

Abstract
Motivation: Numerous competing algorithms for prediction in high-dimensional settings have been developed in the statistical and machine-learning literature. Learning algorithms and the prediction models they generate are typically evaluated on the basis of cross-validation error estimates in a few exemplary datasets. However, in most applications, the ultimate goal of prediction modeling is to provide accurate predictions for independent samples obtained in different settings. Cross-validation within exemplary datasets may not adequately reflect performance in the broader application context.
Methods: We develop and implement a systematic approach to ‘cross-study validation’, to replace or supplement conventional cross-validation when evaluating high-dimensional prediction models in independent datasets. We illustrate it via simulations and in a collection of eight estrogen-receptor positive breast cancer microarray gene-expression datasets, where the objective is predicting distant metastasis-free survival (DMFS). We computed the C-index for all pairwise combinations of training and validation datasets. We evaluate several alternatives for summarizing the pairwise validation statistics, and compare these to conventional cross-validation.
Results: Our data-driven simulations and our application to survival prediction with eight breast cancer microarray datasets, suggest that standard cross-validation produces inflated discrimination accuracy for all algorithms considered, when compared to cross-study validation. Furthermore, the ranking of learning algorithms differs, suggesting that algorithms performing best in cross-validation may be suboptimal when evaluated through independent validation.
Availability: The survHD: Survival in High Dimensions package (http://www.bitbucket.org/lwaldron/survhd) will be made available through Bioconductor.
Contact: levi.waldron@hunter.cuny.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Cross-validation and related resampling methods are de facto standard for ranking supervised learning algorithms. They allow estimation of prediction accuracy using subsets of data that have not been used to train the algorithms. This avoids over-optimistic accuracy estimates caused by ‘re-substitution’. This characteristic has been carefully discussed in Molinaro et al. (2005), Baek et al. (2009) and Simon et al. (2011). It is common to evaluate algorithms by estimating prediction accuracy via cross-validation for several datasets, with results summarized across datasets to rank algorithms (Boulesteix, 2013; Demšar, 2006). This approach recognizes possible variations in the relative performances of learning algorithms across studies or fields of application. However, it is not fully consistent with the ultimate goal, in the development of models with biomedical applications, of providing accurate predictions for fully independent samples, originating from institutions and processed by laboratories that did not generate the training datasets.
It has been observed that accuracy estimates of genomic prediction models based on independent validation data are often substantially inferior to cross-validation estimates (Castaldi et al., 2011). In some cases this has been attributed to incorrect application of cross-validation; however even strictly performed cross-validation may not avoid over-optimism resulting from potentially unknown sources of heterogeneity across datasets. These include differences in design, acquisition and ascertainment strategies (Simon et al., 2009), hidden biases, technologies used for measurements, and populations studied. In addition, many genomics studies are affected by experimental batch effects (Baggerly et al., 2008; Leek et al., 2010). Quantifying these heterogeneities and describing their impact on the performance of prediction algorithms is critical in the practical implementation of personalized medicine procedures that use genomic information.
There are potentially conflicting, but valid, perspectives on what constitutes a good learning algorithm. The first perspective is that a good learning algorithm should perform well when trained and applied to a single population and experimental setting, but it is not expected to perform well when the resulting model is applied to different populations and settings. We call such an algorithm specialist, in the sense that it can adapt and specialize to the population at hand. This is the mainstream perspective for assessing prediction algorithms and is consistent with validation procedures performed within studies (Baek et al., 2009; Molinaro et al., 2005; Simon et al., 2011). However, we argue that it does not reflect the reality that ‘samples of convenience’ and uncontrolled specimen collection are the norm in genomic biomarker studies (Simon et al., 2009).
We promote another perspective: a good learning algorithm should be generalist, in the sense that it yields models that may be suboptimal for the training population, or not fully representative of the dataset at hand, but that perform reasonably well across different populations and laboratories employing comparable but not identical methods. Generalist algorithms may be preferable in important settings, for instance when a researcher develops a model using samples from a highly controlled environment, but hopes the model to be applicable to other hospitals, labs, or more heterogeneous populations.
In this article we systematically use independent validations for the comparison of learning algorithms, in the context of microarray data for disease-free survival of estrogen receptor-positive breast cancer patients. Although concern has been often expressed about the lack of independent validation of genomic prediction models (Micheel et al., 2012; Subramanian and Simon, 2010), independent validation has not been systematically adopted in the comparison of learning algorithms. This deficiency cannot be addressed for prediction contexts where related, independent datasets are unavailable. For many cancer types, however, several micro-array studies have been performed to develop prognostic models. These datasets pave the way for a systematic approach based on independent validations. For instance, a recent meta-analysis of prognostic models for late-stage ovarian cancer provides a comparison of publicly available microarray datasets (Waldron et al., 2014). Furthermore, Riester et al. (2014) showed that combining training datasets can increase the accuracy of late-stage ovarian cancer risk models. Thus situations exist in genomic data analysis where comparable, independent datasets are available, and these present an opportunity to use independent validation as an explicit basis for assessing learning algorithms.
We propose what we term ‘leave-one-dataset-in’ cross-study validation (CSV) to formalize the use of independent validations in the evaluation of learning algorithms. Through data-driven simulations, and an example involving eight publicly available estrogen receptor-positive breast cancer microarray datasets, we assess established survival prediction algorithms using our ‘leave-one-dataset-in’ scheme and compare it to conventional cross-validation.
