

Abstract
Motivation: Understanding and predicting an individual’s response in a clinical trial is the key to better treatments and cost-effective medicine. Over the coming years, more and more large-scale omics datasets will become available to characterize patients with complex and heterogeneous diseases at a molecular level. Unfortunately, genetic, phenotypical and environmental variation is much higher in a human trial population than currently modeled or measured in most animal studies. In our experience, this high variability can lead to failure of trained predictors in independent studies and undermines the credibility and utility of promising high-dimensional datasets.
Methods: We propose a method that utilizes patient-level genome-wide expression data in conjunction with causal networks based on prior knowledge. Our approach determines a differential expression profile for each patient and uses a Bayesian approach to infer corresponding upstream regulators. These regulators and their corresponding posterior probabilities of activity are used in a regularized regression framework to predict response.
Results: We validated our approach using two clinically relevant phenotypes, namely acute rejection in kidney transplantation and response to Infliximab in ulcerative colitis. To demonstrate pitfalls in translating trained predictors across independent trials, we analyze performance characteristics of our approach as well as alternative feature sets in the regression on two independent datasets for each phenotype. We show that the proposed approach is able to successfully incorporate causal prior knowledge to give robust performance estimates.
Contact: daniel.ziemek@pfizer.com
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
With our increasing understanding of the etiology and heterogeneity of complex diseases comes the realization that therapeutic drugs might need to be tailored to specific subpopulations of patients. Our current inability to predict such subpopulations has contributed to the rising cost of drug development and overall health-care expenditure. One aspect of this problem is the identification of patient populations that respond to an experimental drug in a clinical trial. It currently becomes feasible to generate multi-omics (e.g. transcriptomics, genetics and metabolomics) datasets for all patients in a clinical trial of hundreds of people for a cost that is only a small percentage of the overall cost of the trial.
Research on Precision Medicine (Mirnezami et al., 2012) has been particularly strong in oncology as many cancers have a strong genetic basis to leverage for this purpose. For instance, the National Cancer Institutes of Health (NCI) in the USA recently outlined their criteria for the use of omics-based predictors (McShane et al., 2013) in NCI-funded clinical trials. They point out the pitfalls of defining omics-based predictors that do not translate well to patient population outside the initial trial, i.e. the problem of overfitting the available data. As a striking example of the care that has to be taken when defining signatures, Venet et al. (2011) compare 47 published gene-expression signatures for breast cancer. The sobering result is that the majority of signatures do not perform better than any randomly picked set of genes of similar size. In our experience, the aspect of replicability in independent datasets has not received enough attention in the current literature on novel methods. It is relatively easy to demonstrate the benefits of a method within one well-controlled study but much harder to show translatability to independent studies. This problem is especially pronounced in human populations in which genetic and environmental diversity is much higher than in animal studies. As this problem has impacted method adoption for our internal research in several cases, we tried to explicitly validate findings in at least two independent cohorts in each response prediction scenario.
In this article, we focus on human clinical trials with patient-level genome-wide gene-expression data. Responders to therapy are identified at the end of the study using disease-specific measures. The question of interest is whether the baseline or early treatment gene-expression data can predict response to treatment. There has been substantial prior work on establishing predictive gene-expression signatures based on data-driven methods alone as well as by leveraging other types of biological information. For instance, Tibshirani et al. (2002) proposed the use of regularization techniques to improve gene selection for predictive signatures early on. Since then, many authors have proposed approaches using different machine-learning techniques including regularized regression, SVMs and random forests. Cun and Fröhlich (2012) give a recent review. One recent example that utilizes prior knowledge is the PARADIGM approach (Vaske et al., 2010) which uses probabilistic models to integrate genetics, epigenetics and transcriptional data with curated pathway information to determine active pathways in cancer patients, but does not directly attempt a prediction of response and non-response status in trial data.
As a common consensus, most methods employ some form of regularization to overcome the problem of many variables but few samples. Furthermore, methods vary in the amount of prior knowledge they employ—from no prior knowledge at all to a mixture of different omics technologies as in the case of PARADIGM. However, most novel methods have not been developed in the setting of human data with high intrinsic levels of phenotypic and genetic heterogeneity and are not evaluated in a truly independent dataset, e.g. a trial conducted at a different clinic, but exclusively rely on cross-validation approaches.
In contrast, we present a method that attempts to define biologically interpretable, yet predictive signatures of diseases progression or response to drug treatment that translate well to new studies. In conjunction with a well-accepted learning algorithm, L1-regularized logistic regression (Friedman et al., 2010), we base our method on a large collection of causal relationships manually curated from the literature. The causal graph consists of ∼450 000 causal relationships, of which >250 000 are unique, between ∼37 000 entities, representing ∼65 000 full-text articles indexed by PubMed. Each causal relationship describes an experimentally observed perturbation experiment leading to a defined transcriptional change. We previously published a Bayesian inference method on this causal graph that given a set of differentially expressed genes, is able to identify potential upstream regulators and their biological context (Zarringhalam et al., 2013). The method is called Bayesian Causal Reasoning Engine (Bayes-CRE) and will serve as a building block in this work. We briefly outline it in Section 2. A prerequisite for the method to work is that relevant biology is captured in the underlying knowledgebase of causal relationships. We found that Bayes-CRE is able to capture the relevant upstream regulators in numerous test cases, indicating that the collection of pairwise causal relations in our network has sufficient level of complexity to be useful. More large-scale dataset with more complex notion of causality can also be incorporated into our methodology as they become available in the future.
