (default: 100) of W-mers at the end of a complete pass through the dataset is satisfied. The Kullback–Leibler divergence between two PFMs A and B is calculated as follows:
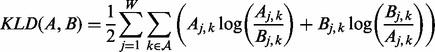 |
(9) |
If convergence is not reached at the end of a pass, the exponent α is updated to the midpoint between α’s current value and one and EXTREME performs another pass through the dataset. EXTREME repeats these steps until the convergence threshold is met.
To accommodate pseudo counts, we modify the indicator function from (6):
| (10) |
By default, EXTREME sets 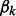 to 0.0001 times the frequency of letter k in the entire dataset.
to 0.0001 times the frequency of letter k in the entire dataset.
To accommodate reverse complements, we also modify the calculation of 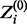 from (3) so that for each Xi, the reverse complement is also evaluated and
from (3) so that for each Xi, the reverse complement is also evaluated and 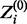 takes the higher of the two values. MEME, in contrast, handles reverse complements by adding a reverse-complemented copy of the data, essentially doubling the size of the data.
takes the higher of the two values. MEME, in contrast, handles reverse complements by adding a reverse-complemented copy of the data, essentially doubling the size of the data.
2.2.2 Seeding
Before running the online EM algorithm, the order of the W-mers Xi is randomized. The online EM algorithm is therefore a stochastic algorithm. This means that different runs of the online EM algorithm can yield different results, even if ran multiple times from the same initial conditions. This can present a problem for seeding because even using the best seed from MEME’s heuristic is not guaranteed to generate the optimal or even consistent solutions, causing EXTREME to converge to local maxima. On the other hand, this also means that seeds that would yield non-optimal solutions in MEME can yield optimal solutions in EXTREME. In fact, local maxima may actually correspond to biologically relevant motifs, especially in datasets that are rich in motifs such as DNase-Seq data. Furthermore, an efficient online EM implementation of MEME offers little benefit if runtimes are dominated by the inefficient seed search.
EXTREME’s seeding strategy applies a search-based motif discovery algorithm to find motifs to initialize the online EM algorithm. Similar to DREME (Bailey, 2011), the seeding algorithm finds words that are enriched in a sequence dataset relative to a negative sequence dataset. We use the same dinucleotide shuffle algorithm used in DREME to generate a dinucleotide-shuffled version of the input sequence set as the negative sequence set. The seeding algorithm counts the number of occurrences of words in the positive sequence set and the negative sequence set and associates a ‘z-score’ with each word. The z-score is given by
| (11) |
where s+ and s− are the number of occurrences of the word in the positive sequence and negative sequence sets, respectively. If s− is zero for a word, it is changed to one to prevent division by zero. Unlike DREME, our seeding algorithm searches for words that are not exact. Each word contains g universal wildcard letters surrounded by flanking sites of l unambiguous letters. For example, TCAGNNGGAC is a word with a gap length, g, of 2 and a half length, l, of 4. The gap length, g, varies between the user-defined parameters gmin and gmax. Z-scores for each value of g are normalized by dividing by the standard deviation of all z-scores for each respective value of g. Words that have a normalized z-score that exceed a user-defined threshold, zthresh, and have at least a user-defined number of occurrences, smin, in the positive sequence set are aligned and grouped together using a hierarchical clustering algorithm we adapted from Xie et al. (2005). Word clusters are converted to frequency count matrices by counting the number of occurrences of each letter at each position along the alignment. The counts are weighted by the normalized z-score of each word in a cluster so that more significant words will contribute more to the count matrix than less significant words. A count matrix is converted to a PFM, 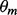 , by dividing each matrix element by its respective row sum. The initial expected counts, c, is initially set to the initial
, by dividing each matrix element by its respective row sum. The initial expected counts, c, is initially set to the initial 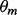 as well.
as well. 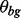 and the expected background counts c0 are set to the nucleotide frequency in the dataset.
and the expected background counts c0 are set to the nucleotide frequency in the dataset. 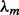 and sm,0 are initialized to the predicted number of motif occurrences divided by n, the total number of W-mers. We predict the number of motif occurrences for a given PFM seed as the number of W-mers that have a goodness-of-fit score >0.7 [see Pan and Phan (2009) for details].
and sm,0 are initialized to the predicted number of motif occurrences divided by n, the total number of W-mers. We predict the number of motif occurrences for a given PFM seed as the number of W-mers that have a goodness-of-fit score >0.7 [see Pan and Phan (2009) for details].