2 METHODS
2.1 Notation and setting
>We consider multiple datasets i = 1, … , I with sample sizes N1, … , NI. Each observation s appears only in one dataset i, i.e. datasets do not overlap, and the corresponding record includes a primary outcome and a vector of predictor variables throughout this article will be gene-expression measurements. Our goal is to compare the performance of different learning algorithms k = 1, … , K that generates prediction models for the primary outcome using the vector of predictors. Throughout this article, the primary outcome is a possibly censored survival time.
We are interested in evaluating and ranking competing prediction methods k = 1, … , K. Since the ranking may depend on the application, the first step is to define the prediction task of interest. We focus on the prediction of metastasis-free survival time in breast cancer patients based on high-throughput gene-expression measurements. Our approach and the concept of CSV, however, can be applied to different types of response variables and any other prediction task.
2.2 Algorithms
We assess six learning algorithms (k = 1, … , 6) appropriate for high-dimensional continuous predictors and possibly censored time-to-event outcomes: Lasso and Ridge regression (Goeman, 2010), CoxBoost (Binder and Schumacher, 2008), SuperPC (Blair and Tibshirani, 2004), Unicox (Tibshirani, 2009) and Plusminus (Zhao et al., 2013). All parameters were tuned either by default methods included in their implementation (Ridge and Lasso regression: R-package glmnet) or by testing a range of parameters in internal cross-validation. Our focus is not to provide a comprehensive array of algorithms, but simply to use a few popular, representative algorithms to investigate CSV.
2.3 CSV matrices
We refer in this article to m-fold cross-validation and related resampling methods collectively as cross-validation (CV). Our ranking procedure for learning algorithms is based on a square matrix Zk of scores (k = 1, … , K). The (i, j) element in the matrix measures how well the model produced by algorithm k trained on dataset i performs when validated on dataset j. Since we consider K methods we end up with K method-specific square matrices We set the diagonal entries of the matrices equal to performance estimates obtained with 4-fold CV in each dataset. We will call Zk the CSV matrix.
Possible definitions for the scores include the concordance index in survival analysis (Harrell et al., 1996), the area under the operating characteristic curve in binary classification problems, or the mean squared distance between predicted and observed values in regression problems.
We use survival models and focus on a concordance index, the C-index, which is a correlation measure (Gnen and Heller, 2005) between survival times and the risk scores, such as linear combinations of the predictors, provided by a prediction model. The heatmap in Figure 1A displays the CSV matrix of C-statistics obtained through validation of eight models trained on the studies in Table 1 with Ridge regression.
Fig. 1.

CSV matrices Zk in simulated and experimental data for Ridge regression. (A) C-indices for training and validation on each pair of actual datasets in Table 1. The diagonal of this matrix shows estimates obtained through 4-fold CV. (B) The heatmap for each pair of studies (i, j), the average C-index when we fit Ridge regression on a simulated dataset generated by resampling gene expression data and censored time to event outcomes from the i-th study in Table 1, and validate the resulting model on a simulated dataset generated by resampling study j. Computation of each diagonal element averages over pairs of independent datasets obtained by resampling from the same study. The heatmaps strongly resemble each other. CAL and MSK are outlier studies: cross-study C-index is ∼0.5 when they are used either for training or validation. The values of the arrays in (A) and (B) that involve these two studies constitute the blue ‘bad performance’ cluster in (C) which contrast the C-indices obtained for study pairs , on simulated data (y-axis) and experimental data (x-axis). Pearson correlation is ∼0.9. The three plots illustrate similarity between our simulation model and the actual datasets in Table 1
Table 1.
Breast cancer microarray datasets curated by Haibe-Kains et al. (2012)
| Number | Name | Adjuvant therapy | Number of patientsa | Number of ER+ | 3Q survival [mo.] | Median follow-up [mo.] | Original identifiersb | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | CAL | Chemo, hormonal | 118 | 75 | 42 | 82 | CAL | Chin et al. (2006) |
| 2 | MNZ | none | 200 | 162 | 120 | 94 | MAINZ | Schmidt et al. (2008) |
| 3 | MSK | combination | 99 | 57 | 76 | 82 | MSK | Minn et al. (2005) |
| 4 | ST1 | hormonal | 512a | 507b | 114 | 106 | MDA5, TAM, VDX3 | Foekens et al. (2006) |
| 5 | ST2 | hormonal | 517 | 325 | 126 | 121 | EXPO, TAM | Symmans et al. (2010) |
| 6 | TRB | none | 198 | 134 | 143 | 171 | TRANSBIG | Desmedt et al. (2007) |
| 7 | UNT | none | 133 | 86 | 151 | 105 | UNT | Sotiriou et al. (2006) |
| 8 | VDX | none | 344 | 209 | 44 | 107 | VDX | Minn et al. (2007) |
Datasets acronyms: CAL, University of California, San Francisco and the California Pacific Medical Center (USA); MNZ, Mainz hospital (Germany); MSK, Memorial Sloan-Kettering (United States). ST1 and ST2 are meta-datasets provided by Haibe-Kains et al. (2012), TRB denotes the TransBIG consortium dataset (Europe), UNT denotes the cohort of untreated patients from the Oxford Radcliffe Hospital (UK), VDX = Veridex (the Netherlands). Number of ER+ is the number of patients classified as Estrogen Receptor positive. 3Q survival indicates the empirical estimate of the 75-th percentile of the distribution of the survival times (in months). Median follow-up (in months) is computed using the reverse Kaplan–Meier estimate to avoid under-estimation due to early deaths (Schemper and Smith, 1996). aNumbers shown are after removal of samples duplicated in the dataset VDX. bDataset identifiers specified in Haibe-Kains et al. (2012).
