

Abstract
We have shown that the receptor tyrosine kinase ErbB4 signals neuregulin1-stimulated proliferation of human cells. Some isoforms of ErbB4 are cleaved to release the soluble intracellular domain p80; however, the function of p80 in cell proliferation remained unclear. Here we propose the possibility for p80 as a negative feedback modulator of ErbB4-mediated cell proliferation. Cells exposed to lower doses of neuregulin1 displayed a stimulated proliferation and contained ErbB4 but barely p80. By contrast, cells exposed to its higher doses displayed a suppressed proliferation and contained p80 but barely ErbB4. Analyses with cells overexpressing the p80 wild type and mutants indicated that nuclear p80 inhibits cell proliferation independently of the tyrosine kinase activity. A screen for a novel protein that interacts with p80 identified α-enolase, which is reported as a transcriptional inhibitor for the proliferation-associated c-myc gene. The c-myc mRNA expression was induced by lower doses of neuregulin1 but was suppressed by its higher doses. Subcellular fractionation demonstrated the localization of not only p80 and α-enolase but also the decrease of the functional c-myc amount in the nuclei of cells exposed to higher doses of neuregulin1. These results suggested that p80, which is generated from ErbB4 and translocates to the nuclei, interacts with α-enolase and inhibits neuregulin1-dependent ErbB4-mediated cell proliferation by impairing the c-myc gene transcription.
Keywords: Growth factor, receptor tyrosine kinase, intracellular domain signaling, cell proliferation, transcriptional regulator
Introduction
Receptor tyrosine kinases of the ErbB family mediate cell proliferation, differentiation, migration, and survival [1]. Not only their deregulated expression but also mutation highly correlates with human tumorigenesis [1]. The family includes ErbB1 or epidermal growth factor (EGF) receptor, ErbB2, ErbB3, and ErbB4 [2-5]. Binding with the EGF family ligands induces the receptor dimerization and activation of the tyrosine kinase activity and then causes the tyrosine autophosphorylation and activation of intracellular signaling pathways that regulate gene transcription, resulting in physiological and pathological outcomes.
ErbB4 is activated by neuregulin (NRG) 1, a member of the neu differentiation factor (heregulin) or NRG subfamily, and then activates the downstream signaling pathways [6]. ErbB4 is widely expressed in human fetal and adult tissues as well as in human breast, ovary, and lung cancers [7-9]. ErbB4 signaling is implicated in the development and homeostasis of the nervous system and the heart [10-12]. ErbB4 can stimulate cell proliferation [13-17]. We have recently demonstrated in human cervical carcinoma HeLa cells a novel ErbB4-mediated signaling pathway that activates the rapid transcription of c-fos gene, a proliferation-associated immediate early gene, through extracellular signal-regulated kinase and the transcription factor serum response factor, and then stimulates cell proliferation in response to NRG1 [18].
Unlike other ErbB proteins, ErbB4 gene generates four isoforms by alternative mRNA splicing. Two of the isoforms differ in the extracellular juxtamembrane region (JM-a and JM-b), and two in the intracellular cytoplasmic domain (CYT-1 and CYT-2) [19,20]. The ErbB4 JM-a isoforms are cleaved within the extracellular juxtamembrane region by tumor necrosis factor-α-converting enzyme and sequentially within the intramembrane region by γ-secretase activity [21,22], releasing the intracellular domain p80. It can translocate to the nuclei and interact with several transcription factors, modulating their activity and then regulating the transcription of target genes [23-29]. In addition, p80 has been shown to control astrogenesis in the developing brain [27] and proliferation and differentiation in the mammary epithelium [30]. Thus, p80 plays a role in a variety of biological processes through transcriptional events. Nevertheless, less is known about how nuclear p80 signaling regulates cellular functions including cell proliferation. In the present study, we found that the accumulation of p80 occurs in parallel with the suppression of cell proliferation in HeLa cells exposed to higher doses of NRG1. We also showed that the suppression of cell proliferation is caused by the overexpression of p80 in the nuclei in a manner that is not dependent on the tyrosine kinase activity. Moreover, we identified with co-immunoprecipitation and proteomics α-enolase that interacts with p80 in the nuclei and suppresses the c-myc gene expression in a manner dependent on higher doses of NRG1. These data suggest a role of p80 in the autoregulation of ErbB4-stimulated cell proliferation, and may provide a novel mechanism for the control of cellular functions by the signaling of an intracellular domain of a receptor tyrosine kinase in the nuclei.
Materials and methods
NRG1 and plasmids
Recombinant NRG1 protein was expressed in bacteria and purified [18]. The expression plasmids for p80 wild type and mutants were constructed [31]: p80, the wild type intracellular domain of the intact ErbB4 and no tags; NLS-p80, a mutant introducing to p80 Flag and hemagglutinin (HA) tags and a nuclear localization signal (NLS) amino-terminally in this order; NLS-p80 KD, the same mutant as NLS-p80 except lacking the tyrosine kinase activity by substituting Lys of residue 751 to Arg.
