

Summary
Non-viral gene delivery systems are promising as they avoid many problems of viral gene therapy by having increased design flexibility, high safety, large DNA cargo capacity, and ease of manufacture. Here, we describe the use of polymeric vectors, in particular biodegradable poly(β-amino esters) (PBAEs), for non-viral gene delivery. These polymers are able to self-assemble with DNA and form positively charged gene delivery nanoparticles. Methods for synthesis of these polymers, particle self-assembly, and transfection using these particles are delineated. A standard protocol is presented as well as a high-throughput screening technique that can be used to more quickly optimize transfection parameters for efficient delivery.
Keywords: Gene therapy, Drug delivery, Polymers, Transfection, High-throughput screening
1. Introduction
Gene delivery is a valuable research tool and also has strong therapeutic potential to treat many human diseases, from monogenic diseases to cancer. However, this potential has not yet been fully realized because a safe and efficient method for gene delivery has not yet been developed. Viral vectors have several safety concerns (e.g., immunogenicity, reversion to the wild type), limited DNA carrying capacity, and production/quality control challenges (1,2). Engineered gene delivery biomaterials have increasingly been shown to address these concerns, but have lower efficiency than viruses (3). Many biomaterials have been used for gene delivery including cationic polymers, liposomes, dendrimers, chitosans, and inorganic nanoparticles (1,4). Cationic polymers, in particular, have been shown to be a flexible system to condense DNA into nanoparticles that are effective for gene delivery (5).
A specific class of cationic polymers, poly(β-amino esters) (PBAEs), are promising as they condense DNA into nanoparticles, are biodegradable via hydrolytic cleavage of their ester groups, and have lower cytotoxicity compared to other cationic polymers such as polyethylenimine (PEI) (6–8). These polymers may act through the “proton sponge mechanism” to enable escape from the endosomal compartment to the cytoplasm (9–11). PBAEs have also been developed as gene delivery systems for the in vivo treatment of prostate cancer (12), as targeted delivery systems using electrostatic coatings of peptide ligands (13), and as transfection agents that rival the efficacy of viral gene delivery systems (14).
Methods to transfect new cell types or quantify the transfection efficiency of novel biomaterials are fundamental to the development of polymeric gene delivery vectors. This chapter discusses high-throughput methods for screening new gene delivery biomaterials while varying important design parameters such as polymer structure, DNA loading basis, and polymer to DNA weight ratio in parallel. These approaches can be used to quickly characterize and optimize new gene delivery formulations.
2. Materials
2.1. Polymer Synthesis
For synthesis of one lead polymer, poly(5-amino-1-pentanol-co-1,4-butanediol diacrylate) (referred to as C32), 5-amino-1-pentanol (Aldrich) and 1,4-butanediol diacrylate (Scientific Polymer Products, Inc.) are required. Additional amine and diacrylate monomers can be purchased and used following the same protocol to create structurally diverse polymers that are useful for gene delivery. These monomers can be purchased from Aldrich (Milwaukee, WI, USA), Scientific Polymer Products, Inc. (Ontario, NY, USA), TCI (Portland, OR, USA), Pfaltz & Bauer (Waterbury, CT, USA), Matrix Scientific (Columbia, SC, USA), and Dajac Monomer–Polymer (Feasterville, PA, USA).
Teflon-lined screw cap glass vials, 8 mL; VWR
Teflon-coated magnetic micro stir bars that fit in vials; VWR
Magnetic stir plate
Incubator/oven (95°C)
2.2. Cell Culture
2.2.1. COS-7 Cells
COS-7 cells, African green monkey kidney fibroblast-like cell line; ATCC #CRL-1651
Medium: 500 mL Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum (50 mL FBS) and 1% Penicillin–Streptomycin (5 mL); Invitrogen
Trypsin; Invitrogen
Phosphate Buffered Saline (PBS); Invitrogen
2.2.2. Human Umbilical Vein Endothelial Cells (HUVECs)
HUVECs, primary human endothelial cells; Lonza #C2519A
Medium: 500 mL Endothelial Cell Growth Medium-2 (EGM-2) medium supplemented with SingleQuot kit; Lonza
ReagentPack kit (Trypsin/EDTA, Trypsin Neutralizing Solution, HEPES Buffered Saline); Lonza
2.2.3. Additional Materials
Hemocytometer; VWR
Incubator (37°C, 5% CO2); Forma Scientific
2.3. Standard Gene Delivery Transfection
Gene delivery polymer(s). From synthesis in Subheading 2.1. and/or bought commercially such as PEI, Mw ~ 25 kDa branched from Sigma or PBAEs can be bought directly from Open Biosystems as Leopard™ Transfection Array polymers.