2.4 Summarization of a CSV matrix
In order to rank learning algorithms k = 1, … , K, we summarize each matrix Zk by a single score. We consider following two candidate approaches.
- The Simple Average of all non-diagonal elements of the Zk matrix:
The Median or more generally a quantile of the non-diagonal entries of Zk. Quantiles offer robustness to outlier values, and the possibility to reduce the influence of those studies that are consistently associated with poor validation scores, both when used for training and validation, and independently of the learning algorithm.
2.5 True global ranking
Throughout our analyses the score is a random variable. First, studies i and j can be seen as randomly drawn from a population of studies. Second, observations within each study can be considered as randomly drawn from the unknown and possibly different distributions Fi and Fj underlying studies i and j. With this view of as random variable, we consider the theoretical counterparts of the empirical aggregating scores (simple average and quantiles) described in Section 2.4 to summarize Zk. The theoretical counterparts are the expected value or quantiles of each score, obtained by integrating the two levels of randomness that we described. The true global ranking of the learning algorithms k = 1, … , K is then defined by these expected values (or quantiles), one for each algorithm. We will call the ranking global because it depends on the super-population (Hartley and Sielken, 1975) and not which populations were sampled by the available datasets.
The true global ranking can be considered as the estimation target of evaluation procedures such as CV or CSV. In Section 2.7 we present the design of a data-driven simulation study in which we can compute the true ranking through Monte Carlo integration. This allows us to evaluate and compare the ability of CV and CSV to recover the true global ranking.
2.6 Datasets
We used a compendium of breast cancer microarray studies curated for the meta-analysis of Haibe-Kains et al. (2012) and available as supplement to their article. We selected all eight datasets (Table 1) for which distant metastasis-free survival (DMFS), the most commonly available time to event endpoint, as well as Estrogen Receptor (ER) status, were available. These studies were generated with Affymetrix HGU GeneChips HG-U133A, HG-U133B and HG-U133PLUS2. We considered exclusively ER-positive tumors. Of these datasets, only one originated from a population-based cohort (Schmidt et al., 2008). Four studies considered only patients who did not receive hormone therapy or adjuvant chemotherapy. Only four provided date ranges of patient recruitment (Chin et al. 2006; Desmedt et al., 2007; Foekens et al., 2006; Schmidt et al., 2008). Table 1 points also to important differences in survival (for instance 3Q survival) that are not easily explicable based on known characteristics of these studies. This variability in design strategies, reporting, as well as outcomes, highlights the prevalence of ‘samples of convenience’ in biomarker studies discussed by Simon et al. (2009).
Samples from dataset ST1 duplicated in dataset VDX were removed. Expression of each gene was summarized using the probeset with maximum mean (Miller et al., 2011). The 50% of genes with lowest variance were removed. Subsequently, gene-expression values were scaled by linear scaling of the 2.5 and 97.5% quantiles as described by Haibe-Kains et al. (2012).
2.7 Simulation design
We simulate heterogeneous datasets with gene-expression profiles and time to event outcomes from a joint probability model. We define the model through a resampling procedure that we apply to the eight breast cancer datasets in Table 1. The resampling scheme is a combination of parametric and nonparametric bootstrap (Efron and Tibshirani, 1993). The goal of our simulation study is to compare CV and CSV when used for ranking and evaluation of competing learning algorithms. Here we use resampling methods to iteratively simulate realistic ensembles of breast cancer datasets from a hierarchical probability model that we define using the actual datasets in Table 1. CV and CSV are then assessed with respect to their ability to recover the true global ranking, which we compute through Monte-Carlo integration.
We will quantify the ability to recover the ranking by using the Kendall correlation between the true global ranking and the estimates obtained with CV or CSV.
For b = 1, … , B = 1000 iterations, we generate a collection of I = 8 datasets as follows. First, we sample eight study labels with replacement from the list of breast cancer studies in Table 1. This step only involves simulations from a multinomial Mult(8,[1/8, … , 1/8]) distribution. We resample the collection of study labels to capture variability in study availability, and heterogeneity of study characteristics. Second, for each of the eight randomly drawn labels, we sample N = 150 patients from the corresponding original dataset, with replacement. If a study is randomly assigned to the j-th label, then each of the N = 150 vectors of predictive variables is directly sampped from the empirical distribution of the j-th study in Table 1. Finally, we simulate the corresponding times to event using a proportional hazards model (parametric bootstrap) fitted to the j-th dataset:
| (1) |
where λj(t|x) is the individual hazard function when the vector of predictors is equal to x and denotes a vector of regression coefficients. We combine the truncated inversion method in Bender et al. (2005) and the Nelson–Aalen estimator for cumulative hazard functions to simulate survival times that reflect survival distributions and follow-up of the real studies. We set the vector to be the coefficients fitted in study j = 1, … , I using the CoxBoost method (Binder and Schumacher, 2008). A different regression method could have been used at this stage. The collections of simulated datasets are then used both (i) to compute by Monte-Carlo method the true global ranking defined in Section 2.5, and (ii) to compute ranking estimates through CV and CSV. Figure 1A displays, for each pair of studies (i, j) in Table 1, the C-index obtained when training a model by Ridge regression on dataset i (rows), and validating that model on dataset j (columns). We computed the diagonal elements (i = j) by 4-fold CV. Figure 1B displays mean C-indices for each (i, j) combination across simulations, when the training and validation studies are generated resampling the i-th and j-th study. Here diagonal elements are computed by averaging C-indices with the training and validation datasets independently generated by resampling from the same study.