We also alter the form of 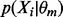 from (4):
from (4):
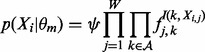 |
(12) |
The bias factor ψ has a value between 0 and 1. A bias factor closer to 0 biases the motif discovery toward subsequences that more closely match the current motif guess, decreasing the number of discovered motif occurrences. A bias factor closer to 1 makes the motif discovery less selective, increasing the number of motif occurrences. After convergence, motif occurrences are identified using (8). ψ is initially set to 1, and its value is varied in a binary search fashion until the number of discovered motif occurrences is between sitesmin (default: 10) and sitesmax (default: 5 times the number of predicted motif sites). Up to 15 different values of ψ are tried before EXTREME stops. Because each initial PFM guess can be tested independently, this seeding strategy can be parallelized to allow multiple motifs to be discovered simultaneously. Hierarchical clustering of the discovered motifs can then identify individual motif classes.
2.2.3 Time complexity
Each pass through the dataset with the online EM algorithm has a time complexity of O(nW). Typically, the online EM algorithm reaches convergence after one to five passes through the data, so the overall time complexity is proportional to the width of the motif and the size of the dataset. The seeding algorithm’s word search also scales linearly with the dataset size, while the hierarchical clustering is inefficient and can scale cubically with the number of words to cluster. In practice, EXTREME as a whole scales linearly in time complexity with the dataset size.
2.2.4 Implementation
EXTREME is written in Python and is available on Github. To calculate E-values, EXTREME uses Cython bindings to the original MEME C source code to call the appropriate functions. EXTREME requires ∼8 GB of memory for a 10 Mb dataset. Most of the memory is devoted to MEME’s E-value calculation, which involves a preprocessing step that does not scale well to large numbers of motif sites.
3 RESULTS
MEME is a popular motif discovery algorithm. It has been a valuable tool in the ongoing challenge of identifying regulatory elements. However, its performance scales poorly with large datasets. Experiments such as ChIP-Seq and DNase-Seq generate data that are too large for MEME to process in a practical amount of time without discarding most of the data. To overcome this challenge, we have developed EXTREME, a motif discovery algorithm that can process ChIP-Seq and DNase-Seq data efficiently without discarding any data. We first show, using simulated datasets, that MEME’s running time scales much faster than EXTREME’s running time with respect to dataset size. Using a ChIP-Seq dataset and a DNase-Seq dataset, we demonstrate that using the entire dataset of sequences is necessary to discover infrequent motifs. We also show that the motifs discovered by EXTREME are similar in quality to the motifs discovered by MEME.
3.1 Comparison of MEME and EXTREME performance
We compare MEME and EXTREME using several simulated datasets. Simulated datasets are generated with the RSAT suite of tools (Thomas-Chollier et al., 2011). We generate four sequence datasets, each containing 1000 random masked hg19 genomic sequences of a single length (100, 200, 300 or 400 bp), using the RSAT random-genome-fragments tool. This masked reference genome was preprocessed with RepeatMasker (Smit et al., 1996–2010) and Tandem Repeats Finder (Benson, 1999) so that repeats (with period of ≤12) are masked by capital Ns. For each of the four sequence datasets, we implant 50, 100, 500 or 1000 instances of the JASPAR (Sandelin et al., 2004) VDR/RXRA heterodimer motif (Supplementary Fig. S1) using the RSAT random-motifs and implant-sites tools, yielding 16 simulated datasets, each containing 1000 sequences of varying lengths and number of motif sites.
For the seeding step of each EXTREME run, we search for words with a half-length l = 6, a gap length g between gmin = 0 and gmax = 2, a normalized z-score greater than the threshold zthresh = 5 and at least smin = 5 occurrences in the positive sequence set. The words are clustered and we select the cluster containing the most words to convert to a PFM seed from which to initialize the online EM algorithm. Because the online EM algorithm is a stochastic algorithm, we repeat the online EM portion of the run 30 times for each dataset with different random seeds to initialize the pseudorandom number generator to get a good estimate of performance. We also run MEME on each of the 16 simulated datasets to find a single motif of a width between 12 and 17 under the two-component model to approximate the same parameters for the EXTREME run. Figure 1 shows that MEME’s running time scales much faster than EXTREME’s running time with respect to the input size for all ‘noise’ levels. Extrapolating from these data, MEME can take weeks to discover a motif in a 10 Mb dataset. EXTREME can complete this same task in hours. With the exception of one of the datasets, MEME is marginally more accurate than EXTREME in each case (Supplementary Fig. S3). In the one exception, MEME fails to converge to the correct motif because there are not enough true motif occurrences relative to the dataset size for MEME’s seeding algorithm to pick a good seed. As the number of motif occurrences increases, both MEME and EXTREME better approximate the true PFM and the relative difference between their results diminish. Although EXTREME’s running time and accuracy vary more as the noise level increases (Supplementary Figs. S2 and S3), EXTREME still consistently generates results comparable with those of MEME in a fraction of MEME’s running time.