Cell culture, transfection, and treatment with NRG1
HeLa cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) at 37°C under an atmosphere of 5% CO2. Cells were plated at approx. 5000 cells/well onto 96-well plates, 1 day later serum-starved for 8 h, and treated for 24 h with or without various doses of NRG1, followed by cell proliferation assay and Western blot analyses. For forced expression of the p80 wild type and mutants, cells were seeded at 50% confluency onto 24-well plates and at approx. 5000 cells/well onto 96-well plates, and 1 day later transfected with 0.4 and 0.1 μg of each of the expression plasmids and the empty vector (mock) in OPTI-MEM (Invitrogen) supplemented with 10% FBS using 1.25 and 0.5 μl of FuGENE HD (Promega), respectively, according to the manufacturer’s protocol. The cells were cultured for 45 h after transfection, followed by cell proliferation assay, Western blot analyses, and immunofluorescence staining.
Cell proliferation assay
The proliferation activity of cells was determined [21]. Briefly, cells were cultured in 100 μl/well of the medium in a 96-well plate for the indicated periods after exposure to NRG1 and transfection with each of the expression plasmids, and incubated for further 2 h after 10 μl/well of Cell Counting Kit-8 reagent (Dojindo) was added. The proliferation was determined by measuring the absorbance of 490 nm in each well using a micro-plate reader (Bio-Rad).
Subcellular fractionation
Confluent cells grown on 35-mm dishes were deprived of serum for 8 h, treated for 24 h with 50 or 200 ng/ml NRG1, and then lyzed for 1 h on ice with 150 μl of cytoplasmic extraction buffer (CEB: 50 mM PIPES/KOH, pH 7.4, 50 mM KCl, 5 mM EGTA, 2 mM MgCl2, 0.1% Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride (PMSF), protease inhibitors (protease inhibitor cocktail, Roche), phosphatase inhibitors (Calbiochem)) under occasional rotation. The extracts were collected and centrifuged at 17900 g for 10 min at 4°C, and the supernatants were used as the cytoplasmic fraction. The nuclei leaving on the same dishes were extracted with 150 μl of SDS-PAGE sample buffer (125 mM Tris/HCl, pH 6.8, 10% sucrose, 4% SDS, 0.08% bromophenol blue, 10% β-mercaptoethanol), and the extracts were used as the nuclear fraction. The cytoplasmic and nuclear fractions were added with equal volumes of SDS-PAGE sample buffer and CEB, respectively, and then the same volumes of the mixtures were loaded onto SDS-PAGE gels, followed by Western blot analyses.
Western blotting
For preparation of whole extracts, cells cultured in 96- and 24-well plates were lyzed with 10 and 50 μl/well of SDS-PAGE sample buffer, respectively. The whole extracts and the subcellular fractions described above (10 μl, each) were subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes (Immobilon-P Transfer Membrane, Millipore). After blocking with 5% non-fat milk or Blocking One-P (1:20) (Nacalai Tesque) in Tris-buffered saline containing 0.1% Tween-20, the membranes were probed with a primary antibody to ErbB4 (1:1000) (Santa Cruz), phosphorylated tyrosine residues (4G10, 1:1000) (Upstate), α-enolase (1:1000) (Santa Cruz), calpain 1 (1:1000) (Sigma), histone H3 (1:3000) (Sigma), c-myc (1:1000) (Takara), or β-actin (1:3000) (Millipore), detected with the appropriate secondary antibodies conjugated with horseradish peroxidase (GE Healthcare), and developed with an enhanced chemiluminescence system (ImmobilonTM Western Chemiluminescence HRP Substrate, Millipore).
Immunofluorescence
Cells grown on coverslips were fixed for 15 min at room temperature with 4% paraformaldehyde in phosphate-buffered saline (PBS), permeabilized for 10 min at room temperature with 0.2% Triton X-100 in PBS, and blocked for 60 min at room temperature with 5% normal swine serum (Vector) in PBS containing 0.3% Triton X-100. The coverslips were incubated for 1 h at room temperature with 2 μg/ml (diluted in PBS containing 0.3% Triton X-100) of the polyclonal rabbit antibody to ErbB4 or normal rabbit immunoglobulin (Dako) as the control. After washing with PBS extensively, the coverslips were incubated with for 1 h at room temperature with a swine anti-rabbit immunoglobulin antibody conjugated with fluorescein isothiocyanate (Dako) (1:500, diluted in PBS containing 0.3% Triton X-100). After washing with PBS, the coverslips were counterstained with 0.5 μg/ml 4’,6-diamidino-2-phenylindole (DAPI) and then observed for fluorescence under a microscope (BX60, Olympus).
Co-immunoprecipitation and proteomics
Immunoprecipitation was performed using FLAG HA Tandem Affinity Purification Kit (Sigma) according to the manufacturer’s instructions. Extracts (approx. 4 mg of protein, each) were prepared from cells transfected with the expression plasmid for NLS-p80, the expression plasmid for NLS-p80 KD, or the empty vector (mock) by using RIPA buffer (50 mM Tris/HCl, pH 8.0, 150 mM NaCl, 1% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS) containing 1 mM PMSF, protease inhibitors, and phosphatase inhibitors, followed by centrifugation at 17900 g for 10 min at 4°C. The supernatants were precleared for 6 h at 4°C with protein G-sepharose beads (GE Healthcare) under constant rotation, and incubated for 16 h at 4°C with anti-Flag M2 resin under constant rotation. After centrifugation at 3000 g for 30 s at 4°C, the immunoprecipitates were washed extensively. Coprecipitated proteins were eluted from the immunoprecipitates with 150 ng/μl of the Flag peptide, resolved on SDS-PAGE, and stained with Oriole Fluorescent Gel Stain (Bio-Rad), followed by proteomics. Proteins within the gel-excised bands were reduced, carboxyamido-methylated, and digested to peptides with trypsin. The resulting peptides were subjected to LC-MS/MS. Fragmentation data were used to search the National Center for Biotechnology Information database using the MASCOT search engine. Probability-based MASCOT scores were used to evaluate identifications of proteins. Only matches with p values < 0.05 for random occurrence were considered significant.