The strong similarity between the two panels is reassuring, in particular with regard to the clear separation of the eight studies into two groups. The first group includes studies MNZ, ST1, ST2, TRP, UNT and VDX, and produces more accurate prediction models than the remaining studies. The datasets in this group are also associated with higher values of the concordance index when used for validation. This difference between the two groups is also illustrated in Figure 1C. It displays the non-diagonal entries of the matrices represented in the left and middle panels, that is the average C-indices from simulated datasets, against the C-indices from real data. This scatterplot shows a clear two-cluster structure: the yellow dots display the 30 training and validation combinations within studies MNZ, ST1, ST2, TRP, UNT and VDX. We will return to this cluster structure in the discussion.
2.8 Evaluation criteria for simulations
In simulation studies we can assess and rank algorithms based on their ability to recover the true underlying models In this subsection, we introduce a criterion that reflects the degree of similarity between the true regression coefficients that were used to simulate the i-th in silico dataset and the coefficients fitted through algorithm k on the j-th simulated dataset. We consider the i = j and cases separately. Similarity between vectors is usually quantified by computing the Euclidean distance between them. However, since our focus is on prediction, we use
the correlation between true and estimated patient-specific prognostic scores, to measure the similarity between the true and estimated regression coefficients Here Xi is the matrix of predictors of dataset i and denotes Pearson’s correlation. The average
| (2) |
over the I studies, provides a measure of the ability of learning algorithm k to recover the model that has generated the training dataset, hence the index self.
Another criterion of interest is the ability of a learning algorithm k to recover the vector of regression coefficients when it is trained on a separate dateset and the unknown models underlying datasets i and j might differ from each other.
This can be quantified with
| (3) |
where the index across emphasizes the focus on cross-study similarity, i.e. on the ability of algorithm k to recover the coefficients when fitted on dataset j, with
In alternative to averaging across studies, or pairs of datasets, as in Equations (2–3) one can also use different summaries, e.g. quantiles, as we do in Section 2.4. Both and are summary statistics to assess and compare learning algorithms. We denote the ranking obtained by ordering the algorithms according to Sself(Sacross) by Rself(Racross). Both and vary across simulations of the datasets ensembles, although the hierarchical simulation model remains fixed and their computations involve the vectors , i = 1, … , I. We will therefore call the rankings Rself and Racross local because they are specific to the collection of datasets at hand.
3 RESULTS
3.1 Simulated data
Our focus in the simulation study is on differences between the rankings and performance estimates obtained by using CV and CSV. We will use and to denote the means of the diagonal and non-diagonal elements of a CSV matrix, respectively. Recall that we compute the diagonal elements through CV.
Figure 2A shows, for K = 6 algorithms, the distributions of and and Figure 2B shows the distribution of the rankings estimates, across 1000 simulated collections of eight datasets. Table 2 compares the medians of the distributions in Figure 2B with the true global rankings that we obtained using the criteria in Section 2.4. The rank of method k is 1 if it outperforms the remaining K – 1 training algorithms. We observe large differences in the distributions of and across simulations (Fig. 1A): the average of the scores, under all the algorithms we considered, is close to 0.65, while the scores are centered at ∼0.55. The variability of and across simulations, however, is comparable.
Fig. 2.

Comparison of CSV and CV on simulated data. Each panel represents evaluations of K = 6 algorithms across 1000 simulations of a compendium of I = 8 datasets. For each simulation the diagonal or off-diagonal elements of the Zk matrix of validation C-statistics is summarized by (A) mean and (B) rank of the mean across algorithms. CV estimates tend to be much higher than the CSV estimates. In most of the simulations Lasso is ranked as one of the worst algorithms, both by CV and CSV, while Ridge and Plusminus are ranked among the best prediction methods
Table 2.
True global rankings and estimates with CV and CSV on simulated data
| Algorithm | Global true ranking |
CSV (median ranks) |
CV (median ranks) |
|||
|---|---|---|---|---|---|---|
| Criterion | Average | Medium | Average | Medium | Average | Medium |
| Ridge | 1 | 2 | 2 | 2 | 1 | 2 |
| Plusminus | 2 | 1 | 2 | 2 | 2 | 2 |
| Superpc | 3 | 3 | 4 | 3 | 4 | 4 |
| Unicox | 4 | 4 | 4 | 4 | 5 | 4 |
| CoxBoost | 5 | 5 | 5 | 5 | 3 | 4 |
| Lasso | 6 | 6 | 6 | 6 | 5 | 6 |
Median estimates across 1000 simulations are displayed for CV and CSV; individual columns refer to summarization of the statistics by using the mean or the median as discussed in Section 2.4. We also computed the true global ranking as well as CV and CSV estimates by using the third quartile of the summaries, and obtained results identical to those displayed for the rankings obtained by summarizing validation results through their median. Both CV and CSV tend to rank Ridge regression and Plusminus as best performing algorithms. Variability of CV and CSV rank estimates across simulations is shown in Figure 2B.