The search for suitable experimental datasets has been surprisingly difficult. Our criteria for inclusion were (i) a dataset of at least 20 human subjects with a defined clinical binary outcome, i.e. responders and non-responders, (ii) at least some detectable difference in gene expression at baseline between the two groups and (iii) the availability of a similar but entirely independent trial for testing purposes. For the purposes of this work, we settled on two appropriate datasets: the studies of Khatri et al. (2013) and Einecke et al. (2010) on acute rejection in kidney transplantation and the work of Arijs et al. (2009) on infliximab treatment in ulcerative colitis.
In the following, we will define the details of our proposed method, compare its performance against alternative feature sets and demonstrate that its application can lead to biologically interpretable predictors that are robust to resampling and, most crucially, seem to translate well to independent patient populations.
2 METHODS
Conceptually, we require a set of features characterizing each patient in the clinical trial which can then be utilized by a classification algorithm for prediction. In the following, we will explore using (i) a significant set of normalized gene expression values, (ii) the set of enriched Gene Ontology (GO) categories (Ashburner et al., 2000) and (iii) significant upstream regulators and their activity scores (Zarringhalam et al., 2013).
2.1 Data processing
We processed gene-expression data from two clinical phenotypes: (i) acute rejection in kidney transplantation (Khatri et al., 2013; Einecke et al., 2010) and (ii) response to infliximab in ulcerative colitis (Arijs et al., 2009). Each phenotype consists of two datasets (GEO accession numbers GSE50058 and GSE21374 in acute rejection and GSE12251 and GSE14580 in response to infliximab). Datasets corresponding to different phenotypes were analyzed separately. For each phenotype, both datasets were combined and RMA (robust multi-array average) normalized. The probes that were absent in all samples—irrespective of response status—were filtered using the mas5calls function from the R Bioconductor package (Gentleman et al., 2004). Differential gene-expression analysis with FDR cutoff of 0.05 and a fold-change cutoff value of 1.3 was performed on the normalized combined dataset as well as individual datasets in each phenotype using the R Bioconductor package. This combined normalization will put all transcript abundance estimates on a similar scale. Note that this part does not use any information on response status from either training or test set. Although this combined normalization may have a slight impact on performance estimates as more test examples are added to the datasets, the performance should be shifted towards better generalizability to the test set. Moreover, this effect equally impacts all tested methods presented in this work as they all use the normalized expression data as a starting point. From a practical point of view, testing data can always be added to the normalization at test time, data re-normalized and the classifiers re-trained on the training data alone.
2.2 Generating differential gene-expression profiles for each individual
In order to obtain enriched GO terms as well as upstream regulators, we need to identify differentially expressed genes per individual. If there are enough replicates and a healthy control group is available, this can be achieved by pairwise comparisons of gene values between the individuals and the controls.
Here, we define differential expression relative to response status of the individual. For example, a gene for a responder is called differentially expressed if it is significantly different from the distribution of gene-expression values in non-responders. More specifically let and denote the mean and standard deviation of normalized gene values for gene g among the responders and non-responders, respectively. If the individual is a responder, the Z-score profile is where xg is the normalized gene value of gene g. If the individual is a non-responder, the Z-profile is then Note that the response status of the individuals in the training set is known and hence the opposite group can be identified. The genes with large Z-score values in the absolute value sense are then declared as differentially expressed. Based on the sign of the Z-scores, we determine the up or down regulation of the gene (up regulated if the Z-score is positive and down regulated if the Z-score is negative). It should be noted that in assigning significance to per-individual gene values, we are making an implicit assumption that zg approximately follows a standard normal distribution. This practical approximation enables us to assess how far a gene value falls from the mean of the distribution and hence generate the profiles of differentially expressed genes per-individual. If extra information (e.g. more replicates per patient or an independent control group) is available, the significance of gene values can be estimated using more direct approaches (such as a standard t-test).
For a new individual or an individual in a test set, the response status is not known a priori. In this case, we have to compute two z-profiles obtained by comparisons with both groups (Fig. 1). The z-profile obtained from the opposite group of the true status of the new individual, will be a ‘correct’ z-profile containing potentially important gene differences that are linked to response. The other z-profile will contain ‘random’ differences of the individual with the rest of the individuals in the same group. Correspondingly, we have two sets of differentially expressed genes. We use an absolute value Z-score cutoff of 2 throughout this work.
Fig. 1.
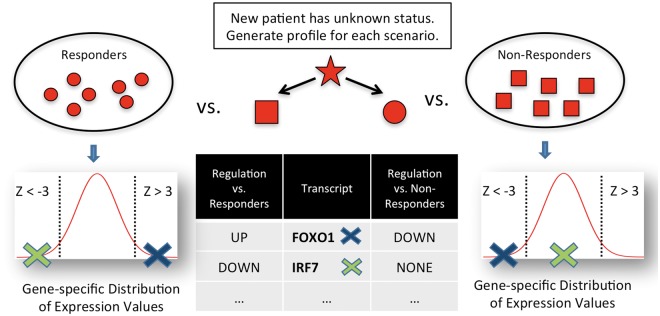
Estimating differential gene expression per individual: For individuals in the training set, response status is known and a Z-score profile is computed with respect to the opposite group. For an individual not in the training set, two Z-score profiles are computed: (i) assuming the individual is a responder and (ii) assuming the individual is a non-responder. Genes with an absolute Z-score greater than a defined cutoff are declared differentially expressed. For instance, the IRF7 transcript would be called differentially down-regulated assuming the new individual is a non-responder, but not differentially regulated assuming responder status
2.3 Feature sets
In the following, we will define five types of feature sets of increasing complexity. In the training phase, a subset of features will be selected by the classifier and only those will be used for prediction. For instance, this corresponds to a subset of differentially expressed genes or a subset of potential upstream regulators. Note that all feature sets that use per-individual differentially expressed genes at prediction time will require two feature profiles assuming, first, that the test subject is a responder and then that the subject is a non-responder based on the procedure outlined above. Our main feature set is Feature Set IV. Prediction accuracy of the feature sets will be discussed in Section 3. Table 1 gives an overview.