RT-PCR
Confluent cells grown on 35-mm dishes were deprived of serum for 8 h and treated for 24 h with or without each of 50 and 200 ng/ml NRG1. Total RNA was extracted from the cells by using ISOGENE (Nippon Gene) and treated with RNase-free DNase I (Takara), and then 1 μg of the total RNA was reverse-transcribed using random hexamers with a reverse transcriptase Superscript III (Invitrogen). In all, 10% of the reaction mixture was subjected as the template to PCR with Ex Taq DNA polymerase (Takara) as follows: for 35 cycles at 95°C for 30 s, at 62°C for 30 s, and at 72°C for 30 s using forward and reverse primers 5’-TCC AGC TTG TAC CTG GAG GAT CTG A-3’ and 5’-CCT CCA GCA GAA GGT GAT CCA GAC T-3’ for c-myc; for 30 cycles at 95°C for 30 s, at 56°C for 30 s, and at 72°C for 30 s using forward and reverse primers 5’-GTC AGT GGT GGA CCT GAC CT-3’ and 5’-TGA GCT TGA CAA AGT GGT CG-3’ for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The PCR products were separated by 1% agarose gel electrophoresis and detected by ethidium bromide staining.
Statistics
Data were presented as the mean ± S.E. For statistical comparison, Student’s t-test was used. P values < 0.05 were considered to be statistically significant.
Others
Protein concentrations were estimated by Coomassie dye binding (Bio-Rad) using bovine serum albumin as a standard. For some experiments, signals detected in the Western blotting and RT-PCR were quantified by densitometry using the Image-J software that had been developed by National Institutes of Health.
Results and discussion
The presence of ErbB4 or p80 confers a positive or negative response of cell proliferation to NRG1
We and others have previously shown that ErbB4 mediates NRG1-stimulated cell proliferation [13-18]. To further clarify the dose response of cell proliferation to NRG1, HeLa cells were cultured with various doses of NRG1 and then the cell proliferation was determined (Figure 1A). We found that ErbB4-mediated cell proliferation was stimulated by lower doses of NRG1 but was inhibited by its higher doses, consistent with our previous data [18]. To dissolve the reason, we examined changes in the endogenous ErbB4 expression and p80 amount to the various doses of NRG1 (Figure 1B). Both ErbB4 and p80 can be detected with the antibody used here because it recognizes the carboxyl-terminal region of ErbB4. At the lower doses exhibiting an enhanced proliferation, ErbB4, but no p80, was detected. By contrast, at the higher doses exhibiting a suppressed proliferation, the ErbB4 expression decreased but the p80 amount increased. Thus, the presence of ErbB4 and p80 is associated with the stimulation and suppression of cell proliferation, respectively. Cleavable isoforms of ErbB4 may be expressed in the plasma membrane of HeLa cells and stimulate NRG1-dependent cell proliferation by activating a downstream signaling pathway [18]. On the other hand, p80 may be released from cleavable isoforms of ErbB4 and accumulate within cells under pathophysiological conditions such as the actions of higher doses and prolonged periods of NRG1 that leads to the excessive activation of ErbB4, resulting in the suppression of NRG1-dependent cell proliferation.
Figure 1.
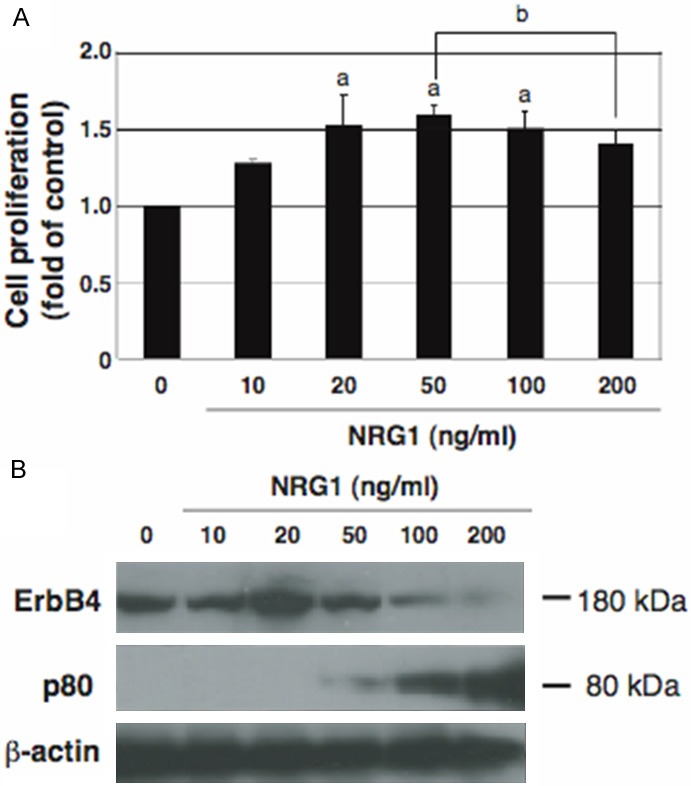
Dose responses of cell proliferation and the ErbB4 and p80 amounts to NRG1. A: Cells were deprived of serum for 8 h and treated for 24 h without (0) or with the indicated doses of NRG1, followed by cell proliferation assay. Data are presented as mean ± S.E. (fold of control) of relative values normalized to the mean value of control cells cultured without NRG1, obtained from three independent experiments. a and b, p < 0.05 vs control cells and cells treated with 50 ng/ml NRG1, respectively. B: The same cells used for cell proliferation assay were analyzed by Western blotting for endogenous ErbB4, p80, and β-actin as the loading control.