Fig. 1.
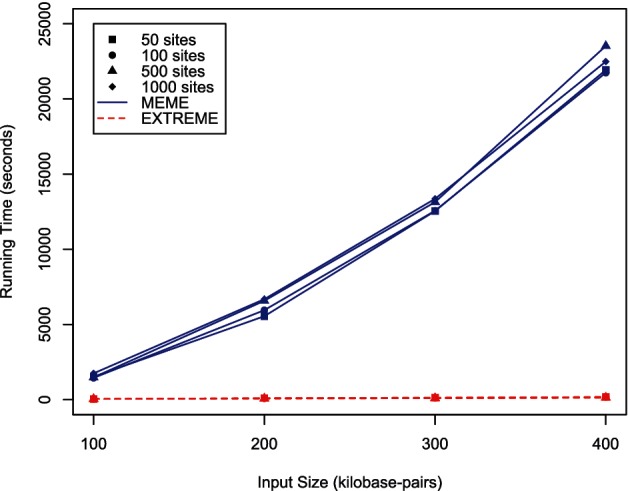
Comparison of MEME and EXTREME performance on simulated datasets of varying sequence length and motif sites. The x-axis is the total number of base pairs in the simulated dataset. The y-axis is the total running time it takes for MEME or EXTREME to complete seeding and reach convergence
3.2 Discovering motifs in ChIP-Seq data
We compare the performance of MEME and EXTREME for discovering motifs in ChIP-Seq data using a dataset generated by the Myers Lab at the Hudson Alpha Institute for Biotechnology (Birney et al., 2007). The ChIP-Seq data correspond to an NRSF ChIP performed on the GM12878 cell line. Peaks were already called and organized into BED files by the authors. We further process the data by intersecting replicates and extracting genomic sequences from the middle 100 bp of the intersected regions from the same hg19 masked reference genome we use for the simulated data. The resulting sequence dataset consists of 2849 sequences and 282 980 bp.
We run EXTREME on the ChIP-Seq dataset to discover multiple motifs. For the seeding step, we search for words with a half-length of 8 bp, a gap length between 0 and 10 bp (inclusive), a normalized z-score >5 and at least five occurrences in the positive sequence set. The word search takes 32 s to find 1248 words. Hierarchical clustering groups these words into 23 clusters, taking 91 s to complete. These 23 clusters are converted to PFMs of widths between 16 and 29 bp, providing seeds for the online EM algorithm. Each seed is processed by the online EM algorithm on a separate core in parallel. Of the 23 seeds, 20 successfully yield motifs within 15 different values of the bias factor ψ (12). The Supplementary Material contains these 20 motifs in MEME Minimal Motif Format. Hierarchical clustering of the 20 motifs groups them into 10 clusters. Online EM running times range from 67 to 859 s, taking an average of 361 s. Running times vary because different seeds can converge to different motifs and may require additional passes through the data to reach convergence. For comparison, we also run MEME on the ChIP-Seq dataset to find a single motif of a width between 16 and 29 bp under the two-component model using a single core. MEME takes 8191 s to find a single motif. While comparison between the multi-core EXTREME run to the single-core MEME run is not straight-forward, it should be noted that the total computing time for EXTREME, which sums the running times for the seeding and each of the online EM runs, is 8305 s. In the computing time it takes for MEME to discover a single motif, EXTREME finds 10 motif clusters in roughly the same amount of time. The disparity between the two programs’ performances is compounded by the fact that MEME discovers multiple motifs in serial, and would require roughly the same running time to find each additional motif.
Many of the discovered motifs are novel, demonstrating varying half-site distances and orientations (Fig. 2). Interestingly, two of the discovered motifs show that the half-sites are reversed. To determine whether the reversed motif is functional, we scan for sequences in the ChIP-Seq dataset matching one of the reversed motifs’ consensus sequence, align these sequences and extract GERP scores (Cooper et al., 2005). Sequences containing this reversed motif are enriched in high GERP scores, showing that these sequences are conserved and possibly functional (Fig. 3).