Table 1.
Classification of feature sets
| Number | Feature Set description | Prior knowledge | Relative to group |
|---|---|---|---|
| Ia | Top 10 most differentially expressed genes | No | No |
| Ib | All differentially expressed genes | No | No |
| II | Z-score of differential expression | No | Yes |
| III | Enriched Gene Ontology terms | Yes | Yes |
| IV | Significant upstream regulators | Yes | Yes |
With respect to (i) their use of prior knowledge independent of the current dataset and (ii) the computation of group-dependent features which will require two feature profiles at test time.
2.3.1 Feature Set I: gene-expression values
The simplest feature set relies on the normalized expression values only and reflects commonly used practice when no additional prior knowledge is available. We define two subtypes, namely normalized gene values of top 10 differentially expressed genes according to P-value (Feature Set Ia or TOP10) and (2) normalized gene values of all differentially expressed genes (Feature Set Ib or ALL).
2.3.2 Feature Set II: gene Z-scores
Feature Set II also does not use any prior knowledge, but tries to exploit differences of individual patients better. At training time, all ‘correct’ individual level Z-score profiles and corresponding differentially expressed genes per individual are computed (Fig. 1). The union of these differentially expressed genes constitutes Feature Set II. In contrast to Feature Set I, the computed Z-scores of, say, a responder contains information as to how differentially each gene is expressed with respect to the non-responder group and not only normalized expression values.
2.3.3 Feature Set III: enriched GO categories or GO terms
GO terms are a commonly used form of prior biological knowledge. Each GO terms is a collection of genes labeled with a specific biologically meaningful term, such as ‘insulin receptor signaling pathway (GO:0008286)’. Feature Set III tries to explore the encoded knowledge to improve response prediction. To determine significantly enriched GO terms, we performed enrichment analysis using the TopGO R package (Alexa et al., 2006) on each individual’s differentially expressed genes at training time. The union of all GO terms with a P-value <10– 6 forms Feature Set III. We use the logarithm of the enrichment P-value as features.
2.3.4 Feature Set IV: significant upstream regulators or Bayes-CRE
Finally, we define a feature set that incorporates relevant upstream regulators of downstream gene expression changes. In Zarringhalam et al. (2013), we introduced a Bayesian framework to identify potential upstream regulators and their biological context. The network relies on causal statements extracted from peer-reviewed PubMed full papers. These causal statements can be used to construct a signed causal graph in which the set of nodes consists of transcripts, proteins, or compounds. A directed edge between nodes indicates the existence of a causal relation between the source and the target nodes while the sign of the edge specifies the direction of regulation. For transcriptomics datasets, we are mostly interested in molecular perturbations leading to transcriptional changes.
From this causal graph a Bayesian network is constructed. The network consists of several layers (Fig. 2). The first layer of the network consists of transcript nodes representing observed differentially expressed genes. To account for the noise in gene-expression data, we introduced a second layer of nodes, called true state nodes that are directly above the transcript nodes reflecting the true state of regulation independent of measurement noise. The next layer of nodes in the network consists of regulators. These are the proteins and compounds in the causal network that potentially regulate the transcripts. The regulator nodes are causally linked to the true state nodes as determined by the causal graph. In order to account for conditions under which the causal relations are relevant, we introduced two additional layers of nodes. For each causal relation in the network, an applicability node was defined which is directly connected to the true state node of the corresponding causal relation. Each causal relation is annotated with the PubMed id of the article reporting the causal relation which in turn is annotated with MeSH (Medical Subject Headings) terms. These MeSH terms, e.g. ‘adipogenesis’ or ‘JAK/STAT signaling cascade’ provide additional biological context and are introduced as context nodes in the network. Context nodes are connected to applicability node of their corresponding causal relation.
Fig. 2.
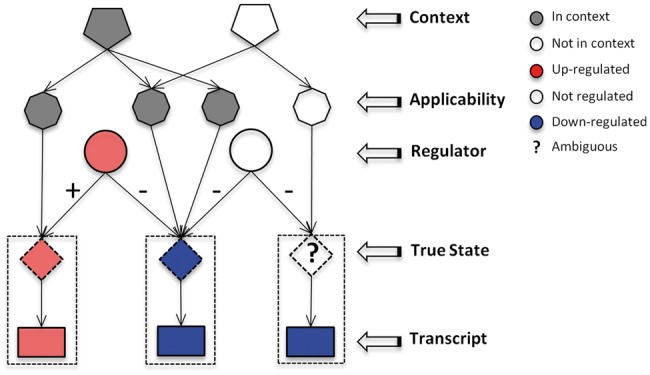
Illustration of the Bayesian Network: for each causal relation, an applicability node is constructed and MeSH terms are used as context nodes. Noise in gene-expression data is accounted for by introducing true state nodes
Based on this network topology, we defined a conditional probability distribution and used a Gibbs-sampling algorithm to query the network and infer upstream regulators, i.e. regulator nodes with high posterior probability given the observed differential gene-expression data. In summary, the input of the inference algorithm are the differentially expressed genes between two conditions and their corresponding direction of regulation (up or down) and the output is the posterior probabilities of regulators, applicability of edges and the contexts with their MeSH terms. More details can be found in Zarringhalam et al. (2013).