Nuclear p80 abrogates cell proliferation independently of the tyrosine kinase activity
Next we tested whether and where p80 suppresses cell proliferation. Because p80 is reported to be localized both in the cytoplasm and in the nuclei [32], we transfected cells with each of the expression plasmids for p80, NLS-p80, and NLS-p80 KD (Figure 2A), and examined their tyrosine autophosphorylation (Figure 2B), subcellular localization (Figure 2C), and effects on cell proliferation (Figure 2D). Normal proliferation was observed in the control cells (mock), where endogenous p80 expression and autophosphorylation were undetectable. Compared to in the control cells, proliferation was drastically suppressed in cells overexpressing p80, where it underwent autophosphorylation and was localized both in the cytoplasm and in the nuclei. Similarly, in cells overexpressing NLS-p80, where it underwent autophosphorylation and was localized only in the nuclei, proliferation was suppressed. Strikingly, in cells overexpressing NLS-p80 KD, where it did not undergo autophosphorylation and was localized in the nuclei, proliferation was suppressed. These results suggested that nuclear p80 is responsible for the suppression of cell proliferation in a manner independent of the tyrosine kinase activity. The activity is required for ErbB4 signaling via the intracellular pathways, but not probably required for p80 signaling, which is consistent with the previous report [26], eliminating the possibility that nuclear p80 regulates cell proliferation by phosphorylating and activating intracellular pathways. Moreover, cells overexpressing each of exogenous p80 wild type and mutants led to a higher level of p80 abundance and a more potent inhibition of proliferation than cells accumulating endogenous p80 upon higher doses of NRG1 (Figures 1A and 2D), implicating a large amount of p80 in the effective suppression of cell proliferation (Figures 1B and 2B).
Figure 2.
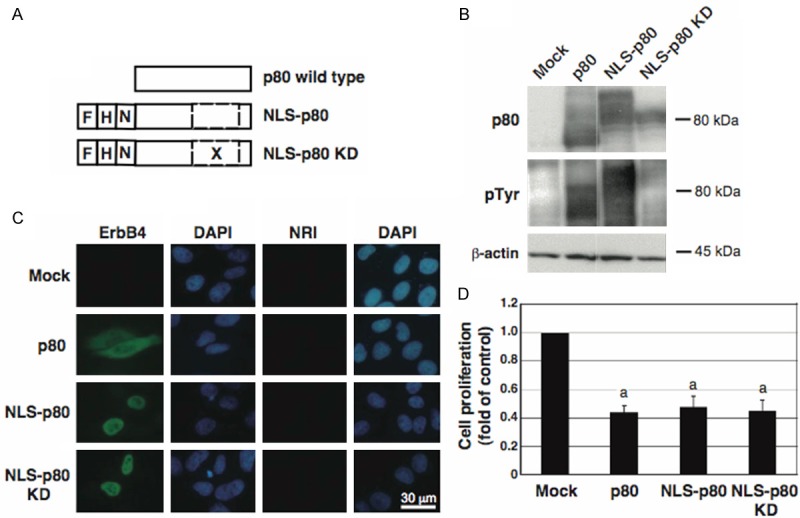
Effects of forced expression of the p80 wild type and mutants on their autophosphorylation and subcellular localization and on cell proliferation. A: The schematic structures of p80, NLS-p80, and NLS-p80 KD. F, a Flag tag; H, a HA tag; N, a NLS; X, a point mutation substituting Lys of residue 751 to Arg; a dashed box, the tyrosine kinase region. B-D: Cells were transfected with each of the expression plasmids for p80, NLS-p80, and NLS-p80 KD, or the empty vector (Mock), and analyzed by Western blotting for p80, phosphorylated tyrosine residues (pTyr), and β-actin (B), by immunofluorescence staining for p80 (green) (C), and by cell proliferation assay (D). C: Cells were incubated with an antibody against ErbB4 or normal rabbit immunoglobulin (NRI), and counterstained with DAPI (blue). D: Data are presented as mean ± S.E. (fold of control) of relative values normalized to the mean value of the control cells, obtained from three independent experiments. a, p < 0.01.