Dimethyl sulfoxide (DMSO), >99.7% and sterile; Sigma
Tissue culture filter unit, 500 mL, 0.2-μ cellulose acetate, sterile; Nalgene
Sodium acetate solution (3 M), pH 5.2, 0.2 μm filtered; Sigma
pEGFP DNA in water (1 mg/mL), stored at –20°C; Elim Biopharmaceuticals
Single channel pipettes (Ex: 1–10 μL, 10–100 μL, and 100–1000 μL)
Eppendorf tubes, 1.5 mL, sterile; VWR
Pipette tips (sterilized by autoclaving for 30 min at 18 psi and at 120°C)
Six-channel aspirator wand (autoclaved); V&P Scientific
Clear, sterile, tissue culture treated multi-well plates (6-well, 12-well, or 24-well, etc.)
Vortex-Genie; VWR
Fluorescent microscope and/or flow cytometer to measure green fluorescent protein (GFP) gene expression.
2.4. High-Throughput Gene Delivery Transfection
Gene delivery polymer(s). These could include materials derived from the synthesis as described in Subheading 2.1. and/or bought commercially, such as PEI, Mw ~ 25 kDa branched from Sigma, or PBAEs bought directly from Open Biosystems as Leopard™ Transfection Array polymers.
DMSO, >99.7% and sterile; Sigma
Tissue culture filter unit, 500 mL, 0.2 μ cellulose acetate, sterile; Nalgene
Sodium acetate solution (3 M), pH 5.2, 0.2 μm filtered; Sigma
pCMV-Luc DNA in water (1 mg/mL), stored at –20°C; Elim Biopharmaceuticals
Single channel pipettes (Ex: 1–10 μL, 10–100 μL, and 100–1000 μL)
Eppendorf tubes, 1.5 mL, sterile; VWR
Twelve-channel pipettes (5–50 μL and 50–300 μL); Finnpipette
Pipette tips (sterilized by autoclaving for 30 min at 18 psi and at 120°C)
Pipetting reservoirs, sterile; VWR
Twelve-channel aspirator wand (autoclaved); V&P Scientific
Clear 96-well half-area plate (sterilized by UV treatment in cell culture hood for at least 1 h); Corning #3695
White, opaque, sterile, tissue culture treated 96-well plate; Costar #3917
Clear 96-well flat-bottom plates with lids, sterile; BD Falcon #353072
Polypropylene 96-well plates (2.4 mL, V-bottom); Sigma #M1561
Bright-Glo™ kits; Promega
Firefly luciferase protein; Promega
Ninety-six-well plate luminometer to measure luciferase gene expression
3. Methods
3.1. Polymer Synthesis
Weigh 400 mg of 5-amino-1-pentanol (or other amine monomer) into a 5 mL sample vial with a teflon-lined screw cap.
Add acrylate monomer to amine monomer at an amine/diacrylate stoichiometric ratio of 1.2:1. For polymer C32, add 640 mg of 1,4-butanediol diacrylate to the 400 mg of 5-amino-1-pentanol.
Add a small teflon-coated magnetic stir bar to the vial.
To polymerize, stir the monomers on a magnetic stir plate in an oven at 95°C for 12 h (Fig. 1).
Remove polymer vial and store in the dark at 4°C until ready to use.
Fig. 1.
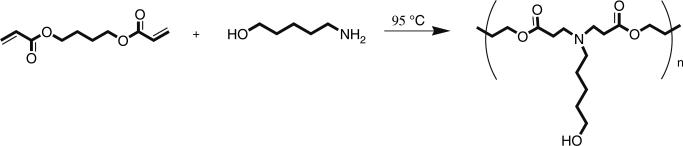
Polymerization of 1,4-butanediol diacrylate and 5-amino-1-pentanol to form gene delivery polymer C32.
3.2. Standard Transfection
Researchers should be familiar with basic sterile cell culture techniques, the ability to grow, passage, and plate cells. All work is conducted in a laminar flow biosafety cabinet using sterilized reagents and equipment.