Feature Set IV is defined as the union of all significant upstream regulators, i.e. posterior probability >0.4, uncovered for each individual’s differentially expressed genes at training time. The posterior probabilities of regulators constitute the features.
2.4 Predicting response via regularized regression
Given any of the input feature sets, we use an L1-regularized logistic regression approach to train a binary classifier using the R glmnet package (Friedman et al., 2010). In case of Feature Sets II, III and IV (Z-scores, Go-terms and Bayes-CRE), the ‘correct’ profile is known during training and used to train the classifier. For a prediction in the testing phase, features derived from both z-profiles are used and two predictions are made, one for each profile. Each prediction is given a probability by the classifier. If the class predictions are cz1 and cz2 with probabilities pz1 and pz2 for the two profiles, a final decision on class label is made using the following decision function: f(i) = cz1 if pz1 > pz2 and f(i) = cz2 if pz2 > pz1, where i indicated the individual for whom the prediction is made. Algorithm 1 summarizes the approach.

2.5 Validation within the dataset: cross validation
We assessed the accuracy of the feature sets by performing a leave-one-out cross validation. In case of Feature Set I, the i-th example was taken out from the dataset and the classifier was trained on the features from the remaining individuals in the set. A prediction was then made on the class label of the i-th individual. In case of Feature Sets II, III and IV, the i-th example was taken out from the dataset and the classifier was trained on the features from the remaining individuals in the set. As response status for the i-th individual is unknown to the classifier, two predictions were made for the i-th individual using both profiles. Using the decision function as described above, a final decision on the class label of the i-th individual was made.
2.6 Training and testing on independent sets
In case of Feature Set I, classifier was trained on features calculated using the training set and predictions were made on subjects in test set. In case of Feature Sets II, III and IV, the classifier was trained on the correct profile in the training set and predictions were made for individuals in the test set using both profiles. The class label was then decided using the decision function.
3 RESULTS
We applied our pipeline using all feature sets to two relevant phenotypes (acute rejection in kidney transplantation and response to infliximab therapy in ulcerative colitis), each containing two independent datasets.
3.1 Predicting acute rejection in kidney transplantation
In Khatri et al. (2013), the authors identified 11 genes that are significantly over expressed in acute rejection from four organs. They report that the identified genes could diagnose acute rejection with high specificity and sensitivity (AUC = 0.8). We analyzed the dataset generated by the authors (GSE50058) as well as an independent dataset [GSE21374 by Einecke et al. (2010)], also analyzed by Khatri et al. (2013) in a similar fashion. The datasets GSE50058 consists of 43 kidney transplant rejection and 54 non-rejection samples. Dataset GSE21347 consists of 76 kidney transplant rejection and 206 non-rejection samples.
The raw data was processed as described in Section 2, leading to a total of 3601 differentially expressed genes in GSE50058 and 454 differentially expressed genes in GSE21374. Among these differentially expressed genes 334 are shared by both datasets. Combining both datasets and filtering for genes with unique entrez id results in 641 differentially expressed genes. These group-wise differentially expressed genes were used as input to Bayes-CRE and upstream regulators and corresponding context (MeSH) terms were identified. Table 2 summarizes the results. Note that this analysis was not used in predictions and was performed for biological interpretation as outlined later in this section.
Table 2.
Top upstream regulators selected by Bayes-CRE in the Acute Rejection study
| Rank | Upstream regulator | Direction | Probability |
|---|---|---|---|
| 1 | IFNG | Up | 1.00 |
| 2 | LPS | Up | 1.00 |
| 3 | SE LPS | Up | 0.99 |
| 4 | HNF1A | Down | 0.98 |
| 5 | IL2 | Up | 0.97 |
| 6 | HNF4A | Down | 0.95 |
| 7 | Beta-estradiol | Up | 0.93 |
| 8 | TNF | Up | 0.90 |
| 9 | E.coli B4 LPS | Up | 0.87 |
| 10 | Alefacept | Down | 0.74 |
| 11 | MYCN | Down | 0.71 |
| 12 | CXCL12 | Up | 0.71 |
| 13 | NKX2-1 | Down | 0.69 |
| 14 | IRF7 | Up | 0.65 |
| 15 | Poly rI:rC-RNA | Up | 0.63 |
Figure 3 (left and middle panels) shows the achieved specificity and sensitivity for all defined feature sets. Here, we focus on real-world performance in an independent test set. Details of the performance differences between cross validation versus independent test set are depicted in Figure 4 and will be discussed in a separate section. The performance of the TOP10 feature is strikingly different depending on the training set used. Whereas it is the top performer when training on the GSE21374 data, it performs worst when roles are switched. This behavior is plausible when considering the different number of differentially expressed genes in the two datasets. When training on GSE21374 chances are that the top 10 genes out of the 454 differentially expressed genes are also contained in the ≈ 3600 genes that are differentially expressed in GSE50058. However, the odds are reversed when training on GSE50058. The ALL Feature Set does poorly in both datasets. In the larger GSE50058 dataset one explanation might be there are a large number of features to pick from which results in overfitting. However, even when training on GSE21374 the ALL feature performs worst. When comparing accuracy of prediction based on cross-validation, TOP10 and ALL perform almost equally well with an accuracy of 0.78 and 0.79, respectively (Supplementary Table S1). It appears that the classification algorithm picked features that gave a slight advantage under training conditions, but which did not generalize at all to the independent test scenario. The Z-score and GO term features also show variable performance in that they are highly sensitive in one run, but highly specific in the other. The Bayes-CRE features show consistent performance across GSE21374 and GSE50058. While accuracy is slightly higher for the TOP10 feature in the first case and about the same for Z-scores in the second case, only the Bayes-CRE feature is able to retain its performance and, as we will see, is also stable when going from cross-validation to independent test set.