Nuclear p80 regulation of α-enolase function represses the c-myc gene expression
To determine how nuclear p80 suppresses cell proliferation, we performed co-immunoprecipitation separately from each of cells overexpressing the two p80 mutants and then proteomics for proteins that interact with nuclear p80 (Figure 3A). Compared with the control cells (mock), some proteins were identified commonly in co-immunoprecipitates from each of the transfectants, and we investigated α-enolase, which is indicated by an arrowhead of No. 4 shown in Figure 3A. α-enolase (47 kDa), also called enolase 1, is well known as a glycolytic enzyme resided in the cytosol, but, interestingly, as well as its alternative translation product c-myc-binding protein-1 (37 kDa) corresponding to the carboxyl-terminal region, is reported as a transcriptional inhibitor resided in the nuclei for c-myc gene, an immediate early gene [33-36]. This gene encodes a transcription factor that regulates transcription of target genes and plays a role in cell proliferation and transformation [37]. Because α-enolase may thus exert transcriptional inhibition of cell proliferation-associated c-myc gene, we proposed a hypothesis that the interaction between p80 and α-enolase in the nuclei causes the inhibition of ErbB4-stimulated cell proliferation by repressing the c-myc gene transcription.
Figure 3.
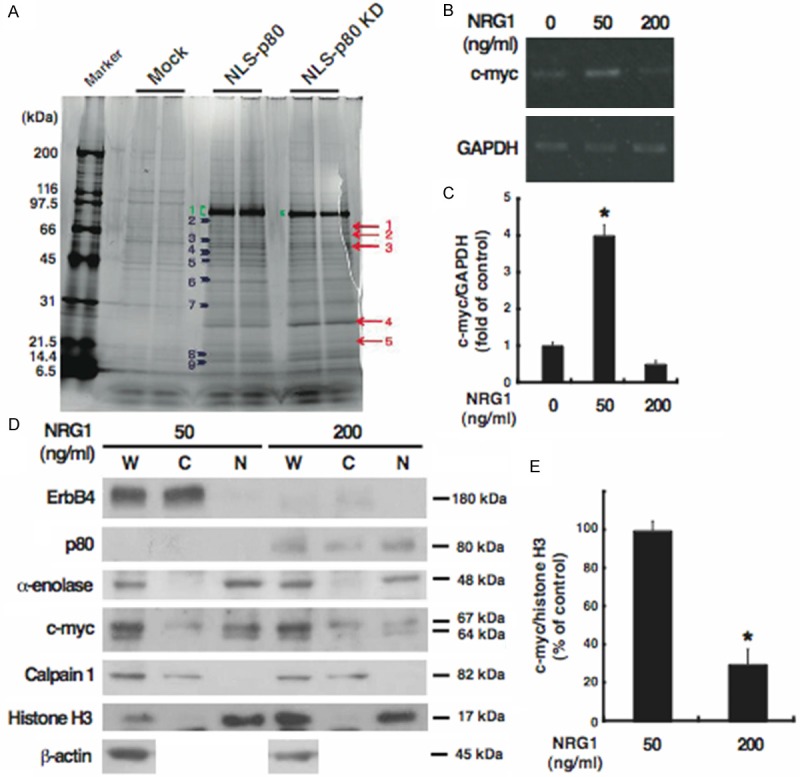
Identification and subcellular distribution of α-enolase as a novel p80-binding protein. A: Cells were transfected with each of the expression plasmids for NLS-p80 and NLS-p80 KD, or the empty vector (Mock), and subjected to immunoprecipitation with p80. Coimmunoprecipitates were loaded onto SDS-PAGE gels and stained by Oriole Fluorescent Gel Stain, followed by proteomics. Proteins detected in both of the cells overexpressing NLS-p80 or NLS-p80 KD but not in the control cells are indicated by arrows; proteins detected more strongly in both of the cells overexpressing NLS-p80 or NLS-p80 KD than in the control cells are indicated by arrowheads; NLS-p80 and NLS-p80 KD are indicated by parentheses. A protein of the band No. 4, indicated by an arrowhead, is α-enolase. B and C: Total RNA was extracted from cells treated for 24 h without (0) or with each of 50 and 200 ng/ml NRG1 and analyzed by RT-PCR for c-myc (338 bp) and GAPDH (212 bp) as the internal control. Results were representative of three independent experiments (B) and quantified by densitometry as a fold (mean ± S.E.) of c-myc expression, normalized to GAPDH expression, in cells treated with NRG1, relative to c-myc expression in untreated cells (C). D and E: Whole extracts (W) and the cytoplasmic (C) and nuclear (N) fractions were prepared from cells treated for 24 h with 50 or 200 ng/ml NRG1 and analyzed by Western blotting for the indicated proteins. For c-myc, the alternative translation initiations from an upstream, in-frame non-AUG (CUG) and a downstream AUG start site result in the production of two isoforms with distinct amino-termini (67 and 64 kDa) [41]. Results were representative of three independent experiments (D) and quantified by densitometry as a percentage (mean ± S.E.) of c-myc amounts (the sum of the 67-kDa and 64-kDa protein), normalized to histone H3 amounts, in the nuclear fraction derived from cells treated with NRG1, relative to c-myc amounts in the nuclear fraction derived from untreated cells (E). *, p < 0.05.