Cell plating. Twenty-four hours prior to transfection, seed cells into a clear tissue culture multi-well plate at 150 cells/μL according to Table 1.
Preparation of polymer stock solutions. Dissolve 50 mg of each synthesized polymer in 500 μL of DMSO in a sterilized eppendorf tube. Vortex to mix to prepare 100 mg/mL polymer stock solutions. For PEI, dissolve 1 mg in 1 mL of water to prepare a 1 mg/mL PEI stock solution.
Preparation of sodium acetate buffer. To prepare sodium acetate (NaAc) buffer (pH 5.2), dilute 4.2 mL of 3 M sodium acetate into 495.8 mL of deionized water. Sterilize by vacuum filtration through a 0.2 μm filter.
Change cell media. On the day of transfection, warm fresh media in a 37°C water bath. Aspirate out the old media from each cell well and replace with equivalent volume of fresh media.
Table 1.
Cell Plating
| Wells/plate | Volume/well | Cells/well |
|---|---|---|
| 6 | 2 mL | 300,000 |
| 12 | 1 mL | 150,000 |
| 24 | 500 μL | 75,000 |
| 96 | 100 μL | 15,000 |
Note: The cells should look healthy as determined by light microscopy. Transfections are typically performed at 70–100% confluence.
-
5
DNA preparation. Thaw the DNA stock solution at room temperature. Dilute an aliquot of the 1 mg/mL DNA stock solution with 25 mM sodium acetate buffer to a final concentration of 0.060 mg/mL. For a 24-well plate, this requires 90 μg of DNA in a final volume of 1.5 mL of 25 mM sodium acetate. Aliquot out 60 μL of diluted DNA into a small volume sterile eppendorf tube for each sample to be made (24 typically).
-
6
Aqueous polymer preparation. Prior to complexation with DNA, each 100 mg/mL polymer/DMSO solution must be diluted in sodium acetate. The dilution is dependent on the final polymer:DNA weight ratios desired. For polymer C32, a weight ratio (w/w) of 30:1 polymer:DNA is typically optimal. However, this may vary with different polymers and different cell systems. Generally, it is advisable to initially try a range of weight ratios and then select the best-performing w/w for future experiments. The aqueous polymer solution should be vigorously mixed prior to use to ensure homogeneity. Table 2 shows a protocol for typical formulations in a 24-well plate format assuming polymer preparation for triplicate samples (200 μL aqueous polymer solution; for different plate formats, all volumes are scaled in proportion to the cell seeding density used in step 1). PEI is prepared analogously except that a low 1:1 polymer to DNA weight ratio is generally optimal and the PEI (and the DNA used to complex PEI) is diluted in 150 mM sodium chloride rather than 25 mM sodium acetate.
-
7
Polymer–DNA complexation/nanoparticle formation. For each polymer replicate sample, add 60 μL of polymer to an eppendorf tube containing 60 μL DNA. Mix by vortexing on a medium setting for 10 s. Time 10 min on a timer to allow for polymer/DNA self-assembly prior to use.
-
8
Add polymer/DNA nanoparticles to the cells. Prior to addition of the polymer/DNA particles, the media over the seeded cells may be optionally removed and replaced with new media with altered composition for the transfection (serum-free or high-serum media for example), although this is not required. Polymeric gene delivery particles are then added to each well according to Table 3. When finished, swirl the plate, return the cells to the incubator, and start a timer.
-
9
Remove polymer/DNA particles from the cells. Typically, particles are incubated on the cells for 1–4 h. Depending on the concentration and the toxicity of materials, in some case overnight incubations are helpful. After incubation, aspirate the polymer/DNA particles from each well using either a 6-channel aspirating wand or a Pasteur pipette. Add a volume of fresh, warm media equal to the initial cell seeding volume used in each well (i.e., 500 μL for a 24-well plate). Return the cells to the incubator.
-
10
GFP expression measurement. Alternate strategies may be used to quantify GFP gene expression. Initial study can be performed using a fluorescent microscope to measure the percentage of green, expressing cells vs. total cells counted in the bright field image. Typically, however, fluorescent activated cell sorting (FACS) is used to quickly count 10,000 or more live cells per sample and gate the GFP positive cells from the GFP negative cells. For analysis of GFP positive cells, it is recommended that two-dimensional gating rather than a one-dimensional histogram is used. For two-dimensional gating, the green channel is plotted on the x-axis and the yellow channel is plotted on the y-axis. In this manner, GFP positive cells can be better differentiated from increased background auto-fluorescence as shown in Fig. 2.