Fig. 3.
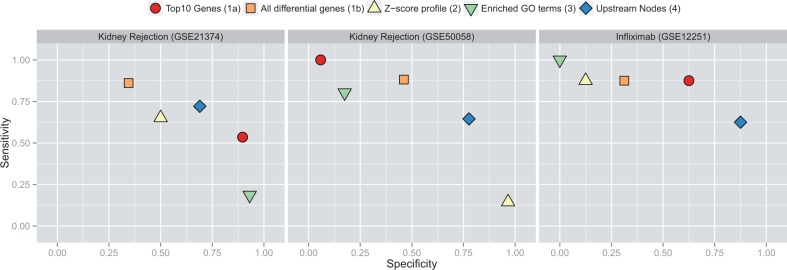
Attained specificity (x-axis) and sensitivity (y-axis) of feature sets in acute rejection datasets (left and middle panels) and response to infliximab in ulcerative colitis (right panel)
Fig. 4.
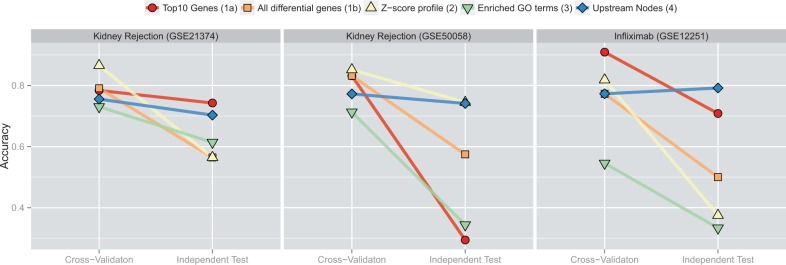
Overall performance comparison of the different feature sets. Each panel shows accuracy of predictions in (left) cross-validation and (right) independent test set
To assess the biological plausibility of the upstream regulators detected by Bayes-CRE, we examined the context of the causal relationships supporting the generated upstream regulators with the aid of the MeSH terms enriched with them. Additionally, we investigated whether the upstream regulators have been previously identified as significant components of acute rejection biology. Table 2 shows the upstream regulators selected by Bayes-CRE on the group-wise comparisons. The direction of regulation of differentially expressed genes in combined normalized datasets was used to generate the table.
Unsurprisingly, several predictive upstream regulator such as upregulation of IFNG, LPS, IL2, TNF, CXCL12 and IRF7 (Table 2) are consistent with heightened immune response and hence higher risk of acute rejection. Furthermore, the associated MeSH terms show abundance of general immunology context some of which can be specifically linked to acute rejection such as MHC Class II, HLA-D antigens and immunodominance. IFNG, one of the most probable upstream regulators, is known for its paramount role in acute rejection. In some skin grafts IFNG has been shown to be necessary for initiating acute graft rejection (Ring et al., 1999). Additionally, IFNG ELISPOT has been proposed as a pre-transplant measurement of donor-specific memory T-cell and subsequently post-transplant risk (Augustine et al., 2005). Alefacept is a medication approved by the FDA for psoriasis and has been suggested as an immunosuppressive agent for kidney transplantation (Cooper and Wiseman, 2010). It is a humanized antibody that is thought to inhibit memory T-cells. Hence, predicted decrease of Alefacept may be a surrogate regulator implying activation of memory T-cells. Once activated, T-cells produce IL2 and other cytokines. In summary, the significant upstream regulators may provide a plausible biological explanation of main events that are predictive of acute rejection, from antigen presentation to T-cell activation and cytokine release.
3.2 Response to infliximab in ulcerative colitis
In Arijs et al. (2009), the authors studied two cohorts (A: GE14580 and B: GE12251) of patients who received treatment of infliximab for refractory ulcerative colitis. They defined response to therapy based on mucosal healing, endoscopic sub-score and histological sub-score at 4 weeks for patients who received a single infusion and 6 weeks for patients who received a loading regimen. In cohort A 24 patients with active ulcerative colitis were studied. Biopsies were collected within a week prior to the first treatment and 4 weeks post-treatment and gene expression was measured. Cohort B was a placebo controlled trial of infliximab therapy in refractory ulcerative colitis where 22 patients with active ulcerative colitis received a loading dose of infliximab and gene expression was measured prior and post treatment. Some key differences between Cohorts A and B include (i) response assessed at week 4 or 6 after infliximab treatment in Cohort A compared to week 8 after infliximab treatment in Cohort B, (ii) Cohort A patients are treated with 5 mg/kg infliximab and Cohort B patients are treated with 5 mg/kg or 10 mg/kg infliximab and (iii) Cohort A patients either have a single infusion or a loading regimen (0, 2 and 6 weeks) while Cohort B patients all received a loading regimen (0, 2 and 6 weeks). The cohorts were independent of one another.
Analysis of differential gene expression resulted in 168 differentially expressed genes in cohort B. Differential gene-expression analysis on cohort A did not result in any significantly expressed genes after FDR correction. Combining the datasets resulted in 280 differentially expressed genes. As in the rejection datasets, these group-wise differentially expressed genes were used as input to Bayes-CRE and upstream regulators and corresponding context (MeSH) terms were identified for biological interpretation purpose. Table 3 summarizes the results of the combined dataset. As before, these results were not used for prediction purposes.