To test this hypothesis, we examined the expression of c-myc mRNA in cells exposed to lower or higher doses of NRG1 (Figure 3B and 3C). Treatment with lower doses of NRG1 stimulated cell proliferation and did not produce p80 whereas treatment with its higher doses suppressed cell proliferation and produced p80 (Figure 1). Compared to in cells cultured without NRG1 (control), the c-myc mRNA expression was enhanced in cells exposed to a lower dose (50 ng/ml) of NRG1. By contrast, the c-myc mRNA expression in cells exposed to a higher dose (200 ng/ml) of NRG1 was attenuated to that in the control cells. The differential responses of the c-myc mRNA expression to NRG1 were consistent with those of p80 accumulation and cell proliferation (Figure 1). Accordingly, to clarify whether the decrease of c-myc mRNA results in the decrease of the functional protein that leads to the decrease of cell proliferation, we fractionated cells exposed to lower or higher doses of NRG1 into the cytoplasmic fraction, including plasma membrane proteins, cytosol proteins, and organelle proteins, and the nuclear fraction, and examined the subcellular distribution of endogenous p80, α-enolase, and c-myc (Figure 3D and 3E). The subcellular fractionation was evaluated using calpain 1 as a cytoplasmic marker [38] and histone H3 as a nuclear marker [39]; β-actin was used as a loading control in the whole extracts [40] (Figure 3D). In cells exposed to a lower dose (50 ng/ml) of NRG1, ErbB4 was detected only in the cytoplasmic fraction, but no p80 was detected in any fractions. By contrast, in cells exposed to a higher dose (200 ng/ml) of NRG1, ErbB4 was barely detected in any fractions, but p80 was detected in both the cytoplasmic and nuclear fractions. These results suggested that endogenous ErbB4 is localized in the plasma membrane of intact cells, but following NRG1 stimulation of the cells p80 is released from ErbB4 and localized to both the cytoplasm and nuclei, consistent with subcellular distribution of exogenous p80 (Figure 2C). On the other hand, strikingly, α-enolase and, expectedly, c-myc were exclusively detected in the nuclear fraction of cells exposed to the lower dose of NRG1. In response to the higher dose of NRG1, the amount of nuclear α-enolase was left unchanged whereas the amount of nuclear c-myc decreased. Therefore, the c-myc gene expression may be controlled by NRG1 at the transcriptional level. These results suggested that endogenous α-enolase and c-myc are predominantly localized in the nuclei of intact cells, but following NRG1 stimulation of the cells the activation of the α-enolase’s transcription-inhibiting activity induces the suppression of the c-myc gene transcription, resulting in the decrease of c-myc protein. This decrease could be caused by the presence of both p80 and α-enolase within the nuclei, but not by the presence of α-enolase only, implying the recruitment of α-enolase by p80 to the promoter region of c-myc gene to repress the α-enolase-mediated c-myc gene transcription. Collectively, p80 may interact with α-enolase mainly within the nuclei in a manner dependent on higher doses of NRG1, downregulating the c-myc gene expression and thereby cell proliferation.
In conclusion, the current study reports a role of the soluble intracellular p80 in the regulation of cell proliferation and some features of the molecular mechanisms: p80 is produced from cleavable isoforms of ErbB4 and translocates to the nuclei in response to NRG1; the interaction of p80 with α-enolase suppresses ErbB4-stimulated cell proliferation through the α-enolase-mediated repression of the c-myc gene expression. Thus, p80 may function as a transcriptional regulator together with α-enolase to control NRG1-dependent ErbB4-mediated cell proliferation properly. Alternately, p80 may block cell proliferation and stimulate cell differentiation in response to NRG1, as supported by previous reports [29,32]. This report might provide first evidence for the presence of a negative feedback regulation to prevent cells from aberrant proliferation by the signaling of an intracellular domain of a receptor tyrosine kinase.
Acknowledgements
We thank Dr. Norie Araki (Kumamoto University, Kumamoto, Japan) for her technical support of proteomics. This work was in part supported by a research grant (121094) from the Sumitomo Foundation, Tokyo, Japan and a Grants-in-Aid for Scientific Research (22247008) from the Ministry of Education, Science, Sports and Culture of Japan.
Disclosure of conflict of interest
None.