Performance differences across algorithms, whether estimated by CV or CSV, are relatively small compared to the overall difference between CV and CSV performance estimates. We also observe differences between the rank distributions produced by CV and CSV. Accordingly, to both CV and CSV, in most of the simulations, Lasso regression is ranked as one of the worst performing algorithms, while Ridge regression and Plusminus are ranked first or second. However, the summaries suggest an advantage of Ridge regression over Plusminus across most of the simulations while rank Plusminus as the best performing algorithm in ∼50% of the simulations. The median rank of CoxBoost across simulations has an improvement of two positions when it is estimated through CV and compared to the summaries; in this case CSV results are more consistent with the true global rankings (Table 2). When we consider the criteria described in Section 2.4, Ridge regression and Plusminus exchange the top-two positions of the true global rankings (see Table 2), although for these two algorithms the Zi,j distributions under our simulation scenario are nearly identical.
The local rankings Racross and Rself of the K = 6 algorithms defined by and in Section 2.8 vary across the 1000 simulated collections of studies. The median Kendall’s correlation between Racross and Rself across simulations is ∼0.5, i.e. the performance measures and tend to define distinct rankings of the competing algorithms, see also the Supplementary Figure S1. We illustrate the extent to which CV and CSV recover the unknown rankings Racross and Rself. The boxplots in Figure 3 display the Kendall’s correlation between local rankings (i) Racross or(ii) Rself , and the rankings estimated through CV (gray boxes) and CSV (white boxes) across simulations. Figure 3C shows the Kendall’s correlation between the true global ranking and the ranking estimates. The median Kendall’s correlation between Rself and the corresponding CSV estimates across simulations is ≈0.5. The CV ranking estimates tend to be less correlated with the local rankings Racross than the CSV estimates. In contrast, the CV estimates tend to be more correlated with Rself than the CSV estimates. We recall that both and Rself are defined summarizing performance measures, and that refer to a single study, while and Racross summarizes performance measures computed using two distinct studies that are used for training and validation.
Fig. 3.

Kendall’s correlation between true global or local rankings and estimates obtained with CSV (white box-plots) or CV (cross-validation, gray box-plots) across simulations. Panels (A) and (B) compare CV and CSV in terms of their correlation to the local rankings (Racross and Rself), while panel (C) considers the true global ranking. Each box-plot represents a correlation coefficient that was computed in each of the 1000 iterations of our simulation study. CSV tend to achieve a higher correlation with the global ranking and Racross than CV. The results displayed have been computed using the mean criterion discussed in Section 2.4
Finally, CSV tends to be more correlated with the true global ranking than CV. This suggests that CSV is more suitable for recovering the true global ranking. When we removed the two outlier studies (CAL and MSK) and repeated the simulation study, the advantage of CSV over CV in recovering the true global ranking was confirmed (median Kendall’s correlation 0.8 versus 0.6, see also Supplementary Figs S2–S4), moreover after their removal Kendall’s correlations between Rself and the CSV estimates tend to be larger than those between Rself and the CV estimates. Overall, as displayed by the Supplementary Figure S3, it appears that, after outlier studies are removed, CSV outperforms substantially CV when used for ranking algorithms.
3.2 Application to breast cancer prognostic modeling
We apply CV and CSV to the I = 8 breast cancer studies described in Section 2. Generally, the results resemble those obtained on simulated data. The top panel in Figure 4 illustrates the distributions of the diagonal and off-diagonal validation statistics in Zk for each of the K = 6 algorithms. Except for the distinctly larger interquartile ranges of the box-plots we observe several similarities with Figure 2. Note that each box-plot represents validation scores within a single Zk-matrix, whereas in Figure 2 each box-plot displays a summary of 1000 Zk matrices, one for each simulation. This explains the higher variance observed in Figure 4. We also observe the following.
estimates are ∼0.06 higher than estimates on the C-index scale. To interpret the magnitude of this shift on the C-index scale consider a population with two groups of patients, high and low risk patients, covering identical proportions 0.5 of the population. A perfect discrimination model that correctly recognizes the subpopulation of each individual, when the hazard ratio between high versus low risk patients is 2.7, achieves on average a C-index of 0.62. It is necessary to double the hazard ratio to 5.4 to increase the average C-index of the perfect discrimination model to 0.68. Thus, it is fair to say that the CV results are considerably more optimistic than the CSV estimates.
The ranking defined by CSV, using median summaries of the scores, is nearly identical to the global ranking in our simulation example (see Supplementary Table ST1 and Table 2). With both, median and third quartile aggregation of the statistics, the rankings defined by CV and CSV differ substantially (Kendall’s correlations 0.6 and 0.07). This is consistent with the results of the simulation study, where median correlation of the rankings estimated through CSV and CV was ∼0.4 (see Supplementary Fig. S1).
The presence of outlier studies (CAL and MSK) has a strong effect on the ranking estimates when we use the mean to summarize the Zk matrices. After aggregating the validation statistics by averaging, both CSV and CV rank Superpc first. This result might be due to the high variability, ∼0.5, of the validation scores corresponding to models trained by outlier studies. In particular, Superpc and Unicox are the only algorithms that produce models with substantial prediction performances when trained on the MSK study. With median summarization the ranking estimates are less influenced by the presence or absence of outlier studies. We therefore recommend the use of the median to summarize Zk matrices.