Table 3.
Top upstream regulators selected by Bayes-CRE in response to infliximab treatment in UC patients
| Rank | Upstream regulator | Direction | Probability |
|---|---|---|---|
| 1 | IFNG | Down | 1.00 |
| 2 | LPS | Down | 1.00 |
| 3 | TNF | Down | 0.99 |
| 4 | Retinoic acid | Down | 0.92 |
| 5 | SE LPS | Down | 0.89 |
| 6 | Poly rI:rC-RNA | Down | 0.86 |
| 7 | Decitabine | Down | 0.82 |
| 8 | Allergens | Down | 0.77 |
| 9 | IL1 | Down | 0.62 |
All Feature Sets were generated as described before. Since differential gene-expression analysis on cohort A did not result in any significantly expressed genes, we only used cohort B as training set and cohort A as testing set. The right panel in Figure 3 depicts attained specificity and sensitivity for this dataset. The GO terms, Z-scores and ALL Feature Sets perform poorly on the test set. Picking the TOP10 genes leads to acceptable performance with an emphasis on sensitivity. The best performance is obtained using the Bayes-CRE features with an accuracy of 0.79. As before, the Bayes-CRE predictions emphasize specificity over sensitivity. It is worth noting that the strategy of picking top differentially expressed genes has been employed in the original paper of this study (Arijs et al., 2009). However, the top five genes were picked from the combined cohort. While that is technically correct, the reported performance of 95% sensitivity and 85% specificity is somewhat misleading as it cannot be attained on the individual cohorts. As we will see in Figure 4, similar values are attained in the training cohort here, but cannot be sustained in the independent test.
Similar to the acute rejection study, we examined the biological plausibility of upstream regulators uncovered by Bayes-CRE. Table 3 shows the top upstream regulators selected by Bayes-CRE on the group-wise comparisons. The direction of regulation of differentially expressed genes in combined normalized datasets was used to generate the table.
Ulcerative colitis is a chronic manifestation of inflammation of the colonic mucosa with TNF playing a central role in the disease. Several anti-TNF therapeutics such as infliximab, Aadilumab, Certolizumab, Golimumab are now used in patients who fail to respond to conventional treatment regimens that include immunosuppressive drugs. However a subset of these patients do not respond to anti-TNF therapeutics as well and the biological mechanisms at play here are poorly understood. Interestingly, one of the significant predicted upstream regulators that distinguishes the infliximab responders from the non-responders here is the TNF pathway itself. The higher expression of the TNF pathway components in non-responders may suggest the lack of response to be due to an inadequate infliximab dosing. Also to note that this study has been done in patients who have not been treated with infliximab prior to this study thus ruling out the non-response to infliximab being due to generation of anti-infliximab antibodies. MeSH terms associated with IFNG regulator such as tryptophan and kynurenine metabolism are supported by an observed dysregulation of enzymes such as IDO1, TDO2 and KYNU in these pathways in ulcerative colitis (data not shown). Another MeSH term, enriched for the LPS hypothesis, is the cyclooxygenase term with established links to the disease. A study by Silverberg et al. (2009) shows a genetic association for IFNG locus with ulcerative colitis. Perturbations in the gut microbial flora and colonic mucosal integrity in ulcerative colitis can result in the dysregulation of predicted pathways such as LPS and Interferon. DSS-induced colitis in LPS sensitive mice treated with LPS exhibit a more severe disease while LPS has no effect in DSS-induced colitis in LPS hyporesponsive mice (Lange et al., 1996). Further evidence for a role for LPS in ulcerative colitis comes from studies showing patients with elevated levels of LPS in disease (Rojo et al., 2007). LPS, present only on Gram-negative bacteria binds to TLR4 and induces pro-inflammatory cytokines thus driving inflammation in disease. Furthermore genetic studies show a significant association for TLR4 with ulcerative colitis and Crohns disease (Shen et al., 2010). It can be inferred that the non-responders have an overall increased level of inflammation despite no significant differences in the disease activity scores resulting from the activation of the LPS-TLR4 pathway triggered by the presence of gram-negative bacteria.
3.3 Analysis of prediction performance
One of the key points of this work is that optimization of methods based on one study can lead to misleadingly high estimates of performance and that the use of appropriate prior knowledge can help to avoid the situation. Figure 4 summarizes the situation based on our data and implemented methods. As expected, performance in cross-validation runs is higher than in the independent test sets. In our examples, the TOP10 feature set has one of the highest declines and, in contrast, Bayes-CRE features have a consistently small drop in performance. Performance even slightly improves in the infliximab dataset. It is also interesting to note the spread in accuracy in the different datasets. When training on GSE21374 in the kidney rejection dataset (left panel in Fig. 4), the classifier can pick from a set of ≈450 differentially expressed genes. Prediction is attempted in GSE50058 which has about eight times more differentially expressed (and therefore predictive) genes. As the underlying biology is shared to some extent, chances are higher to pick useful features. This situation reverses when training on GSE50058 as can be observed in the large spread of accuracies in the middle panel of Figure 4.
Unfortunately, it is also not obvious from our data how much a given feature will deteriorate in performance in an independent set. For instance, the TOP10 differentially expressed genes approach works well for two of the three datasets, but does very poorly on the third (middle panel of Fig. 4). Some feature sets oscillate between high specificity and high sensitivity as evident for the Z-score feature in Figure 3. As accuracy is just one well-accepted measure of performance, we also analyzed results based on F-measure and balanced accuracy (see Supplementary Figs S1 and S2) with broadly similar conclusions. As desired, the Bayes-CRE approach seems to be able to use prior knowledge to focus on biologically relevant features that translate well to independent studies.