Table 2.
Polymer Preparation
| Polymer: DNA (w/w) | Polymer (μg) | Polymer/DMSO (μL) | NaAc (μL) | Polymer concentration (μg/μL) |
|---|---|---|---|---|
| 1 | 12 | 1.2 | 199 | 0.06 |
| 5 | 60 | 6.0 | 194 | 0.30 |
| 10 | 120 | 12 | 188 | 0.60 |
| 20 | 240 | 24 | 176 | 1.20 |
| 30 | 360 | 36 | 164 | 1.80 |
| 50 | 600 | 60 | 140 | 3.00 |
| 100 | 1200 | 120 | 80 | 6.00 |
Table 3.
Gene Delivery Nanoparticle Dose
| Wells/plate | Volume/well | Particle volume/well (μL) | DNA/well (μg) |
|---|---|---|---|
| 6 | 2 mL | 400 | 12 |
| 12 | 1 mL | 200 | 6 |
| 24 | 500 μL | 100 | 3 |
| 96 | 100 μL | 20 | 0.6 |
Fig. 2.

Representative HUVEC transfection efficacy of C32 and method for FACS gating. The one-dimensional histogram of C32/DNA transfected cells (56.5% positive) (left) includes some falsely positive cells that are excluded during the two-dimensional analysis of the same data set as shown by the FACS density plot gating for the negative control (0% positive) (middle) and the C32/DNA transfection (44.7% positive) (right). Ratio of GFP fluorescence (x-axis) to background fluorescence (y-axis) is used to accurately gate positive cells.
3.3. High-Throughput Screening (15)
The following protocol is for a 96-well plate testing 11 different polymers (columns 1–11 are for each polymer and column 12 is for control). Each polymer is tested in quadruplicate at two different polymer to DNA w/w ratios (20 w/w is tested in the top-half of the plate (rows A–D) and 100 w/w is tested in the bottom-half of the plate (rows E–H)).
Cell plating. Twenty-four hours prior to transfection, seed 15,000 cells/well into a white 96-well tissue culture plate.
Preparation of polymer stock solutions. Dissolve 50 mg of each synthesized polymer in 500 μL of DMSO in a sterilized eppendorf tube. Vortex to mix to prepare 100 mg/mL polymer stock solutions. For PEI, dissolve 1 mg in 1 mL of water to prepare a 1 mg/mL PEI stock solution.
Preparation of sodium acetate buffer. To prepare sodium acetate (NaAc) buffer (pH 5.2), dilute 4.2 mL of 3 M sodium acetate into 495.8 mL of deionized water. Sterilize by vacuum filtration through a 0.2 μm filter.
Plating sodium acetate buffer. First, transfer 30 mL of sodium acetate buffer to a pipetting reservoir. Using a multi-channel pipette, add 900 μL/well NaAc to row A of a 2.4 mL deep 96-well plate (Fig. 3, plate #1). Next, in a clear 96-well plate, add 176 μL/well NaAc to row A (for 20 w/w polymer to DNA particles) and 80 μL/well NaAc to row B (for 100 w/w polymer to DNA particles) (Fig. 3, plate #2).
DNA preparation and plating. Thaw the pCMV-Luc DNA stock solution at room temperature. Dilute 600 μL of the 1 mg/mL DNA stock with 9.4 mL of sodium acetate buffer in a 15 mL sterile tube. Transfer the diluted DNA solution to a pipetting reservoir and add 25 μL/well to all wells of a 96-well half-area plate (Fig. 3, plate #3).
Plating media. Warm 25 mL of media in a 37°C water bath, transfer the media to a pipetting reservoir, and add 200 μL/well to each well of a new clear 96-well plate (Fig. 3, plate #4).
Aqueous polymer preparation. For each 100 mg/mL polymer/DMSO solution and the positive control, add 100 μL of concentrated polymer to a single well (#1–12) in row A of the 2.4 mL deep 96-well plate containing 900 μL of sodium acetate buffer (Fig. 3, plate #1). Vigorously pipette the solution several times to ensure that the polymers are dissolved fully in the buffer.