References
- 1.Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 2.Kraus MH, Issing W, Miki T, Popescu NC, Aaronson SA. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: evidence for overexpression in a subset of human mammary tumors. Proc Natl Acad Sci U S A. 1989;86:9193–7. doi: 10.1073/pnas.86.23.9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prigent SA, Lemoine NR. The type 1 (EGFR-related) family of growth factor receptors and their ligands. Prog Growth Factor Res. 1992;4:1–24. doi: 10.1016/0955-2235(92)90002-y. [DOI] [PubMed] [Google Scholar]
- 4.Sarkar FH, Ball DE, Li YW, Crissman JD. Molecular cloning and sequencing of an intron of Her-2/neu (ERBB2) gene. DNA Cell Biol. 1993;12:611–5. doi: 10.1089/dna.1993.12.611. [DOI] [PubMed] [Google Scholar]
- 5.Plowman GD, Culouscou JM, Whitney GS, Green JM, Carlton GW, Foy L, Neubauer MG, Shoyab M. Ligand-specific activation of HER/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc Natl Acad Sci U S A. 1993;90:1746–50. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter G. ErbB-4: mechanism of action and biology. Exp Cell Res. 2003;284:66–77. doi: 10.1016/s0014-4827(02)00100-3. [DOI] [PubMed] [Google Scholar]
- 7.Srinivasan R, Poulsom R, Hurst HC, Gullick WJ. Expression of the c-erbB-4/HER4 proteins and mRNA in normal human fetal and adult tissues and in a survey of nine solid tumour types. J Pathol. 1998;185:236–45. doi: 10.1002/(SICI)1096-9896(199807)185:3<236::AID-PATH118>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 8.Aguilar Z, Akita RW, Finn RS, Ramos BL, Pegram MD, Kabbinavar FF, Pietras RJ, Pisacane P, Sliwkowski MX, Slamon DJ. Biologic effects of heregulin/neu differentiation factor on normal and malignant human breast and ovarian epithelial cells. Oncogene. 1999;18:6050–62. doi: 10.1038/sj.onc.1202993. [DOI] [PubMed] [Google Scholar]
- 9.Al Moustafa AE, Alaoui-Jamali M, Paterson J, O’Connor-McCourt M. Expression of P185-erbB-2, P160erbB-3, P180erbB-4, and heregulin alpha in human normal bronchial epithelial and lung cancer cell lines. Anticancer Res. 1999;19:481–6. [PubMed] [Google Scholar]
- 10.Rio C, Rieff HI, Qi P, Corfas G. Neuregulin and erbB receptors play a critical role in neuronal migration. Neuron. 1997;19:39–50. doi: 10.1016/s0896-6273(00)80346-3. [DOI] [PubMed] [Google Scholar]
- 11.Chen S, Rio C, Ji RR, Dikkes P, Coggeshall RE, Woolf CJ, Corfas G. Disruption of ErbB4 receptor signaling in adult non-myelinating Schwann cells causes progressive sensory loss. Nat Neurosci. 2003;6:1186–93. doi: 10.1038/nn1139. [DOI] [PubMed] [Google Scholar]
- 12.Bersell K, Arab S, Harling B, Kühn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–70. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 13.Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. 1994;91:7727–31. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, Su Huang HJ. A common mutant epidermal growth factor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996;56:5079–86. [PubMed] [Google Scholar]
- 15.Russell KS, Stern DF, Polverini PJ, Bender JR. Neuregulin activation of ErbB receptors in vascular endothelium leads to angiogenesis. Am J Physiol Heart Circ Physiol. 1999;277:H2205–H2211. doi: 10.1152/ajpheart.1999.277.6.H2205. [DOI] [PubMed] [Google Scholar]
- 16.Kainulainen V, Sundvall M, Maatta JA, Santiestevan E, Klagsbrun M, Elenius K. A natural erbB4 isoform that does not activate phosphoinositide 3-kinase mediates proliferation but not survival or chemotaxis. J Biol Chem. 2000;275:8641–9. doi: 10.1074/jbc.275.12.8641. [DOI] [PubMed] [Google Scholar]
- 17.Gilmour L, Macleod KG, McCaig AGullic WJ, Smyth JF, Langdon SP. Expression of erbB-4/HER-4 growth factor receptor isoforms in ovarian cancer. Cancer Res. 2001;61:2169–76. [PubMed] [Google Scholar]
- 18.Eto K, Hommyo A, Yonemitsu R, Abe S. ErbB4 signals neuregulin1-stimulated cell proliferation and c-fos gene expression through phosphorylation of serum response factor by mitogen-activated protein kinase cascade. Mol Cell Biochem. 2010;339:119–25. doi: 10.1007/s11010-009-0375-z. [DOI] [PubMed] [Google Scholar]
- 19.Elenius K, Corfas G, Paul S, Choi CJ, Rio C, Plowman GD, Klagsbrun M. A novel juxtamembrane domain isoform of HER4/ErbB4. Isoform-specific tissue distribution and differential processing in response to phorbol ester. J Biol Chem. 1997;272:26761–8. doi: 10.1074/jbc.272.42.26761. [DOI] [PubMed] [Google Scholar]
- 20.Elenius K, Choi CJ, Paul S, Santiestevan E, Nishi E, Klagsbrun M. Characterization of a naturally occurring ErbB4 isoform that does not bind or activate phosphatidyl inositol 3-kinase. Oncogene. 1999;18:2607–15. doi: 10.1038/sj.onc.1202612. [DOI] [PubMed] [Google Scholar]
- 21.Rio C, Buxbaum JD, Peschon JJ, Corfas G. Tumor necrosis factor α-converting enzyme is required for cleavage of erbB4/HER4. J Biol Chem. 2000;275:10379–87. doi: 10.1074/jbc.275.14.10379. [DOI] [PubMed] [Google Scholar]