Fig. 2.
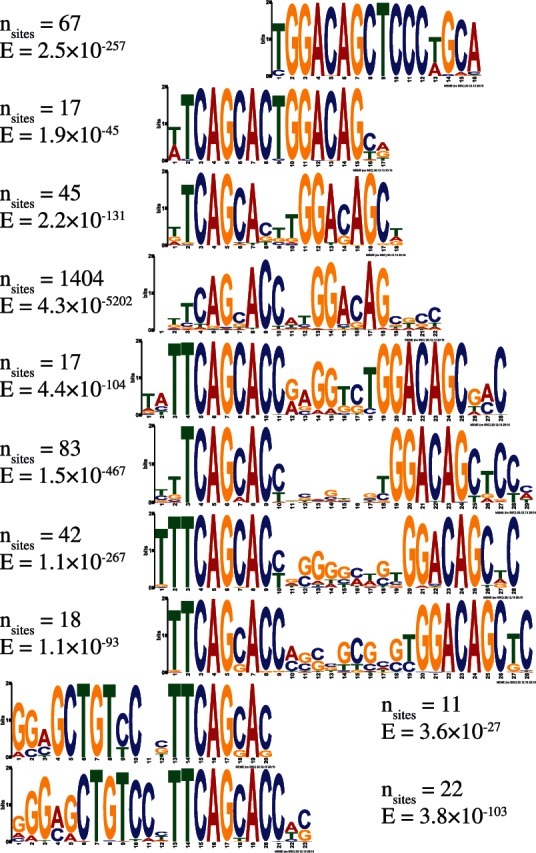
Motifs discovered by EXTREME in the GM12878 NRSF ChIP-Seq dataset. Each motif comes from one of the 10 motif clusters. Motifs are aligned to highlight the varying distances and orientations between the half-sites. Number of non-overlapping motif sites in non-repetitive regions and E-values shown next to each motif. E-values are calculated according to MEME’s heuristic
Fig. 3.
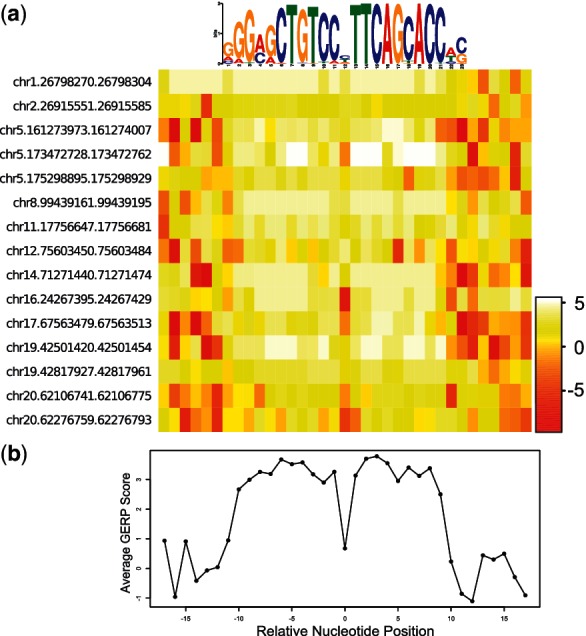
Conservation analysis of the reversed NRSF motif. Sequences in the GM12878 NRSF ChIP-Seq dataset containing the consensus sequence GCTGTCCNTTCAGCA or its reverse complement are aligned with 10 bp flanking sequences. (a) GERP scores are plotted for each nucleotide in a heatmap. The motif’s sequence logo is aligned at the top for reference. (b) The average GERP score plotted against the genomic positions, relative to the center of the alignment
Some of the motifs discovered in this dataset have a low number of occurrences. One of the motifs, for example, only has 11 sites in the data. It would be unlikely to discover these infrequent motifs if the majority of sequences are discarded. This highlights the importance of using the entire dataset for thorough motif discovery.
3.3 Discovering motifs in DNase-Seq data
To assess the performance of MEME and EXTREME for DNase-Seq data, we use a DNase-Seq FP dataset generated by the Stamatoyannopoulos Lab at the University of Washington (Neph et al., 2012). The DNase-Seq data correspond to a footprinting experiment performed on the K562 cell line. FPs are already organized into a BED file by the authors. We further process the FP data by extending each FP by 5 bp on each side and then merging any intersecting regions. Genomic sequences are extracted from the masked hg19 reference genome. The resulting dataset consists of 198 527 sequences and 10 487 345 bp.