Figure 4B illustrates lack of agreement between CSV and CV performance estimates. The black digits contrast, for each dataset i, the CSV summary
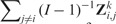 versus the CV summary Performance measures refer to Ridge regression. Similarly, the gray digits in this panel contrast
versus the CV summary Performance measures refer to Ridge regression. Similarly, the gray digits in this panel contrast 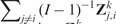 with The CV performance statistics are only moderately correlated with the CSV statistics
with The CV performance statistics are only moderately correlated with the CSV statistics  (correlation = 0.2), and negatively correlated with the CSV summaries
(correlation = 0.2), and negatively correlated with the CSV summaries 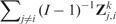 (correlation = −0.33).
(correlation = −0.33).
Fig. 4.

Panel (A) describes the CSV and CV statistics in Zk, separately for each of the six algorithms that we considered. Each box-plot represents the variability of CV or CSV performance statistics from a single Zk matrix. The CV statistics tend to be higher than the CSV statistics. Panel (B) contrasts with black digits, for each study i, the CSV summary 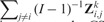 with the CV summary Similarly, with gray digits it contrasts the CSV summary
with the CV summary Similarly, with gray digits it contrasts the CSV summary 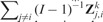 with the CV summary This panel shows results for the learning algorithm Ridge regression and the displayed numbers refer to Table 1 (outliers CAL and MSK were removed). Cross-validation statistics on the y-axis are moderately correlated to the CSV summaries on the x-axis; identical considerations hold for all K = 6 algorithms that we used
with the CV summary This panel shows results for the learning algorithm Ridge regression and the displayed numbers refer to Table 1 (outliers CAL and MSK were removed). Cross-validation statistics on the y-axis are moderately correlated to the CSV summaries on the x-axis; identical considerations hold for all K = 6 algorithms that we used
3.3 CV and CSV summaries
Correlation between CSV and CV summary statistics, as displayed in Figure 4B, suggests that cross- and within-study performances are less redundant than one might expect. In Figure 4B study specific CSV summaries are plotted against CV for Ridge regression. For each study we have a single CV statistic and two CSV statistics obtained by averaging the Z-matrix column- and row-wise. In the column-wise case correlations, between CSV and CV summaries, vary across algorithms ∼0.5, while in the row-wise case all the correlations are negative. Overall, we can consider cross- and within-study prediction as two related but distinct problems.
We also noted that CV is less suitable for detection of outlier studies than CSV; in particular CV can estimate encouraging prediction performances even on studies associated, under each training algorithm, with poor CSV summaries For instance, with the SuperPC algorithm all but one C-index estimates obtained with CV are above 0.6.
3.4 Specialist and generalist algorithms
Our analyses lead to the question of whether some algorithms can be considered as generalist or specialist procedures according to our definitions. Our examples are not exhaustive and additional comparisons, within the development of new prognostic models, are necessary in order to determine ‘specialist’ or ‘generalist’ tendencies of these algorithms. However, the fact that Ridge regression, Lasso regression and CoxBoost are ranked distinctly better accordingly to CV than CSV, in most iterations of our simulation study, suggests that these algorithms might be specialist procedures and adapt to the specific properties of the individual dataset. The status of generalist versus specialist, for each algorithm, can be discussed using the local performance criteria Sself and Sacross, which are conceived to measure within-single-studies and generalizable prediction performances. We note that CoxBoost and Ridge regression tend to achieve better ranks in Rself than in Racross. In particular CoxBoost improves its position by 1 or 2 ranks in most simulations, which is similar to what we observed comparing CoxBoost’s CSV and CV rankings. In summary, in our study, these two algorithms seem to have—accordingly to all the criteria that we considered—a tendency to specialize to the dataset at hand. We mention that, as one can expect, for all the algorithms Sself is consistently higher than Sacross. We also compared CV to independent within-study validation using our simulation model. For the independent within-study validation, we iteratively pair two datasets generated using identical regression coefficients and gene expression distributions. Subsequently, we train a model on the first dataset and evaluate it on the second one. As can be seen in Supplementary Figure S5, CV values, as expected, are slightly smaller than for the independent within-study validations.
4 DISCUSSION AND CONCLUSION
In applying genomics to clinical problems, it is rarely safe to assume that the studies in a research environment faithfully represent what will be encountered in clinical application, across a variety of populations and medical environments. From this standpoint, study heterogeneity can be a strength, as it allows to quantify the degree of generalizability of results, and to investigate the sources of the heterogeneity. This aspect has long been recognized in meta-analysis of clinical trials (Moher and Olkin, 1995). Therefore, we expect that an increased focus on quantifying cross-study performance of prediction algorithms will contribute to the successful implementation of the personalized medicine paradigm.
In this article we provide a conceptual framework, statistical approaches and software tools for this quantification. The conceptual framework is based on the long-standing idea that finite populations of interest can be viewed as samples from an infinite ‘super-population’ (Hartley and Sielken, 1975). This concept is especially relevant for heterogeneous clinical studies originating from hospitals that sample local populations, but where researchers hope to make generalizations to other populations.
As an illustrating example, we demonstrate CSV on eight independent microarray studies of ER-positive breast cancer, with metastasis-free survival as the endpoint of interest. We also develop a simulation procedure involving two levels of non-parametric bootstrap (sampling of studies and sampling of observations within studies) in combination with parametric bootstrap, to simulate a compendium of independent datasets with characteristics of predictor variables, censoring, baseline hazards, prediction accuracy and between-dataset heterogeneity realistically based on available experimental datasets.