To analyze why Bayes-CRE features are able to predict stably in this scenario, we performed a bootstrapping analysis by randomly selecting 2/3 of samples as training and 1/3 as testing on each dataset. The process was repeated 100 times and the number of times that the predictive upstream regulator were selected by L1 regularization was recorded. Figure 5 shows how often an upstream regulator was picked in dataset GSE21374 on the x-axis and GSE50058 on the y-axis. In addition, the color indicates whether a upstream regulator plays a role in the combined analysis as referenced in Table 2. Clearly, the most prevalent upstream regulators of IFNG, LPS and TNF are relevant in both datasets. In addition, more dataset specific features can be seen towards the x- and y-axes of the plot. As the underlying biology relating to immune response and inflammation seems to be shared among the datasets, the Bayes-CRE approach is able to integrate the signals from downstream transcripts into a feature, i.e. the posterior probability of regulation that serves well as a predictor. That does not seem to be possible based on the gene-expression data alone. Finally, we also ran permutation experiments to ensure that results are due to the phenotype labels, i.e. (non-) rejection of kidney transplants and response to infliximab treatment, and not random artifacts. Permuted datasets did not result in any or only very few upstream regulators in all permutations. As a consequence, we did not proceed to any higher level feature generation.
Fig. 5.
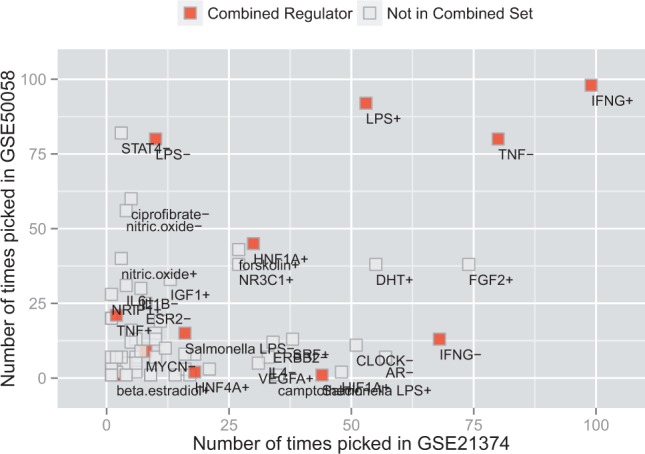
Number of times each upstream regulator was picked as a top predictor by the classifier in dataset GSE21374 (x-axis) and dataset GSE50058 (y-axis) from the acute kidney rejection datasets. Red color indicates whether an upstream regulator plays a role in the combined analysis as referenced in Table 2
4 DISCUSSION
Patient populations exhibit high genetic, environmental and phenotypic heterogeneity. This makes the search for robust predictors in clinical trials a difficult endeavor. If we rely on classical (low-dimensional) biomarkers, prediction performance might not be sufficient. Using high dimensional datasets, e.g. transcriptomics datasets, can easily lead to predictors that have stellar performance in the training trial, but poor performance in an independent study (Fig. 4). This seems to be the case even when all proper considerations of cross-validation or a test/training split within one clinical trial are observed—at least given current sample sizes. Many methods have been developed to avoid overfitting of the data, but as we demonstrate in this work, even a well-accepted regularized classifier will not always be able to pick out the biologically robust features based on expression data alone. We found that any of the defined feature sets is able to predict with high accuracy under a certain set of circumstances. For instance, the Z-score profiles perform best when trained on GSE50058, but very poorly in all other cases (Fig. 4). In contrast, picking the TOP10 genes does not work at all when training on GSE50058, but well in the other cases. The Bayes-CRE feature set retained performance across all scenarios tested and was among the top predictors in independent test sets.
It should be noted that the performance of this method is strongly dependent on the prior knowledge encoded in the underlying knowledgebase. If no relevant upstream regulators are available in the knowledgebase that can aggregate the downstream transcriptional signal, performance will be poor as no useful features can be generated. At the same time, the number of generated features can easily be checked. If there are a number of differentially expressed genes present in a dataset, but regulator nodes receive only weak posterior probabilities, the knowledgebase is likely to contain no relevant biology and other approaches should be pursued. As demonstrated by our results, generalization to independent datasets in the presence of high levels of confounding factors (such as clinical site, exact composition of trial population, etc.) is very difficult and research should be invested to not only find better mathematical approaches to exploit the dataset at hand, but incorporate other prior knowledge in an optimal way. The knowledgebase that we utilize in this work may contain noise as well, however, this noise should be independent of a particular trial population as it tries to describe general biological facts.
Another potential criticism of the Bayes-CRE method is the focus on IFNG, LPS and TNF as key regulators in the assessed datasets. Clearly, these nodes are well-known regulators of immune system and inflammatory processes and would have been picked as relevant by experts in the fields. We evaluated the performance of only using IFNG downstream genes as defined by our knowledgebase. The results are slightly inferior to Bayes-CRE features, but roughly comparable (Supplementary Fig. S3). In contrast to the manual trial and error approach of defining potential downstream genes based on expert opinion per dataset, the Bayes-CRE approaches provides a comprehensive assessment of relevance of all encoded knowledge in the knowledgebase. In our cases, it correctly identified many relevant potential regulators of involved processes facilitating acceptance of the derived classifiers. This points to a need for public, readily available repositories of causal biological knowledge to derive better classifiers and interpret biological datasets more quickly. Protein–protein-interaction databases like IntAct (Orchard et al., 2014) are in the process of adopting their curation process to include causal relationships (S. Orchard, personal communication).