We first discover motifs in the DNase-Seq dataset using MEME. We do not run MEME on the whole dataset because we know MEME can take months to complete for a dataset of this size. We therefore run MEME-ChIP on the dataset, which runs MEME on 600 randomly selected sequences. For the data subset, MEME discovers two motifs that strongly resemble previously discovered motifs (CTCF and SP1). The other discovered motifs are repetitive or fail to meet our E-value threshold of 0.01.
In the seeding step of EXTREME, we first search for words with a half-length of 4 bp, a gap length between 0 and 10 bp (inclusive), a normalized z-score >5 and at least 10 occurrences in the positive sequence dataset. The word search takes 836 s to yield 761 words. Hierarchical clustering of the words takes 23 s to group the words into 129 clusters. We then convert the clusters to 129 PFM seeds of widths between 8 and 19 bp. Each seed is processed independently by the online EM algorithm on a separate core in parallel. Running times vary for each of the online EM runs, ranging from 4475 to 18300 s, completing in an average of 7390 s. Hierarchical clustering groups the discovered motifs into 22 distinct clusters.
To discover additional motifs in the DNase-Seq data, we mask the seven most abundant motifs from different motif clusters by replacing instances of those motifs with capital Ns and restart the motif discovery. We remove these motif instances because the first round of motif discovery shows that many different seeds can converge to the same motif, and we want to bias the motif discovery toward different motifs. Based on TOMTOM (Gupta et al., 2007) analysis, the seven motifs strongly match known motifs (TOMTOM E < 0.01): CTCF, SP1, SRF, NRF1, JUNDM2, ZNF143 and TAL1/GATA1. In this second round of motif discovery, we search for words with a half-length of 5 bp, a gap length between 0 and 10 bp (inclusive), a normalized z-score > 8 and at least 10 occurrences in the positive sequence dataset. The word search takes 888 s to yield 1187 words. Hierarchical clustering of the words completes in 102 s and yields 357 clusters, which are then converted to PFM seeds of widths between 10 and 21 bp. Each seed is independently processed by the online EM algorithm on a separate core in parallel. Online EM run times range from 3330 to 22 702 s, completing in an average of 7605 s. Hierarchical clustering condense the motifs into 131 clusters.
Examples of motifs discovered in the K562 dataset are shown in Figure 4. All motifs discovered by EXTREME in the K562 dataset are available in MEME Minimal Motif Format in the Supplementary material. Many of the motifs discovered by EXTREME have a low number of occurrences relative to the total size of the dataset. One motif only has 464 occurrences in the 10.5 Mb dataset (Fig. 4e). These kinds of motifs are too infrequent to be discovered in subsets of the data. Discovering motifs in a subset of the data is only possible for motifs that are present in high abundance, such as the ones shown in Figure 4a and b, which are also the motifs discovered by the MEME run on the data subset. Again, this highlights the importance of using the whole dataset for motif discovery. Using MEME to discover these infrequent motifs is not practical because MEME can take months to discover a motif in a dataset as large as the K562 dataset. Furthermore, the number of occurrences for motifs is less than expected. For example, EXTREME only finds 1771 occurrences of the CCAAT box motif (Fig. 4f), even though the ENCODE NFYA ChIP-Seq data indicate it should be present in at least a third of all human promoters. The reason for the discrepancy is likely due to the way Neph et al. (2012) called FPs. Neph et al. (2012) reported high-confidence FPs at an FDR of 1%. This is a stringent threshold and we therefore expect their footprinting algorithm to call many false negatives as a result.
Fig. 4.
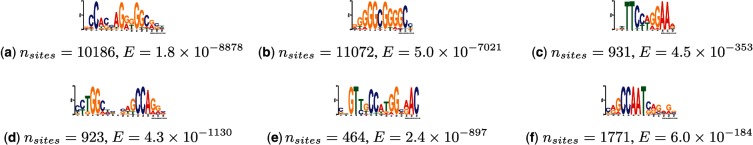
Six examples of motifs discovered by EXTREME in the K562 dataset. Number of non-overlapping motif sites in non-repetitive regions and E-values shown below each motif. The E-values show how significant the motifs are, calculated according to MEME’s heuristic
3.4 Comparison to known motifs
We assess the similarity of the motifs discovered by EXTREME in the DNase-Seq and ChIP-Seq datasets to known motifs using TOMTOM. Some of the motifs discovered by EXTREME have highly significant matches to known motifs (Fig. 5). Many of the motifs discovered, however, are novel and fail to meet our TOMTOM E-value threshold of 0.01. Validating these novel motifs requires further computational or experimental scrutiny.