Cross-validation is the dominant paradigm for assessment of prediction performance and comparison of prediction algorithms. The perils of inflated prediction-accuracy estimations by incorrectly or incompletely performed cross-validation are well known (Molinaro et al., 2005; Subramanian and Simon, 2010; Simon et al., 2011; Varma and Simon, 2006). However, we show that even strictly performed cross-validation can provide optimistic estimates relative to CSV performance. All algorithms, in simulation and example, showed distinctly decreased performance in CSV compared to cross-validation. Although it would be possible to further reduce between-study heterogeneity, for example by stricter filtering on clinical prognostic factors, we believe this degree of heterogeneity reflects the reality of clinical genomic studies and likely other applications. Some sources of biological heterogeneity are unknown, and it is impossible to ensure consistent application of new technologies in laboratory settings. Prediction models are used in presence of unknown sources of variation. Formal CSV provides a means to assess the impact of unknown or unobserved confounders that vary across studies.
In simulations, the ranking of algorithms by CSV was closer to the true rankings defined by cross-study prediction, both when we considered Racross and the global true ranking. Surprisingly, CSV was also competitive with CV for recovering true rankings based on within-study prediction, such as Rself. Although the performance differences we observed between algorithms were smaller than the difference between CV and CSV, Lasso consistently compared poorly with most of the competing algorithms, both under CV and CSV evaluations. Lasso, and other algorithms that ensure sparsity have been shown to guarantee poor prediction performances in previous comparative studies (Bøvelstad et al., 2007; Waldron et al., 2011).
Systematic CSV provides a means to identify relevant sources of heterogeneity within the context of the prediction problem of interest. By simple inspection of the CSV matrix we identified two outlier studies that yielded prediction models no better than random guessing in new studies. This may be related to known differences in these studies: smaller numbers of observations, higher proportions of node positive patients, different treatments and larger tumors (Supplementary Figs S6–S9). Conversely, other known between-study differences do not seem to have created outlier studies or clusters of studies as seen in the Z matrix, such as between studies where all or no patients received hormonal treatment. We note that incorporation of clinical prognostic factors into genomic prognostic models could likely produce gains in CSV accuracy, and that such multi-factor prognostic models could also be assessed by the proposed matrix of CSV statistics.
In practice it is neither possible nor desirable to eliminate all sources of heterogeneity between studies and between patient populations. The adoption of ‘leave-one-in’ CSV, in settings where at least two comparable independent datasets are available, can provide more realistic expectations of future prediction model performance, identify outlying studies or clusters of studies, and help to develop ‘generalist’ prediction algorithms which will hopefully be less prone to fit to dataset-specific characteristics. Further work is needed to formalize the identification of clusters of comparable studies, to develop databases for large-scale cross-study assessment of prediction algorithms, and to develop better ‘generalist’ prediction algorithms. Appropriate curated genomic data resources are available in Bioconductor (Gentleman et al., 2004) through the curatedCRCData, curatedBladderData and curatedOvarianData (Ganzfried et al., 2013) packages, and in other common cancer types through InSilicoDB (Taminau et al., 2011). In realms where such curated resources are available, CSV is in practice no more difficult or CPU-consuming than cross-validation, and should become an equally standard tool for assessment of prediction models and algorithms.
ACKNOWLEDGEMENT
We wish to thank Benjamin Haibe-Kains for making the curated breast cancer datasets used in this study publicly available.
Funding: German Science Foundation [BO3139/2-2 to A.L.B.]. National Science Foundation [grant number CAREER DBI-1053486 to C.H. and DMS-1042785 to G.P.]; National Cancer Institute [grant 5P30 CA006516-46 to G.P. and 1RC4 CA156551-01 to L.W. G.P. and L.T.].
Conflict of interest: none declared.
REFERENCES
- Baek S, et al. Development of biomarker classifiers from high-dimensional data. Brief. Bioinform. 2009;10:537–546. doi: 10.1093/bib/bbp016. [DOI] [PubMed] [Google Scholar]
- Baggerly KA, et al. Run batch effects potentially compromise the usefulness of genomic signatures for ovarian cancer. J. Clin. Oncol. 2008;26:1186–1187. doi: 10.1200/JCO.2007.15.1951. [DOI] [PubMed] [Google Scholar]
- Bender R, et al. Generating survival times to simulate Cox proportional hazards models. Stat. Med. 2005;24:1713–1723. doi: 10.1002/sim.2059. [DOI] [PubMed] [Google Scholar]
- Binder H, Schumacher M. Allowing for mandatory covariates in boosting estimation of sparse high-dimensional survival models. BMC Bioinform. 2008;9:14. doi: 10.1186/1471-2105-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair E, Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2:511–522. doi: 10.1371/journal.pbio.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulesteix AL. On representative and illustrative comparisons with real data in bioinformatics: response to the letter to the editor by smith et al. Bioinformatics. 2013;29:2664–2666. doi: 10.1093/bioinformatics/btt458. [DOI] [PubMed] [Google Scholar]
- Bøvelstad HM, et al. Predicting survival from microarray data–a comparative study. Bioinformatics. 2007;23:2080–2087. doi: 10.1093/bioinformatics/btm305. [DOI] [PubMed] [Google Scholar]