- 22.Ni CY, Murphy MP, Golde TE, Carpenter G. α-Secretase cleavage and nuclear localization of ErbB4 receptor tyrosine kinase. Science. 2001;294:2179–81. doi: 10.1126/science.1065412. [DOI] [PubMed] [Google Scholar]
- 23.Komuro A, Nagai M, Navin NE, Sudol M. WW domain-containing protein YAP associates with ErbB-4 and acts as a cotranscriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J Biol Chem. 2003;278:33334–41. doi: 10.1074/jbc.M305597200. [DOI] [PubMed] [Google Scholar]
- 24.Williams CC, Allison JG, Vidal GA, Burow ME, Beckman BS, Marrero L, Jones FE. The ERRB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5 nuclear chaperon. J Cell Biol. 2004;167:469–78. doi: 10.1083/jcb.200403155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y, Sullivan LL, Nair SS, Williams CC, Pandey AK, Marrero L, Vadlamudi RK, Lones FE. Coregulation of estrogen receptor by ERBB4/HER4 establishes a growth-promoting autocrine signal in breast tumor cells. Cancer Res. 2006;66:7991–8. doi: 10.1158/0008-5472.CAN-05-4397. [DOI] [PubMed] [Google Scholar]
- 26.Linggi B, Carpenter G. ErbB-4 s80 intracellular domain abrogates ETO2-dependent transcriptional repression. J Biol Chem. 2006;281:25373–80. doi: 10.1074/jbc.M603998200. [DOI] [PubMed] [Google Scholar]
- 27.Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G. Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell. 2006;127:185–97. doi: 10.1016/j.cell.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 28.Sundvall M, Veikkolainen V, Kurppa K, Salah Z, Tvorogov D, van Zoelen EJ, Aqeilan R, Elenius K. Cell death or survival promoted by alternative isoforms of ErbB4. Mol Biol Cell. 2010;21:4275–86. doi: 10.1091/mbc.E10-04-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paatero I, Jokilammi A, Heikkinen PT, Iljin K, Kalliontemi OP, Jones FE, Jaakkola PM, Elenius K. Interaction with ErbB4 promotes hypoxia-inducible factor-1α signaling. J Biol Chem. 2012;287:9659–71. doi: 10.1074/jbc.M111.299537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muraoka-Cook RS, Sandahl MA, Strunk KE, Miraglia LC, Husted C, Hunter DM, Elenius K, Chodosh LA, Earp HS 3rd. ErbB4 splice variants Cyt1 and Cyt2 differ by 16 amino acids and exert opposing effects on the mammary epithelium in vivo. Mol Cell Biol. 2009;29:4935–48. doi: 10.1128/MCB.01705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishibashi K, Fukumoto Y, Hasegawa H, Abe K, Kubota S, Aoyama K, Kubota S, Nakayama Y, Yamaguchi N. Nuclear ErbB4 signaling through H3K9me3 that is antagonized by EGFR-activated c-Src. J Cell Sci. 2013;126:625–37. doi: 10.1242/jcs.116277. [DOI] [PubMed] [Google Scholar]
- 32.Linggi B, Cheng QC, Rao AR, Carpenter G. The ErbB4-s80 intracellular domain is a constitutively active tyrosine kinase. Oncogene. 2006;25:160–3. doi: 10.1038/sj.onc.1209003. [DOI] [PubMed] [Google Scholar]
- 33.Subramanian A, Miller DM. Structural analysis of alpha-enolase. Mapping the functional domains involved in down-regulation of the c-myc protooncogene. J Biol Chem. 2000;275:5958–65. doi: 10.1074/jbc.275.8.5958. [DOI] [PubMed] [Google Scholar]
- 34.Chang YS, Wu W, Walsh G, Hong WK, Mao L. Enolase-alpha is frequently down-regulated in non-small cell lung cancer and predicts aggressive biological behavior. Clin Cancer Res. 2003;9:3641–4. [PubMed] [Google Scholar]
- 35.Perconti G, Ferro A, Amato F, Rubino P, Randazzo D, Wolff T, Feo S, Giallongo A. The Kelch protein NS1-BP interacts with alpha-enolase/MBP-1 and is involved in c-Myc gene transcriptional control. Biochim Biophys Acta. 2007;1773:1774–85. doi: 10.1016/j.bbamcr.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Hsu KW, Hsieh RH, Lee YH, Chao CH, Wu KJ, Tseng MJ, Yeh TS. The activated Notch1 receptor cooperates with α-enolase and MBP-1 in modulating c-myc activity. Mol Cell Biol. 2008;28:4829–42. doi: 10.1128/MCB.00175-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuhrmann G, Rosenberger G, Grusch M, Klein N, Hofmann J, Krupitza G. The MYC dualism in growth and death. Mutat Res. 1999;437:205–17. doi: 10.1016/s1383-5742(99)00084-8. [DOI] [PubMed] [Google Scholar]
- 38.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 39.Lai S, O’Callaghan B, Zoghbi H, Orr HT. 14-3-3 binding to ataxin-1 (ATXN1) regulates its dephosphorylation at Ser-776 and transport to the nucleus. J Biol Chem. 2011;286:34606–16. doi: 10.1074/jbc.M111.238527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yilmaz A, Fernandez S, Lairmore MD, Boris-Lawrie K. Coordinate enhancement of transgene transcription and translation in a lentiviral vector. Retrovirology. 2006;3:13. doi: 10.1186/1742-4690-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hann SR, King MW, Bentley DL, Anderson CW, Eisenman RN. A non-AUG translational initiation in c-myc exon 1 generates a N-terminally distinct polypeptide whose synthesis is disrupted in Burkitt’s lymphoma. Cell. 1998;52:185–95. doi: 10.1016/0092-8674(88)90507-7. [DOI] [PubMed] [Google Scholar]