Fig. 5.
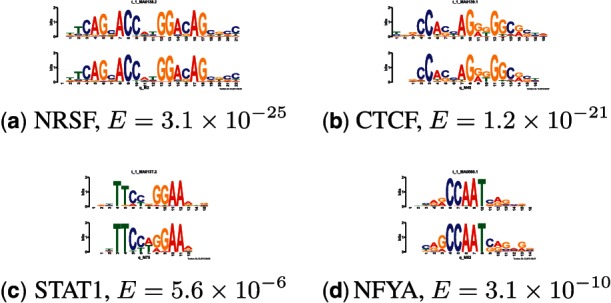
TOMTOM comparisons of motifs discovered by EXTREME with motifs in databases. Each panel shows the logo of the motif discovered by EXTREME (lower logo) aligned with the best matching motif in the databases (upper logo), along with the name of the best matching motif and significance value of the match
4 DISCUSSION
A search-based seeding strategy combined with the online EM algorithm is effective for efficient de novo motif discovery in large datasets. EXTREME uses the online EM algorithm to discover motifs that closely match motifs discovered by MEME. MEME can take months to discover even a single motif in a large dataset like the DNase-Seq dataset. While strategies such as discarding sequences are effective for quickly discovering abundant motifs, it is insufficient for finding infrequent motifs, which are numerous in DNase-Seq data. EXTREME can quickly process entire large datasets without discarding sequences or using specialized hardware. If available, EXTREME can take advantage of parallelized hardware configurations, which is useful for rapidly discovering multiple motifs in large datasets. Although such configurations are not available to all researchers, EXTREME can still be used with more traditional configurations to serially discover multiple motifs at a substantially faster rate than MEME can.
We expect EXTREME to be a valuable tool for thorough motif discovery in large datasets. Its ability to discover multiple motifs in DNase-Seq data will be especially useful for understanding transcriptional regulation. Because motifs discovered by EXTREME closely match motifs discovered by MEME, the results can be used to reliably associate FPs with well-studied TFs. This also means that any discovered novel motifs can confidently be associated with TFs. This is especially useful for the study of TFs that lack a suitable antibody for ChIP experiments.
While EXTREME is effective in motif discovery, there is still much room for improvement. EXTREME’s performance can be vastly improved if it were reimplemented in C. Future implementations of EXTREME can also incorporate more MEME elements such as the one occurrence per sequence and zero or one occurrence per sequence models. To encourage further investigation, we have made EXTREME publicly available at the Github repository http://github.com/uci-cbcl/EXTREME.
Funding: National Institute of Biomedical Imaging and Bioengineering, National Research Service Award (EB009418) from the University of California, Irvine, Center for Complex Biological Systems.
Conflict of Interest: none declared.
REFERENCES
- Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Mach. Learn. 1995a;21:51–80. [Google Scholar]
- Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995b;3:21–29. [PubMed] [Google Scholar]
- Bailey TL, et al. The value of position-specific priors in motif discovery using MEME. BMC Bioinformatics. 2010;11:179. doi: 10.1186/1471-2105-11-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappé O, Moulines E. On-line expectation–maximization algorithm for latent data models. J. R. Stat. Soc. Series B Stat. Methodol. 2009;71:593–613. [Google Scholar]
- Cooper GM, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster AP, et al. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Series B Methodol. 1977;39(1):1–38. [Google Scholar]
- Gupta S, et al. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Phan S. Threshold for positional weight matrix. Eng. Lett. 2009;16:498–504. [Google Scholar]
- Reid JE, Wernisch L. STEME: efficient EM to find motifs in large data sets. Nucleic Acids Res. 2011;39:e126. doi: 10.1093/nar/gkr574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, et al. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandve GK, et al. Algorithms in Bioinformatics. Berlin Heidelberg: Springer; 2006. Accelerating motif discovery: motif matching on parallel hardware; pp. 197–206. [Google Scholar]
- Smit A, et al. 1996–2010 RepeatMasker Open-3.0. [Google Scholar]
- Thomas-Chollier M, et al. RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res. 2011;39:W86–W91. doi: 10.1093/nar/gkr377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, et al. Systematic discovery of regulatory motifs in human promoters and 3’ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]