The use of other (non-causal) prior knowledge sources is certainly promising as well. However, we feel that the causal relationships used in this study are well-suited to summarize downstream transcriptional activity into fewer features that are biologically relevant. This might be harder to achieve based on protein–protein-interaction data as no directionality exists in the network and the relationships are not directly related to transcriptional activity. In future work we plan to add large-scale datasets with more complex notion of causality into our methodology as they become available.
It was surprisingly hard to collect public data for this study as we required trials of at least 20 human subjects with a defined clinical binary outcome, i.e. (i) responders and non-responders, (ii) at least some detectable difference in gene expression at baseline between the two groups and (iii) the availability of a similar but entirely independent trial for testing purposes. We expect this situation to change over the coming years as more and more trials generate relevant molecular data and are made available for public research. However, the size of the trials will remain limited as the generation of molecular data is only a small fraction of the cost and overall costs of large clinical trials remain astronomical. This situation will make approaches using prior knowledge even more relevant.
In future work, we plan to test the methods on more datasets as they become available, extend the method to allow for continuous phenotypes and take placebo response for drug trials into account. The latter should be possible by integrating placebo arms of clinical trials into the regularized regression framework. However, we were not able to find suitable publicly available datasets to test such extensions at this point. This will be important to distinguish diagnostic signatures that are predictive of disease progression from signatures that are predictive of drug response itself.
In summary, we have presented a method for prediction of clinical phenotypes based on genome-wide expression data that makes use of a large collection of causal relationships defined from the literature. Features selected by the L1-regularized regression method correspond to upstream molecular entities that can readily be interpreted biologically and subsume sets of transcriptional changes in a useful manner. The method performs well in the analyzed datasets and, importantly, gives stable performance estimates across cross-validation as well as independent test set runs. Given that more and more clinical trials involving heterogeneous populations will become available, methods such as the one presented here can help to make the vision of precision medicine a reality.
Conflict of Interest: none declared.
REFERENCES
- Alexa A, et al. Improved scoring of functional groups from gene expression data by decorrelating go graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- Arijs I, et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut. 2009;58:1612–1619. doi: 10.1136/gut.2009.178665. [DOI] [PubMed] [Google Scholar]
- Ashburner M, et al. Gene ontology: tool for the unification of biology. the gene ontology consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine JJ, et al. Pre-transplant ifn-gamma elispots are associated with post-transplant renal function in african american renal transplant recipients. Am. J. Transplant. 2005;5:1971–1975. doi: 10.1111/j.1600-6143.2005.00958.x. [DOI] [PubMed] [Google Scholar]
- Cooper JE, Wiseman AC. Novel immunosuppressive agents in kidney transplantation. Clin. Nephrol. 2010;73:333–343. doi: 10.5414/cnp73333. [DOI] [PubMed] [Google Scholar]
- Cun Y, Fröhlich HF. Prognostic gene signatures for patient stratification in breast cancer: accuracy, stability and interpretability of gene selection approaches using prior knowledge on protein-protein interactions. BMC Bioinform. 2012;13:69. doi: 10.1186/1471-2105-13-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einecke G, et al. A molecular classifier for predicting future graft loss in late kidney transplant biopsies. J. Clin. Invest. 2010;120:1862–1872. doi: 10.1172/JCI41789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, et al. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, et al. A common rejection module (crm) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J. Exp. Med. 2013;210:2205–2221. doi: 10.1084/jem.20122709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S, et al. The role of the lps gene in experimental ulcerative colitis in mice. APMIS. 1996;104:823–833. doi: 10.1111/j.1699-0463.1996.tb04948.x. [DOI] [PubMed] [Google Scholar]
- McShane LM, et al. Criteria for the use of omics-based predictors in clinical trials. Nature. 2013;502:317–320. doi: 10.1038/nature12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnezami R, et al. Preparing for precision medicine. N. Engl. J. Med. 2012;366:489–491. doi: 10.1056/NEJMp1114866. [DOI] [PubMed] [Google Scholar]
- Orchard S, et al. The mintact project–intact as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring GH, et al. Interferon-gamma is necessary for initiating the acute rejection of major histocompatibility complex class ii-disparate skin allografts. Transplantation. 1999;67:1362–1365. doi: 10.1097/00007890-199905270-00012. [DOI] [PubMed] [Google Scholar]
- Rojo OP, et al. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2007;13:269–277. doi: 10.1002/ibd.20019. [DOI] [PubMed] [Google Scholar]
- Shen X, et al. The toll-like receptor 4 d299g and t399i polymorphisms are associated with crohn’s disease and ulcerative colitis: a meta-analysis. Digestion. 2010;81:69–77. doi: 10.1159/000260417. [DOI] [PubMed] [Google Scholar]
- Silverberg MS, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat. Genet. 2009;41:216–220. doi: 10.1038/ng.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, et al. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl Acad. Sci. USA. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaske CJ, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using paradigm. Bioinformatics. 2010;26:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venet D, et al. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput. Biol. 2011;7:e1002240. doi: 10.1371/journal.pcbi.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarringhalam K, et al. Molecular causes of transcriptional response: a bayesian prior knowledge approach. Bioinformatics. 2013;29:3167–3173. doi: 10.1093/bioinformatics/btt557. [DOI] [PMC free article] [PubMed] [Google Scholar]