Fig. 3.

High-throughput screening method.
Note: For steps #8–13, all pipetting is done using 12-channel pipettes. Change pipette tips after each use.
-
8
Polymer dilutions (Fig. 3, step 1). Add 24 μL of polymer solution from row A of the 2.4 mL deep 96-well plate (plate #1) to row A of the polymer dilution plate (plate #2) containing 176 μL of sodium acetate buffer. Similarly, add 120 μL of polymer solution from row A of the 2.4 mL deep 96-well plate (plate #1) to row B of the polymer dilution plate (plate #2) containing 80 μL of sodium acetate buffer. Vigorously pipette the solutions multiple times to ensure that they are well mixed.
-
9
Polymer–DNA complexation/nanoparticle formation(Fig. 3, step 2). For quadruplicate samples, add 25 μL of polymer from row A of the polymer dilution plate (plate #2) to 25 μL of DNA in rows A–D of the DNA plate (plate #3). Add 25 μL of polymer from row B of the polymer dilution plate (plate #2) to 25 μL of DNA in rows E–H of the DNA plate (plate #3). For each addition, vigorously pipette the solution several times to promote polymer/DNA self-assembly. To generate reproducible particles, it is important to be consistent with the mixing technique. Start a 5 min timer once the plate is finished.
-
10
Polymer/DNA particles dilution (Fig. 3, step 3). At the end of 5 min, add 30 μL of polymer/DNA particles from row A of the DNA plate (plate #3) to row A of the media plate (plate #4) containing 200 μL of media. Repeat for rows B–H.
-
11
Add polymer/DNA particles to the cells (Fig. 3, step 4). Use a 12-channel aspiration wand to remove the media from the cells seeded into the white 96-well plate previously (step #1). Add 150 μL of polymer/DNA particles from row A of the media plate (plate #4) to row A of the cell plate. Repeat for rows B–H, being careful to pipette against the side of the wells as opposed to directly over top the cells which can dislodge the cells if not careful.
-
12
Remove polymer/DNA particles from the cells. After a 1–4 h incubation time, aspirate the polymer/DNA solution from the cells and add 100 μL of warm fresh media to each well. Return the cells to the incubator.
-
13
Luciferase protein assay. This assay is generally performed 2–3 days post-transfection. Thaw the Bright-Glo™ Luciferase Assay Kit by warming the buffer in a room temperature water bath. Remove the cells from the incubator and allow them to equilibrate to room temperature as well. Add the buffer to the substrate vial, re-cap, invert to mix, and then dispense into a reservoir. Using a multi-channel pipette, add 100 μL of the solution to each well of the cell plate. After the last addition, time 2 min on a timer. Place the plate on an orbital shaker or manually shake to promote mixing during this time. After 2 min, measure the luminescence in a plate reader using an integration time of 1 s/well. Compare the relative luminescence to a standard curve to obtain the mass of luciferase protein in each well.
4. Notes
In general, any adherent cell type may be used following these same protocols. COS-7 and HUVEC cell protocols are shown as examples of an easier-to-transfect mammalian cell line and more difficult-to-transfect human primary cells, respectively. Typical transfection of COS-7 cells using polymer C32 is 90–100% whereas for HUVECs it is 40–50%. ~—100% where for HUVECs it is~—50%.
Different-sized plasmid DNA may be used to create these polymer/DNA particles. For easy visual inspection of gene expression and quantitative cell population information enhanced green fluorescent protein (EGFP) DNA is often used. For high-throughput screening applications, luciferase (Luc) DNA is often used instead. For high-throughput screening applications, luciferase (Luc) DNA is often used instead.
It is important that the cationic polymers are added to the anionic DNA, rather than the reverse. It is also important that the two components are very well mixed. We suggest vortexing at medium speed rather than simply pipetting up and down. The optimal self-assembly incubation time is between 10 and 15 min. Leaving the assembled polymer/DNA particles for longer than 40 min before use may reduce activity. A slightly shorter self-assembly waiting time is typically used when performing high-throughput methods.
If for a given application transfection is lower than desired, increase the polymer to DNA weight ratio and/or the DNA loading basis per cell well. For example, in a 24-well plate use a 6 μg DNA/well basis instead of a 3 μg DNA/well basis. Additionally, increasing the time that the polymer/DNA particles incubate with the cells or reducing the serum concentration can increase efficacy. Lastly, reducing the cell confluence to 50–70% confluent at the time of transfection can increase efficacy.