- Castaldi PJ, et al. An empirical assessment of validation practices for molecular classifiers. Brief. Bioinform. 2011;12:189–202. doi: 10.1093/bib/bbq073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10:529–541. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Demšar J. Statistical comparisons of classifiers over multiple data sets. J. Mach. Learn. Res. 2006;7:1–30. [Google Scholar]
- Desmedt C, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the transbig multicenter independent validation series. Clin. Cancer Res. 2007;13:3207–3214. doi: 10.1158/1078-0432.CCR-06-2765. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani RJ. An Introduction to the Bootstrap. New York: Chapman and Hall; 1993. [Google Scholar]
- Foekens JA, et al. Multicenter validation of a gene ExpressionBased prognostic signature in lymph NodeNegative primary breast cancer. J. Clin. Oncol. 2006;24:1665–1671. doi: 10.1200/JCO.2005.03.9115. [DOI] [PubMed] [Google Scholar]
- Ganzfried BF, et al. curatedOvarianData: clinically annotated data for the ovarian cancer transcriptome. Database. 2013 doi: 10.1093/database/bat013. [Epup ahead of print, doi: 10.1093/database/bat013, April 2, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeman J. l1 penalized estimation in the cox proportional hazards model. Biometr. J. 2010;52:70–84. doi: 10.1002/bimj.200900028. [DOI] [PubMed] [Google Scholar]
- Gnen M, Heller G. Concordance probability and discriminatory power in proportional hazards regression. Biometrika. 2005;92:965–970. [Google Scholar]
- Haibe-Kains B, et al. A three-gene model to robustly identify breast cancer molecular subtypes. J. Natl Cancer Inst. 2012;104:311–325. doi: 10.1093/jnci/djr545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell FE, et al. Multivariate prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stati. Med. 1996;15:361–387. doi: 10.1002/(SICI)1097-0258(19960229)15:4<361::AID-SIM168>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Hartley HO, Sielken RL., Jr A ‘Super-Population viewpoint’ for finite population sampling. Biometrics. 1975;31:411–422. [PubMed] [Google Scholar]
- Leek JT, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010;11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheel C, et al. Evolution of Translational Omics: Lessons Learned and the Path Forward. Wahington, D.C: National Academies Press; 2012. [PubMed] [Google Scholar]
- Miller JA, et al. Strategies for aggregating gene expression data: the collapserows R function. BMC Bioinform. 2011;12:322. doi: 10.1186/1471-2105-12-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc. Natl Acad. Sci. USA. 2007;104:6740–6745. doi: 10.1073/pnas.0701138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moher D, Olkin I. Meta-analysis of randomized controlled trials: A concern for standards. JAMA. 1995;274:1962–1964. [PubMed] [Google Scholar]
- Molinaro AM, et al. Prediction error estimation: a comparison of resampling methods. Bioinformatics. 2005;21:3301–3307. doi: 10.1093/bioinformatics/bti499. [DOI] [PubMed] [Google Scholar]
- Riester M, et al. Risk prediction for Late-Stage ovarian cancer by meta-analysis of 1525 patient samples. JNCI J Natl Cancer Inst. 2014 doi: 10.1093/jnci/dju048. [Epup ahead of print, doi:10.1093/jnci/dju048, April 3, 2014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schemper M, Smith TL. A note on quantifying follow-up in studies of failure time. Clinical Trials. 1996;17:343–346. doi: 10.1016/0197-2456(96)00075-x. [DOI] [PubMed] [Google Scholar]
- Schmidt M, et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68:5405–5413. doi: 10.1158/0008-5472.CAN-07-5206. [DOI] [PubMed] [Google Scholar]
- Simon RM, et al. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J. Natl Cancer Inst. 2009;101:1446–1452. doi: 10.1093/jnci/djp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon RM, et al. Using cross-validation to evaluate predictive accuracy of survival risk classifiers based on high-dimensional data. Brief. Bioinform. 2011;12:203–217. doi: 10.1093/bib/bbr001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J. Natl Cancer Inst. 2006;98:262–272. doi: 10.1093/jnci/djj052. [DOI] [PubMed] [Google Scholar]
- Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J. Natl Cancer Inst. 2010;102:464–474. doi: 10.1093/jnci/djq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmans WF, et al. Genomic index of sensitivity to endocrine therapy for breast cancer. J. Clin. Oncol. 2010;28:4111–4119. doi: 10.1200/JCO.2010.28.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taminau J, et al. inSilicoDb: an R/Bioconductor package for accessing human affymetrix expert-curated datasets from GEO. Bioinformatics. 2011;27:3204–3205. doi: 10.1093/bioinformatics/btr529. [DOI] [PubMed] [Google Scholar]
- Tibshirani R. uniCox: Univarate shrinkage prediction in the Cox model. 2009. R package version 1.0. [Google Scholar]
- Varma S, Simon R. Bias in error estimation when using cross-validation for model selection. BMC Bioinformatics. 2006;7:91. doi: 10.1186/1471-2105-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron L, et al. Optimized application of penalized regression methods to diverse genomic data. Bioinformatics. 2011;27:3399–3406. doi: 10.1093/bioinformatics/btr591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron L, et al. Comparative meta-analysis of prognostic gene signatures for Late-Stage ovarian cancer. JNCI J Natl Cancer Inst. 2014 doi: 10.1093/jnci/dju049. [Epub ahead of print, doi:10.1093/jnci/dju049, April 3, 2014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, et al. 2013. Mas-o-menos: a simple sign averaging method for discrimination in genomic data analysis. http://biostats.bepress.com/harvardbiostat/paper158/ (24 October 2014, date last accessed) [DOI] [PMC free article] [PubMed] [Google Scholar]