If for a given application cytotoxicity occurs, decrease the polymer to DNA weight ratio and/or the DNA loading basis per cell well.
High-Throughput Screening Advantages/Limitations:
4.1. Advantages
Allows for testing of hundreds of polymers, in quadruplicate, in a single day
Minimizes amount of reagents used
4.2. Limitations
Many pipette tip boxes and plate types must be stocked and sterilized
Mixing, during parallel particle formation, by pipettes may not be as vigorous as done by vortexing. This can affect particle self-assembly in some instances.
References
- 1.Partridge KA, Oreffo ROC. Gene delivery in bone tissue engineering: Progress and prospects using viral and nonviral strategies. Tissue Eng. 2004;10:295–307. doi: 10.1089/107632704322791934. [DOI] [PubMed] [Google Scholar]
- 2.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 3.Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 4.Merdan T, Kopecek J, Kissel T. Prospects for cationic polymers in gene and oligonucleotide therapy against cancer. Adv. Drug Delivery Rev. 2002;54:715–758. doi: 10.1016/s0169-409x(02)00046-7. [DOI] [PubMed] [Google Scholar]
- 5.Putnam D. Polymers for gene delivery across length scales. Nat. Mater. 2006;5:439–451. doi: 10.1038/nmat1645. [DOI] [PubMed] [Google Scholar]
- 6.Lynn DM, Langer R. Degradable poly(beta-amino esters): Synthesis, characterization, and self-assembly with plasmid DNA. J. Am. Chem. Soc. 2000;122:10761–10768. [Google Scholar]
- 7.Green JJ, Shi J, Chiu E, Leshchiner ES, Langer R, Anderson DG. Biodegradable polymeric vectors for gene delivery to human endothelial cells. Bioconjug. Chem. 2006;17:1162–1169. doi: 10.1021/bc0600968. [DOI] [PubMed] [Google Scholar]
- 8.Anderson DG, Lynn DM, Langer R. Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Ang. Chem. Int. Edn. 2003;42:3153–3158. doi: 10.1002/anie.200351244. [DOI] [PubMed] [Google Scholar]
- 9.Akinc A, Langer R. Measuring the pH environment of DNA delivered using nonviral vectors: Implications for lysosomal trafficking. Biotechnol. Bioeng. 2002;78:503–508. doi: 10.1002/bit.20215. [DOI] [PubMed] [Google Scholar]
- 10.Sonawane ND, Szoka FC, Verkman AS. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J. Biol. Chem. 2003;278:44826–44831. doi: 10.1074/jbc.M308643200. [DOI] [PubMed] [Google Scholar]
- 11.Akinc A, Thomas M, Klibanov AM, Langer R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005;7:657–663. doi: 10.1002/jgm.696. [DOI] [PubMed] [Google Scholar]
- 12.Anderson DG, Peng WD, Akinc A, Hossain N, Kohn A, Padera R, Langer R, Sawicki JA. A polymer library approach to suicide gene therapy for cancer. Proc. Natl. Acad. Sci. U.S.A. 2004;101:16028–16033. doi: 10.1073/pnas.0407218101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green JJ, Chiu E, Leshchiner ES, Shi J, Langer R, Anderson DG. Electrostatic ligand coatings of nanoparticles enable ligand-specific gene delivery to human primary cells. Nano Lett. 2007;7:874–879. doi: 10.1021/nl062395b. [DOI] [PubMed] [Google Scholar]
- 14.Green JJ, Zugates GT, Tedford NC, Huang Y, Griffith LG, Lauffenburger DA, Sawicki JA, Langer R, Anderson DG. Combinatorial modification of degradable polymers enables transfection of human cells comparable to adenovirus. Adv. Mater. 2007;19(19):2836–2842. [Google Scholar]
- 15.Zugates GT, Anderson DG, Langer R. High-throughput methods for screening polymeric transfection reagents. In: Friedmann T, Rossi J, editors. Gene Transfer: Delivery and Expression of DNA and RNA. Cold Spring Harbor Laboratory Press; New York, NY: 2007. pp. 547–54. [DOI] [PubMed] [Google Scholar]